Method Article
Flusso di lavoro completo per l'analisi dell'istone modificazioni post-traduzionali Utilizzando Bottom-up Spettrometria di Massa: Da istone estrazione all'analisi dei dati
In questo articolo
Riepilogo
Questo protocollo descrive un flusso di lavoro completamente integrato per la caratterizzazione di istone modificazioni post-traslazionali utilizzando la spettrometria di massa (MS). Il flusso di lavoro prevede la purificazione istone da colture di cellule o tessuti, istone derivatizzazione e la digestione, analisi MS utilizzando nano-flusso cromatografia liquida e le istruzioni per l'analisi dei dati. Il protocollo è stato progettato per il completamento entro 2 - 3 giorni.
Abstract
Nucleosomi sono la più piccola unità strutturale della cromatina, composto da 147 coppie di basi di DNA avvolte attorno ad un ottamero di proteine istoni. funzione Histone è mediata da una vasta modificazione post-traduzionale da una miriade di proteine nucleari. Queste modifiche sono fondamentali per l'integrità nucleare come regolano cromatina struttura e reclutare enzimi coinvolti nella regolazione genica, la riparazione del DNA e cromosomi condensa. Anche se una gran parte della comunità scientifica adotta tecniche a base di anticorpi per caratterizzare istone PTM abbondanza, questi approcci sono bassi throughput e prevenuto contro le proteine hypermodified, come l'epitopo potrebbe essere ostacolato da modifiche nelle vicinanze. Questo protocollo descrive l'uso della cromatografia liquida nano (nLC) e spettrometria di massa (MS) per un'accurata quantificazione modificazioni degli istoni. Questo metodo è stato progettato per caratterizzare una grande varietà di PTM istone e la relativa abbondanza di diversi istone varianti all'interno di sanalisi ingle. In questo protocollo, gli istoni sono derivatizzati con anidride propionica seguita da digestione con tripsina per generare peptidi di 5-20 aa di lunghezza. Dopo la digestione, il che viene mostrata N-terminali dei peptidi istone sono derivatizzato per migliorare la ritenzione cromatografica durante NLC-MS. Questo metodo consente la quantificazione relativa dei PTM istone abbracciano quattro ordini di grandezza.
Introduzione
Epigenetica è definita come lo studio delle variazioni ereditarie nella espressione genica che sorgono da meccanismi diversi da alterare la sequenza sottostante 1. regolazione epigenetica è un fattore critico durante lo sviluppo come l'organismo subisce drammatico fenotipica cambia anche se il suo contenuto di DNA non cambia. Ci sono diversi componenti critici necessari per una corretta manutenzione epigenetica, tra cui istone modificazioni post-traduzionali (PTM), le varianti istone, RNA non codificanti, la metilazione del DNA e fattori di legame al DNA, ognuno dei quali influenzano l'espressione genica attraverso diversi meccanismi 2. Per esempio, mentre la metilazione del DNA è una modificazione altamente stabile che reprime traduzione gene 3, varianti istoni e PTMs istoni sono molto più dinamico e può influenzare cromatina in una varietà di modi 4.
PTM istoni sono prevalentemente localizzati sulle code N-terminale, in quanto sono la regione più esposta e flessibiledella proteina. Tuttavia, il nucleo nucleosoma è anche fortemente modificata rispetto alle proteine medi 5. Anche se i marchi istoni sono stati ampiamente caratterizzato negli ultimi dieci anni, molti collegamenti tra marchi istoni noti e la loro funzione non sono ancora chiare. Ciò è dovuto al fatto che la maggior parte PTMs istone non operano da soli, ma funzioni in tandem con altri PTM ( "cross-talk") alterare un processo specifico come trascrizione 6,7. Ad esempio, il contrassegno H3S10K14ac combinatoria sul gene p21 attiva la trascrizione, che non si verificherebbe con uno solo dei due PTM 8. I patti proteina HP1 cromatina riconoscendo H3K9me2 / ME3 e diffondere la modifica nucleosomi vicini. Tuttavia, HP1 non può legare H3K9me2 / 3 quando l'adiacente S10 è fosforilata 9. Acetilazione di H3K4 inibisce il legame della proteina spChp1 a H3K9me2 / ME3 in SCHIZOSACCHAROMYCES POMBE 10. Inoltre, l'istone lisina demethylase PHF8 ha la più alta efficienza di legame nucleosoma quando tre PTM H3K4me3, K9ac e K14ac sono presenti 11. Questi esempi mettono in evidenza l'importanza di raggiungere una visione globale dei cambiamenti PTM istone piuttosto che concentrarsi su singole modifiche.
La presenza della sequenza di varianti aumenta anche la complessità dell'analisi istoni, come isotipi istoni hanno generalmente sequenze altamente simili, ma spesso hanno ruoli diversi nella cromatina. Ad esempio, H2A.x ha una sequenza C-terminale che viene più facilmente fosforilata sul danno al DNA rispetto al H2A canonica 12, ed è necessario per l'inattivazione dei cromosomi sessuali in meiosi maschile del mouse 13; Allo stesso modo, CENP-A sostituisce canonica H3 istone in centromeri 14. Nonostante le loro diverse funzioni, queste varianti condividono una grande parte della loro sequenza aminoacidica con la rispettiva istone canonica, rendendo difficile identificare e quantificare separatamente.
tecniche a base di anticorpi, come western blotting sono stati ampiamente adottati per caratterizzare istoni. Tuttavia, approcci basati su anticorpi sono limitati per i seguenti motivi: (i) possono confermare la presenza di una modifica e non possono identificare PTMs sconosciuti; (Ii) sono polarizzati a causa della presenza di segni coesistenti, che può influenzare affinità di legame; (iii) non possono identificare segni combinatori, come solo pochissimi anticorpi sono disponibili per tale scopo e (iv) attraversare-reagiscono tra le varianti istoni altamente simili o PTM simili (ad esempio, di- e trimethylation di residui di lisina). Egelhofer et al. ha descritto che più del 25% degli anticorpi commerciali non superano i test di specificità di Dot Blot o Western Blot, e tra gli anticorpi specifici oltre il 20% non in esperimenti di immunoprecipitazione della cromatina 15. La spettrometria di massa (MS) è convertita in uno analitico più adatto per studiare nuovi e / o combinatori PTM,ed è stato ampiamente implementato per istoni (recensiti 16). Ciò è dovuto principalmente alla elevata sensibilità ed accuratezza di massa di MS, e la possibilità di eseguire analisi su larga scala.
La strategia bottom-up è il più comunemente usato strategia proteomica MS-based per la caratterizzazione degli istoni e loro PTM, in cui la proteina intatta è enzimaticamente digerito in peptidi brevi (5-20 aa). Questo facilita sia la digestione separazione LC e la rilevazione MS. Masse nell'intervallo 600 - 2.000 Da sono comunemente più facilmente ionizzati e identificati con maggiore precisione massa e risoluzione di grandi masse. MS / MS frammentazione è anche migliorata, come peptidi sono generalmente ben adatti per la dissociazione di collisione indotta (CID). Tuttavia, istoni rappresentano una sfida per la bottom-up MS in quanto sono altamente arricchito nei residui di aminoacidi essenziali, vale a dire lisina ed arginina. Pertanto, la digestione tripsina porta alla generazione di peptidi che sono troppo smtutto per la conservazione e la localizzazione LC inequivocabile del PTM. Per aggirare questo problema, il nostro protocollo comprende lisina e peptide chimico N-terminale derivatizzazione 17. L'uso di anidride propionica è raccomandato efficiente derivatizzazione chimica rispetto ad altri reagenti 18. Tali blocchi derivatizzazione dei gruppi Ɛ-amino di residui di lisina non modificate e monometilici, permettendo tripsina eseguire proteolisi solo al C-terminale di residui di arginina. ammine derivatizzato non possono scambiarsi protoni con la soluzione e quindi i peptidi sono generalmente solo doppiamente o triplamente carica, facilitando MS e rilevazione MS / MS. Inoltre, derivatizzazione N-terminale aumenta peptide idrofobia e la ritenzione cromatografica quindi in fase inversa. Qui, descriviamo il flusso di lavoro per purificare istoni e li prepara per l'analisi PTM tramite proteomica bottom-up (Figura 1). Questa strategia realizza quantificazione dei marchi istoni singole e marchi combinatori Fo PTMs istoni che sono relativamente vicini nella sequenza aminoacidica.
Protocollo
1. Raccolta delle cellule dalla cultura
- Se le cellule sono state coltivate in sospensione, raccogliere le cellule per centrifugazione a 300 rcf per 5 min. Se aderenti, aspirare e scartare medio delle cellule. Lavare le cellule attaccate con PBS senza Ca 2+ e Mg 2+ (denominato PBS andare avanti). Incubare le cellule in entrambi tripsina o tripsina-EDTA (0,025% - 0,5% a seconda della linea cellulare) con un volume sufficiente a coprire la superficie delle piastre a 37 ° C finché le cellule staccano (tempo varia per differenti linee cellulari).
- Raccogliere le cellule per centrifugazione a 300 rcf per 5 min. Lavare le cellule altre due volte in PBS e raccogliere per centrifugazione.
- Stimare l'ematocrito circa dalle graduazioni segnate sui tubi 1,8 ml o 15 ml provette coniche.
Nota: Le celle della cultura possono essere snap-congelati in azoto liquido e conservati a -80 ° C a tempo indeterminato in questa fase.
2. Isolamento dei nuclei da cellule intatte
- cellule Scongelare su ghiaccio.
- Scongelare buffer di isolamento nucleare (NIB, Tabella 1).
- Preparare circa 5 ml NIB tampone (Tabella 1) per ogni ematocrito 100 ml. Per ogni 1 ml NIB tampone, aggiungere inibitori della proteasi e agenti stabilizzanti come segue: 1 ml di 1 M DTT, 2,5 ml di 200 mm AEBSF, 2 ml di 2,5 micron microcystin e 2 ml di 5 M butirrato di sodio. NIB con inibitori verrà indicato come NIB da questo punto in avanti.
Nota: Se istone fosforilazione è studiato, includere EDTA proteasi gratuito e cocktail di inibitori di fosfatasi. - Rimuovere un quinto volume di tampone NIB così preparata e aggiungere NP-40 rinnovabili (Tabella 1) ad una concentrazione finale di 0,2%. Il volume restante quattro quinti sarà utilizzato per lavaggi.
- Lavare pellet cellulare 1:10 pellet cellulare a NIB senza NP-40 Rapporto rinnovabili (v / v). Rimuovere il surnatante per centrifugazione a 700 rcf per 5 min.
- Lyse il cell pellet appoggiandolo su ghiaccio e aggiungendo pellet 01:10 cella NIB con 0,2% NP-40 rinnovabili (v / v).
- Se l'estrazione da campioni di tessuto, omogeneizzare con omogeneizzatori mortaio e pestello o Dounce. cellule in coltura possono essere omogeneizzati delicatamente pipettando.
- Incubare cellule omogeneizzati in ghiaccio per 5 - 10 minuti. Le cellule saranno lisi e rilasciare i nuclei.
- Centrifugare a 1000 rcf per 5 - 10 minuti a 4 ° C. Il pellet contiene principalmente cella nuclei, mentre il surnatante contiene componenti prevalentemente citoplasmatici. Salvare la frazione citoplasmatica se lo si desidera.
- Lavare il pellet nuclei risospendendo delicatamente 1:10 (v / v) NIB senza NP-40 Alternative.
Nota: Questa fase di lavaggio è unicamente quello di rimuovere le tracce di detergenti prima di estrarre istoni da nuclei. - Centrifugare a 1000 rcf per 5 minuti a 4 ° C e rimuovere il surnatante.
- Ripetere il passo 2.10 - 2.11 almeno due volte per rimuovere completamente NP-40 Alternative. Rimozione di NP-40 Alternative è evidente uns delicato pipettaggio durante la fase di lavaggio non costituisce più bolle.
- Per l'estrazione istone dai tessuti:
- Risciacquare tessuto congelato fresco o congelato in ghiacciata NIB.
- Trasferire il tessuto di una capsula di Petri immessi sul ghiaccio con NIB, quel tanto che basta per mantenere il tessuto bagnato.
- Dice in pezzi più piccoli (<1 mm) con lama di rasoio per aumentare la superficie di contatto per l'isolamento dei nuclei.
- Trasferire il tessuto tritato ad un omogeneizzatore pre-raffreddata e lavare in NIB pipettando su e giù.
- Rimuovere tampone mediante centrifugazione a 300 rcf per 5 min.
- Aggiungere NIB contenente NP-40 Alternative alle cellule in cellule: buffer rapporto 1:10 (v / v) e omogeneizzare per 5 - 10 colpi.
- Verificare la lisi cellulare e ripetere l'omogeneizzazione, se necessario. Un buon indicatore che le cellule sono state lisate è la riduzione del volume pellet. Il pellet deve contenere solo i nuclei.
- Centrifugare a 700 rcf per 5 minuti e salvare pellet. Questo pellet può essere estratto 1 - 2 più tempos 1:10 (v / v) di NIB contenente NP-40 Alternative; in questa fase, gli istoni sono estratti dalla cromatina e il pellet è rimpicciolito notevolmente.
- Lavare due volte con 2 - 3 ml di NIB senza NP40 alternativo per rimuovere le tracce di detergente.
Nota: punto di arresto intermedio: campione può essere risospeso nel volume minimo di NIB + 5% glicerolo, e conservato a -80 ° C.
3. Estrazione e purificazione di istoni da Nuclei
Nota: Gli istoni sono molto ricchi di residui di aminoacidi essenziali, permettendo loro di strettamente interagire con la spina dorsale acido fosforico del DNA. Gli istoni sono tra le proteine più elementari nel nucleo, consentendo loro di essere estratti in acido solforico ghiacciata (0,2 MH 2 SO 4) con la contaminazione minima da proteine non istoni, che precipitano in acido forte. TCA alta concentrazione (ad una concentrazione finale del 33%) può quindi essere utilizzato per precipitare istoni dalla solforicoacido. TCA è memorizzato come 100% in bottiglia scura a 4 ° C.
- Nuclei delle cellule Risospendere in 1: 5 (v / v) refrigerati 0.2 MH 2 SO 4 (Tabella 1) delicatamente pipettando.
- Incubare il campione con rotazione costante o delicata agitazione per 2 - 4 ore a 4 ° C. Tipicamente, per campioni con più di 500 microlitri pellet cellulare, un'estrazione 2 ore è sufficiente estrarre istoni; incubazione più lungo può comportare estrazione di altre proteine basiche. Per pellet nucleari più piccole (<200 ml), 4 ore di estrazione fornisce una resa migliore.
- Centrifugare a 3.400 rcf a 4 ° C per 5 min.
- Trasferire il surnatante in una nuova provetta.
- Ripetere i passaggi 3,3-3,4 per rimuovere il materiale insolubile.
- Per precipitare istoni, aggiungere refrigerate 100% TCA (Tabella 1) per il supernatante raccolto (contenente ora istoni) nel rapporto di 1: 3 (v / v), in modo da ottenere una concentrazione TCA finale del 33%. Mescolare invertendo il tubo di un paio di tIME.
Nota: I campioni si accende nuvoloso su aggiunta di TCA, che indica la presenza di istoni. - Incubare la miscela in ghiaccio per almeno 1 ora. Per i più piccoli formati di partenza pellet, si consiglia di precipitazioni durante la notte.
- Centrifugare a 3.400 rcf per 5 min. Il mantello istoni lati dei tubi e anche depositare sul fondo. Un pellet insolubili bianco costituisce anche nella parte inferiore del tubo, che contiene principalmente proteine non-istoni e altre biomolecole. Rimuovere il surnatante tramite aspirazione, con attenzione senza raschiare i lati o il pellet.
- Usando una pipetta Pasteur di vetro, risciacquare il tubo con ghiacciata acetone + 0,1% HCl (Tabella 1) in modo da coprire le proteine precipitate rivestimento lati e sul fondo.
- Centrifugare a 3.400 rcf per 2 minuti e aspirare il surnatante, con attenzione senza raschiare i lati o il pellet.
- Ripetere i passaggi 3,9-3,10 utilizzando il 100% di acetone ghiacciata.
- pellet secco con flusso d'aria o con un vacuum centrifugare, o semplicemente lasciando il tubo aperto. Acetone evapora rapidamente.
- Sciogliere gli istoni con DDH 2 O (acqua bidistillata) a volume minimo possibili per sciogliere completamente lo strato bianco. Gli istoni sono facilmente solubili in acqua. Per pellets in una provetta da 1,5 ml microcentrifuga, 100 ml DDH 2 O è di solito sufficiente per raccogliere istoni.
- Centrifugare a 3.400 rcf per 2 minuti e trasferire il surnatante in una nuova provetta.
4. Stima della concentrazione della proteina e Purezza
- Per la misura di concentrazione di proteine, utilizzare BCA, Metodo di Bradford o l'analisi degli aminoacidi (AAA). Non usare tecniche che adottano assorbanza a 280 nm, come gli istoni sono poveri in residui di amminoacidi aromatici.
- Verificare la purezza degli istoni estratti mediante analisi SDS-PAGE con un gel di acrilammide 15% e Coomassie colorazione (opzionale).
- Se elevata purezza varianti singolo istoni si desiderano, continuare a frazionamento HPLC-UV divarianti istone (sezione 5). In caso contrario, passare direttamente alla preparazione del campione per l'analisi istone PTM bottom-up (sezione 6).
5. Separazione delle istone varianti da HPLC in fase inversa (opzionale)
Nota: varianti istoni elevata purezza possono essere ottenuti per frazionamento miscela istone grezza usando HPLC a fase inversa accoppiata ad un rivelatore UV. Questi istoni purificati sono utili per studi che richiedono una maggiore sensibilità e purezza. Tuttavia, per la caratterizzazione istone PTM di serie, questo passaggio può essere saltato perché l'analisi è sufficientemente sensibile ed esaustivo. Frazionamento di istone intatto varianti idealmente richiede almeno 100 - 300 mg di materiale di partenza.
- Collegare un'apposita colonna C 18 5 micron a un HPLC a seconda della concentrazione partendo istone: con circa 100 mg di istoni, UTILIZZO 2.1 mm x 250 mm di colonna con una portata di 0,2 ml / min; con circa 300 mcg istoni, utilizzare colonna 4,6 x 250 mmuna portata di 0,8 ml / min. Preparare tampone A e B utilizzando vetreria dedicata come segue:
- Preparare tampone A: 5% per HPLC acetonitrile, 0,1% TFA in acqua di grado HPLC.
- Preparare tampone B: 95% per HPLC acetonitrile, 0,1% TFA in acqua di grado HPLC.
- Collegare la colonnina di un rivelatore UV, e impostare l'assorbanza a 210-220 nm.
- Acidificare campione istone disciolto in acqua con 100% TFA per ottenere una concentrazione finale di 0,1-1% TFA.
- Equilibrare la colonna con 100% tampone A per almeno 15 min alla portata raccomandata, che corrisponde approssimativamente a tre volumi di colonna. Utilizzare questo segnale per impostare il livello zero di assorbanza del rivelatore UV.
- Preparare le provette di dimensioni appropriate per raccogliere frazioni manualmente o in un raccoglitore automatico di campioni.
- Iniettare campione ad una concentrazione di circa 1 mg / mL o superiore. I campioni disciolti in volumi più grandi potrebbero alterare l'equilibrio della colonna duanello di carico e portare a ritenzione inferiore.
- Eseguire il gradiente, programmato come segue: da 0 a 30% B in 1 min, 30 a 60% B in 90 min, e da 60 a 90% B in 1 min.
- Raccogliere frazioni (esempio cromatogramma mostrati in figura 2) a intervalli di 1 minuto usando un collettore automatica frazione. Raccogliere frazioni in tubi di dimensioni adeguate a contenere l'intero volume.
- Asciugare giù campioni frazionati in un concentratore a vuoto.
Nota: tappa intermedia: frazioni istoni secchi possono essere conservati a temperatura ambiente per periodi brevi (1 - 2 giorni) o in freezer -80 ° C per periodi di lunga durata.
6. chimica Derivatizzazione degli istoni Utilizzando propionico Anidride per l'analisi bottom-up
- (Quantità consigliata: 50 - 100 mg) Sciogliere campioni istone in 40 ml di 50 mM NH 4 HCO 3, pH 8,0. Se i campioni sono stati in puro DDH 2 O, aggiungere concentrato NH 4 HCO 3 per compensare 50 mm, pH 8.0.
- Bagnare una punta P10 pipetta nel campione per controllare il pH utilizzando strisce indicatore di pH, senza perdite di campione. NH 4 OH e acido formico possono essere usati per regolare il pH a 8,0.
Nota: La seguente parte del protocollo (passi 6.3 - 6.7) dovrebbe essere fatto in lotti di massima tre a quattro campioni, al fine di mantenere anidride propionica reattiva. - Utilizzare cappa per le fasi successive in cui viene utilizzato anidride propionica. Preparare reagente propionylation fresca miscelando anidride propionica con acetonitrile in rapporto 1: 3 (v / v). Aggiungere propionylation reagente al campione in 1: 4 (v / v). Per 40 istoni microlitri, aggiungere 10 ml di reagente propionylation.
Nota: È possibile osservare detriti bianco in questa fase. Tuttavia, questo contiene principalmente sali e acido propionico, e quindi nessuna azione specifica deve essere presa. - Aggiungere rapidamente NH 4 OH ristabilire pH 8,0 alla soluzione. Nota: anidride propionica reagire con le ammine libere dei peptidi produce propacido ionico che diminuisce il pH. Solitamente, aggiungendo NH 4 OH al campione con un rapporto di 1: 5 (v / v) è appropriato per ristabilire pH 8,0, ad esempio, 8 ml di NH 4 OH al 40 microlitri di campione.
- Mescolare immediatamente nel vortex.
- Controllare il pH con la stessa procedura Fase 6.2.
Attenzione: Quando il pH è superiore a 10,0, etichettatura di altri residui amminoacidici con maggiore pKa è possibile. - Incubare campioni a temperatura ambiente per 15 min.
- Ripetere i passaggi 6,3-6,7, rigorosamente eseguendo la reazione per non più di 3 o 4 campioni per lotto di reagenti propionylation.
- campioni a secco fino al 10 - 20 ml in un concentratore a vuoto. Questo evapora reagita anidride propionica, acetonitrile, acido acetico e ammoniaca come gas liberato dal NH 4 OH. Se i campioni asciugano completamente, nessuna perdita campione significativo si verificano.
Nota: isopropanolo può essere usato al posto di acetonitrile. Tuttavia, acetonitrile ha bassa tensione superficiale e quindi piùrapida evaporazione. - Campioni Risospendere o diluire con DDH 2 O fino a 40 ml di volume finale si ottiene.
- Ripetere i passaggi 6,2-6,9. Un doppio giro di istone propionylation assicura> 95% di completamento reazione.
- Riempire la bottiglia anidride propionica con gas argon in modo da evitare la formazione di acido acetico a contatto con l'umidità nella bottiglia.
Nota: tappa intermedia: Il campione può essere conservato a -80 ° C ricostituito in DDH 2 O o secchi.
7. proteolitici digestione con tripsina
- Istoni Risospendere in 50 mM NH 4 HCO 3 per ottenere una concentrazione ottimale di 1 mg / mL o superiore. Più campioni diluiti inducono una diminuzione dell'efficienza tripsina.
Nota: I istoni in questa fase devono essere a pH 8,0 Se ancora acida, quindi aggiungere NH 4 HCO 3 sale a campione utilizzando punta della pipetta. - Aggiungere tripsina i campioni istoni in un rapporto 1:10 (peso / peso).
- incubate a 37 ° C per 6 - 8 ore.
- Fermare la digestione congelando in -80 ° C.
- Essiccare giù il campione a 10 - 20 microlitri in un concentratore a vuoto.
Nota: tappa intermedia: Il campione può essere conservato a -80 ° C.
8. Propionylation dell'istone peptidi a N-Termini
Nota: Questa sezione descrive la derivatizzazione di peptide N-termini generata dalla tripsina digest. Tale procedura migliora la ritenzione HPLC dei peptidi più brevi (ad esempio, amminoacidi 3 - 8 di istone H3), come il gruppo propionile aumenta peptide idrofobicità.
- Campioni Risospendere in 30 ml di 100 mM NH 4 HCO 3.
- Ripetere i passaggi 6,1-6,9.
Nota: È normale che l'essiccazione dei campioni sotto vuoto richiede un tempo più lungo in questa fase. - Risospendere o diluire i campioni con 50 - 100 ml DDH 2 O + o 0,1% TFA o 0,5% di acido acetico. Nota: L'acido acetico è consigliato per lunghi depositi, come TFA agevolarnees ossidazione metionina nel lungo termine. D'altra parte, TFA è raccomandato se viene eseguita stage-ribaltamento (sezione 9) lo stesso giorno, come TFA assiste una migliore ritenzione cromatografica.
Nota: tappa intermedia: Il campione può essere conservato a -80 ° C.
9. Desalting campione con Stage-punte
Nota: In questa fase, c'è il sale presente nel campione. Sali impediscono analisi HPLC-MS perché ionizzano durante elettrospray, sopprimendo il segnale da peptidi. Sali possono anche formare addotti ionici su peptidi, riducendo l'intensità del segnale per il peptide non-addotto. Come il peptide addotto avrà una massa diversa, il peptide non sarà adeguatamente identificati o quantificare.
- Usando un puntale P1000, pugno un disco di C 18 materiale da un disco di estrazione in fase solida. Premere il minidisco dalla punta P1000 usando un capillare di silice fusa e depositare il minidisc al fondo di un P100 / 200 punta della pipetta. Assicurarsi che il disk è incuneata saldamente nella parte inferiore della punta (Figura 3).
Nota: La punta P1000 ha un piuttosto piccolo foro per perforare il disco C 18. È opportuno tagliare all'ultimo centimetro della punta in modo da avere un foro di diametro maggiore. - Utilizzare due C 18 colpi nella stessa punta P100 / P200 se la dissalazione più di 25 mg di campione.
- Utilizzare un adattatore centrifuga per tenere stage punte in atto in 1,5 ml o 2 tubi ml microcentrifuga. Utilizzare lento - rotazione (300 400 RCF); i solventi normalmente passano attraverso la resina in meno di un minuto.
- Lavare la resina mediante filatura con 50 ml di acetonitrile 100% per attivare il materiale C 18 e rimuovere eventuali contaminazioni.
- Equilibrare disk da vampate di calore 80 ml di 0,1% TFA per lenta centrifugazione.
- Acidificare il campione a pH 4,0 o inferiore con acido acetico. Controllare il pH con le strisce di pH per ridurre al minimo la perdita di campione. campione del carico sul disco da una lenta centrifugazione.
- lavaggiocampione irrigando 70 - 80 ml di 0,1% TFA lenta centrifugazione.
- Eluire campione da vampate di calore 70 microlitri 75% acetonitrile e acido acetico 0,5% in lenta centrifugazione. Raccogliere il campione in una provetta da 1,5 ml.
- campione secco in un concentratore a vuoto.
Nota: tappa intermedia: Il campione può essere conservato a -80 ° C.
10. Analisi dell'istone Peptidi
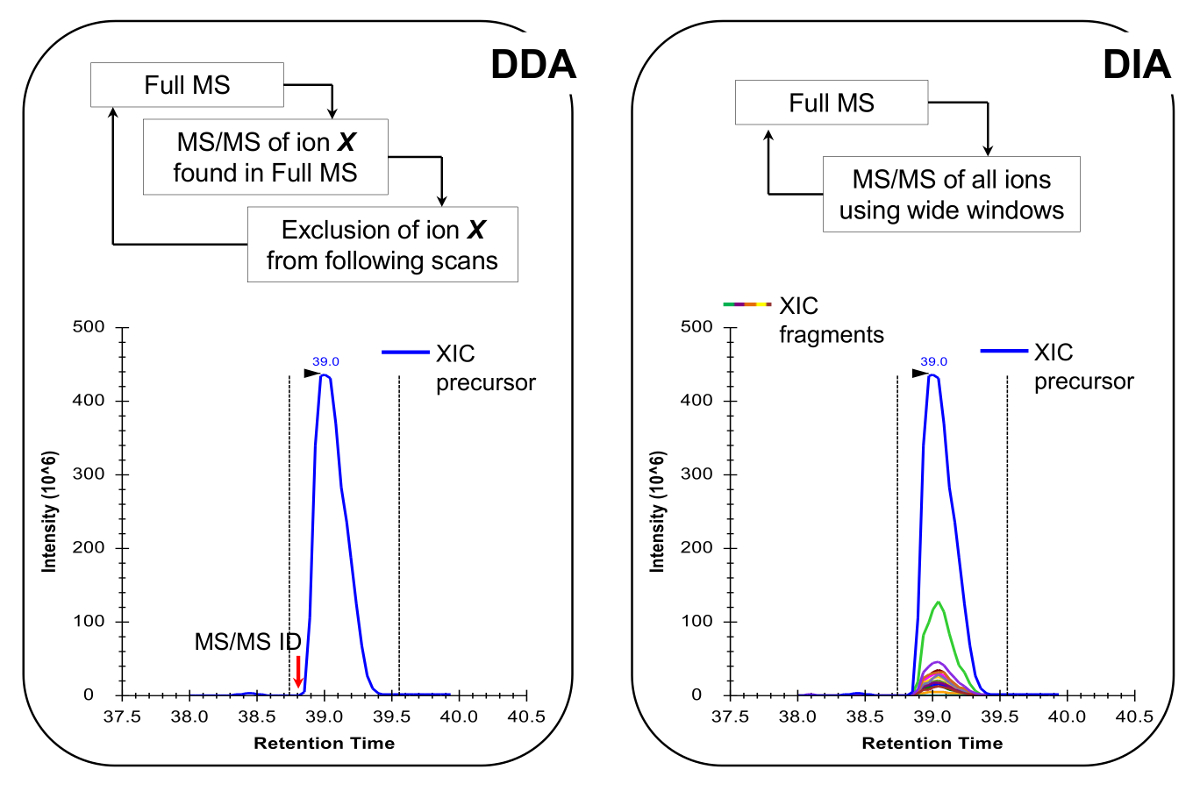
Nota: La piattaforma NLC-MS dovrebbe essere impostato come fatto in analisi peptide tradizionale. Si raccomanda 300 colonna flusso nl (75 micron ID colonna analitica, C 18 particelle), in quanto sono un ottimo compromesso tra sensibilità e stabilità - L'uso di 200. Il metodo di acquisizione MS può essere una combinazione di acquisizione dati-dipendente (DDA) con scansioni mirate 19 o un data-indipendente acquisizione (DIA) 20,21, entrambi descritti in Risultati rappresentativi e Figura 4.
- Preparare buffer HPLC - A: acido formico 0,1% inacqua HPLC-grade; B: acido formico 0,1% in HPLC-grade acetonitrile.
- Programmare il metodo HPLC come segue: da 0 a 30% tampone B in 30 min, dal 30 al 100% B per il prossimo 5 min ea isocratica 100% B per 8 min. Se la HPLC non è programmato per equilibrazione colonna automatico prima caricamento del campione, quindi includere il seguente: gradiente da 100 a 0% B in 1 min e flusso isocratica allo 0% B per 10 min. Impostare la portata dell'analisi a 250 - 300 nl / min.
- Programmare il metodo di acquisizione MS per eseguire una DDA combinata con scansioni mirate 19 o DIA 20,21 (Figura 4). Assicurarsi che il ciclo di lavoro MS permette piena MS scansione ogni ~ 2 sec, per avere punti dati sufficienti per tutto il picco cromatografico, che consente una quantificazione più accurata. Nota: Con C 18 cromatografia, la larghezza media basale di picco è di circa 30 secondi per il gradiente sopra descritto.
- Caricare circa 1 mg di campione sul HPLC column.
- Eseguire il metodo HPLC-MS / MS come programmato.
Nota: Nel protocollo si sconsiglia specifiche di colonne, strumenti MS o MS particolari parametri, come qualsiasi setup ottimale che un individuo proteomica laboratorio sviluppato sarà adatto per il metodo. laboratori Proteomica dovrebbero usare la loro messa a punto ottimizzata, in quanto peptidi istoni separato come peptidi tradizionali.
Analisi 11. Dati
- Importare i file grezzi MS in software per eseguire integrazione dell'area di picco. Nota: EpiProfile 22 è consigliata, in quanto è ottimizzato per i peptidi istone; utilizzando la conoscenza del tempo di ritenzione eluizione cromatografica esegue affidabile estrazione di peptidi istone noti area del picco. In alternativa, Skyline 23 è un altro software ideale per lo scopo.
- Calcolare l'abbondanza relativa di un dato peptide dividendo la sua superficie per la superficie totale di detto peptide in tutte le sue forme modificate. Nota: In caso di scoperta analysis Mascot Si raccomanda di identificare gli spettri di peptidi istoni modificati. Le prestazioni di questo strumento è stato descritto di recente 24. Tutti gli altri motori di ricerca di database per la proteomica sono anche utilizzabili, ma dopo i nostri test hanno fornito prestazioni inferiori.
Risultati
A titolo di esempio, abbiamo analizzato gli istoni estratti da cellule staminali embrionali umane (hESC) con e senza acido retinoico (RA) stimolazione, a partire da 200 microlitri pellet cellulari. La presenza di RA in coltura cellulare porta alla differenziazione ESC. Dal pellet cellulare, circa 50 - 100 mg di istoni sono stati estratti, che è più che sufficiente per eseguire iniezioni multiple LC-MS di peptidi istoni. Dopo derivatizzazione, la digestione, e dissalazione, i campioni sono stati caricati su un 75 micron x 15 centimetri C 18 colonna (diametro delle particelle 3 micron, dimensioni dei pori 300 A) in modalità seriale con un sistema di liquido nano cromatografia ad alte prestazioni con i chip microfluidica accoppiate a un ibrido lineare trappola quadrupolo - spettrometro di massa Orbitrap. acquisizione MS è stata effettuata utilizzando DIA. In parallelo, i campioni sono stati anche analizzati con un metodo DDA utilizzando un UHPLC nano-flusso accoppiato ad uno spettrometro di massa trap-Orbitrap ione ibrido (dati non mostrati). Inogni ciclo, una rilevazione completa MS Orbitrap è stata effettuata con la gamma di scansione di 290 a 1.400 m / z, una risoluzione di 60.000 (a 200 m / z) e AGC di 10 6. Poi, i dati modalità di acquisizione dipendente è stato applicato con una dinamica esclusione di 30 sec. scansioni MS / MS sono stati seguiti in ioni genitore da quelle più intense. Gli ioni con stato di carica di uno sono stati esclusi da MS / MS. È stato utilizzato un window isolamento di 2 m / z. Gli ioni erano frammentati utilizzando collisione indotta dissociazione (CID) con l'energia di collisione del 35%. Rilevamento trappola ionica è stato utilizzato con modalità normale intervallo di scansione e velocità di scansione normale con AGC di 10 4.
I dati grezzi MS sono stati analizzati adottando il software per l'estrazione di precursori e di ioni frammento cromatogrammi, vale a dire Skyline 23 e 22 EpiProfile. EpiProfile è stato ottimizzato per i peptidi istoni, in quanto integra intelligente estrazione area del picco a causa di una precedente conoscenza di Peptempo di ritenzione marea. D'altra parte, Skyline è ottimizzato per le analisi DIA, e quindi le figure DIA visualizzati (Figure 4 e 5A) sono schermate da questo software. Dal cromatogramma ionico estratta, l'area sotto la curva viene recuperato, e questo viene utilizzato per stimare l'abbondanza di ciascun peptide. L'area del picco cromatografico è stato calcolato per il [M + H] + [M + 2H] 2+, e [M + 3H] 3+ ioni dello stesso peptide, sebbene nella maggior parte dei casi la [M + 2H] 2+ era la forma prevalente. Questo fornisce l'abbondanza grezzo di una data forma modificata di un peptide. Al fine di raggiungere la relativa abbondanza di PTM, la somma di tutte le diverse forme modificate di un peptide istone stato considerato come 100%, e la zona della particolare peptide è stato diviso per l'area totale di tale peptide istone in tutte le sue forme modificate .
peptidi istoni sono presenti in un VArie di forme isobariche (Figura 5). Peptidi isobariche, ad esempio, K18ac e K23ac, possono essere quantificati solo a livello MS / MS, in cui vengono utilizzati i loro frammenti di ioni uniche per determinare il rapporto della specie isobariche (Figura 5A e 5B). Questo rapporto è utilizzato per dividere l'area del picco cromatografico tra le due specie. Quando si utilizzano DDA, queste forme isobariche sono stati inclusi in un elenco di masse mirati, perché questi peptidi hanno bisogno di essere selezionati per la frammentazione attraverso tutta la loro eluizione, che non si verificherebbe in un esperimento di serie DDA. La discriminazione di abbondanza relativa delle specie isobariche viene eseguita controllando il profilo di eluizione dei frammenti di ioni. D'altra parte, DIA tipo di acquisizione non richiede alcuna elenco di inclusione. Tuttavia, questo tipo di metodo di acquisizione non è compatibile con la ricerca di database tradizionali, e quindi potrebbe impedire la scoperta di peptidi modificati sconosciuti.
lisina acetilazione (+ 42,011 Da) è stata discriminata dalla trimethylation quasi isobarica (+ 42,047 Da) utilizzando alta risoluzione di acquisizione MS (> 30.000). Inoltre, acetilazione è più idrofobico di trimethylation, portando a eluizione di peptidi acetilati entro tale quelli Trimethylated. La forma non modificata dello stesso peptide eluisce anche in seguito, a causa del fatto che la lisina propionylated. In sintesi, l'ordine delle idrofobicità per un peptide con un sito modificabile è di- e Trimethylated
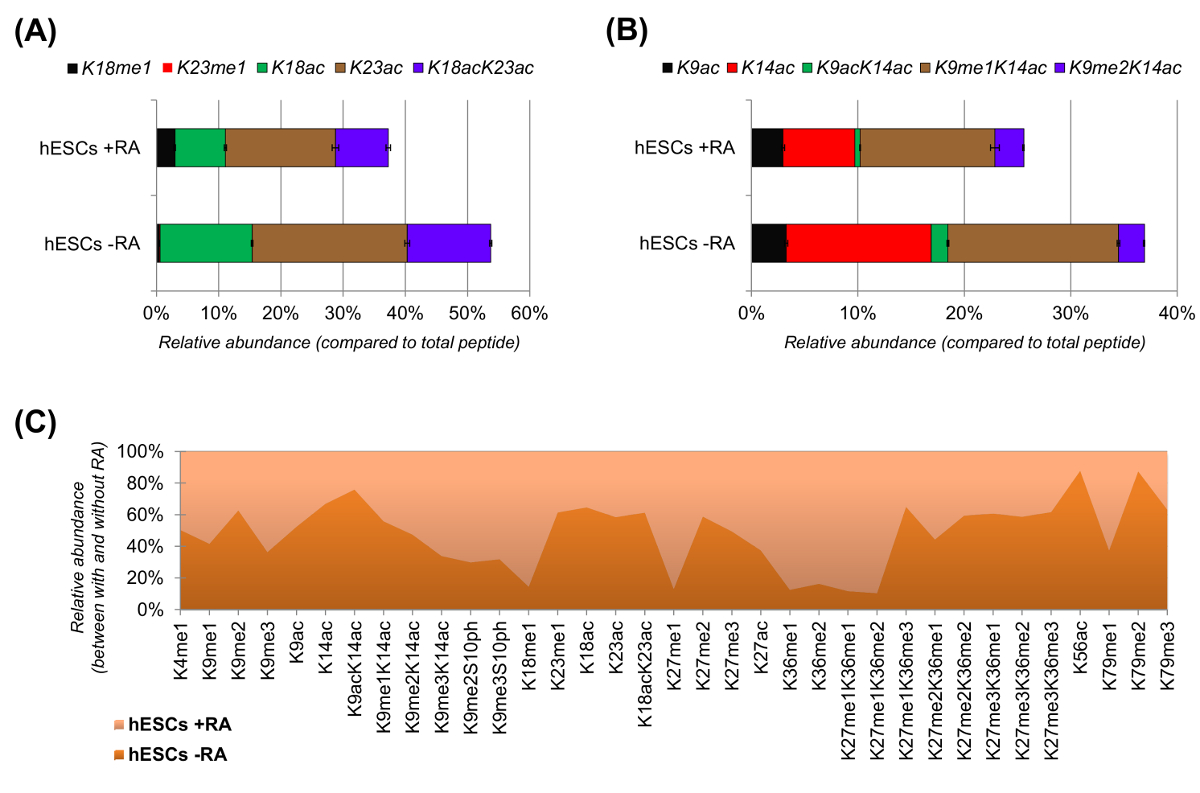
hESC hanno mostrato una netta riduzione dei peptidi acetilati quando stimolato per la differenziazione (Figura 6A e 6B). Questo non è sorprendente, come i risultati precedenti riportati superiore acetilazione in CES rispetto a quelli di differenziazione 25,che riflette la natura in generale permissivo della cromatina pluripotenti. Focalizzando l'attenzione su istone H3, 35 diverse forme modificate sono stati quantificati (Figura 6C). Tuttavia, tutti proteoforms istoni che possono essere studiati con questo approccio sono più di 200, comprese tutte le varianti e modifiche istoni abbondanza basse (dati non mostrati). Inoltre, la nostra analisi ha mostrato che un'alta riproducibilità può essere ottenuta tra le repliche tecniche, come dimostra la piccola dimensione delle barre di errore rappresentano (± deviazione standard). Nel loro insieme, questa sezione descrive come estrarre la relativa abbondanza di istoni modificati peptidi utilizzando i dati NLC-MS.

Figura 1:. Flusso di lavoro per MS / MS istoni analisi bottom-up I dieci passi per l'analisi degli istoni sono mostrati, tra cui una stima del tempo necessario a ogni passo. Il numero di sezione è dato tra parentesi come presente nel manoscritto. Sezione 5, descrivendo campione frazionamento di isolare le diverse varianti degli istoni, può essere omesso se non vi è la necessità di analisi ad alta sensibilità di un dato variante. Clicca qui per vedere una versione più grande di questa figura.
{kind=link}

Figura 2: in fase inversa ad alto flusso LC per istone Variante Frazionamento e Coomassie Gel (A) cromatogramma LC-UV che rappresenta intatta la separazione degli istoni.. varianti dell'istone H3 possono essere discriminati gli uni dagli altri in base al loro tempo di eluizione. Le frazioni possono essere raccolti manualmente o utilizzando un raccoglitore di frazioni automatizzato. (B) Coomassie del gel di tre repliche di purificazione istoni.= "Https://www.jove.com/files/ftp_upload/54112/54112fig2large.jpg" target = "_ blank"> Clicca qui per vedere una versione più grande di questa figura.

Figura 3:. Making of Stage-ribaltamento spina con un puntale P1000, pugno un disco fatto di C 18 materiale da un disco di estrazione in fase solida (secondo pannello). Il minidisk si attacchi nella punta (pannello centrale), in modo che possa essere spinto fuori in un più piccolo P100 / 200 puntale utilizzando qualsiasi tipo di piccole capillare. In questo esempio, abbiamo usato un diametro esterno tubo di silice fusa 700 micron. Il minidisk deve essere spinto verso il fondo del P100 / 200 punta della pipetta finché non può andare oltre (ultimo pannello). La punta palcoscenico è pronto per l'istone dissalazione, in quanto ha una capacità sufficiente per mantenere il campione di materiale sufficiente per numerose repliche. In particolare, uno minidisk è sufficiente per 15 - 20 mg di sample. Se è necessario più del campione, più dischi possono essere imballati gli uni sugli altri. Si prega di cliccare qui per vedere una versione più grande di questa figura.
{kind=link}

Figura 4:. Rappresentazione schematica della DDA e DIA metodi quando utilizza DDA, il ciclo di scansione SM è caratterizzata dalla selezione sequenziale di ioni precursori per MS / MS frammentazione base alla loro intensità e stato di carica. Una volta che uno ione precursore è stato frammentato è ubicato all'interno di un elenco di esclusione per evitare selezione ripetitiva dello stesso peptide, in modo che la MS può "scavare" in segnali meno abbondanti. Questo metodo di acquisizione è la tecnica di scelta nella proteomica per la modalità di scoperta. La quantificazione è ottenuta integrando il segnale di scansione completa di un determinato ione accanto alle MS identificati /spettro MS. In DIA, l'intera gamma di m / z è frammentato ad ogni ciclo di scansione. Questo approccio è meno adatto per la modalità di scoperta, ma produce un profilo cromatografico di tutte ioni, precursori e prodotti. Questo porta ad una quantificazione più sicuri e la discriminazione delle forme isobariche. Clicca qui per vedere una versione più grande di questa figura.
{kind=link}

Figura 5: Quantificazione di isobariche peptidi (A) Esempio di due peptidi isobariche comunemente abbondanti nell'analisi istone.. Il cromatogramma ionico estratto (XIC) della loro massa e relativi precursori isotopi (sopra) è identico. Tuttavia, la XIC degli ioni prodotto (sotto) consente la discriminazione delle due forme isobariche. In particolare, gli ioni frammento solo unico deve essere noiEd per stimare l'abbondanza relativa delle due specie. (B) Rappresentazione degli ioni frammento uniche per i due peptidi descritti (evidenziato in rosso). (C) Lista dei peptidi comunemente analizzati in Homo sapiens che hanno almeno un equivalente isobarica. Sequenza varianti tra i peptidi istone elencati sono indicati. Si prega di cliccare qui per vedere una versione più grande di questa figura.
{kind=link}

Figura 6: Risultati rappresentativi di cellule staminali embrionali umane con e senza retinoico trattamento acido (A) quantificazione relativa della istone H3 peptide KQLATKAAR (aa 18 - 26), in tutte le sue proteoforms modificati.. La relativa abbondanza è stato stimato utilizzando tutti proteoforms come 100% (relpercentuale ativo del peptide non modificato non è mostrato) (B) quantificazione relativa della istone H3 peptide KSTGGKAPR (aa 9 -.. 17) (C) abbondanza relativa dei peptidi identificati per canonica H3 istone con e senza trattamento delle cellule con acido retinoico. La figura indica in quale delle due trattamenti le date modifiche sono più abbondanti (> 50%). Nel complesso, abbiamo dimostrato che l'acetilazione dell'istone H3 diminuisce nella maggior parte dei residui di lisina su induzione della differenziazione cellulare. Si prega di cliccare qui per vedere una versione più grande di questa figura.
{kind=link}
| soluzione # | Composizione | ||||||
| 1 | Isolamento nucleare Buffer (NIB) stock è composto come segue e conservato congelato a 100 ml aliquote a -20 ° C; scongelatiNIB può essere conservato a 4 ° C per alcune settimane: 15 mM Tris, 60 mM KCl, 15 mM NaCl, 5 mM MgCl 2, 1 mM CaCl 2, e 250 mM di saccarosio. Il pH del tampone viene regolato a 7,5 con HCl. | ||||||
| 2 | Inibitori della proteasi (aggiungere fresco di buffer prima dell'uso): 1 M ditiotreitolo (DTT) in DDH 2 O (1,000x); 200 mm AEBSF in DDH 2 O (400x) | ||||||
| 3 | inibitore fosfatasi (aggiungere fresco di buffer prima dell'uso): 2,5 micron microcistina in 100% di etanolo (500x) | ||||||
| 4 | inibitori HDAC (aggiungere fresco di buffer prima dell'uso): 5 M di sodio butirrato, realizzati da una titolazione di acido butirrico 5 M con NaOH a pH 7.0 (500x) | ||||||
| 5 | NP-40 Alternativa: 10% v / v in DDH 2 O | ||||||
| 6 | 0.2 MH 2 SO 4 in DDH 2 O | ||||||
| 7 | Acido tricloroacetico (TCA): 100% w / v in DDH 2 O | ||||||
| 8 | Acetone + 0,1% di acido cloridrico (HCl): 0,1% v / v HCl in acetone | ||||||
Tabella 1. Solutions.
Discussione
Il protocollo qui descritto è ottimizzato considerare i costi, il tempo e le prestazioni. Altre preparazioni sono possibili, ma hanno limitazioni, soprattutto nel caso di accoppiamento con analisi MS. Per esempio, il protocollo di estrazione ad alta sale può essere usato per purificare istoni 26 invece di TCA precipitazioni (punto 3). protocollo di alto sale è intrinsecamente più lieve, in quanto non utilizza acido forte. Questo conserva PTM acido-labili e aumenta il rendimento degli istoni estratti, come TCA precipitazioni co-precipitati molte altre proteine di legame della cromatina. Tuttavia, l'estrazione alto contenuto di sale porta a campioni contenenti il sale troppo concentrato per HPLC-MS / MS. In una preparazione alternativa, istone digestione può essere eseguita senza propionylation (sezione 6 - 8), ad esempio riducendo il tempo tripsina incubazione e il rapporto enzima / substrato 27 o usando ARGC la digestione enzimatica 28-30. Tuttavia, si raccomanda derivatizzazione con anidride propionica, come hot porta alla generazione di peptidi più idrofobi, che sono meglio mantenuti durante cromatografia liquida.
Per derivatizzazione chimica, una varietà di anidridi di acidi organici sono stati valutati e loro meriti ampiamente discusso 18. Tuttavia, anidride propionica dimostrò il miglior compromesso tra efficienza, prodotti laterali attenuate e migliore idrofobicità peptide. Potenzialmente, anidride propionica può essere acquistato in forma marcati con isotopi; ciò consente un'analisi multiplexing a causa della possibilità di miscelazione più campioni e discriminare al livello MS sulla base delle differenti masse impartiti dall'etichetta pesante. Tuttavia, tale analisi porta ad una maggiore complessità del cromatogramma LC-MS e riduce la quantità di campione che può essere iniettato per ogni singola condizione.
A questo proposito, alcuni aspetti critici del protocollo devono essere evidenziati. Il seguente dovrebbe essere utilizzato come checklist per trovare errori nello svolgimento della procedura nel caso in cui si ottengono risultati negativi. In primo luogo, dopo precipitazione nuclei il pellet deve essere accuratamente lavato con NIB senza NP-40 Alternative (sezione 2.10) fino alla rimozione completa del detergente (evidente dalla mancanza di bolle durante la miscelazione). Non riuscendo a farlo comprometterebbe l'estrazione istone con acidi. In secondo luogo, dopo l'istone precipitazione con TCA (punto 3.9) lavaggi del pellet con acetone è cruciale. La presenza di acido concentrato avrebbe danneggiato il passo successivo, se propionylation e la digestione (paragrafo 6.1) vengono eseguite direttamente. Sarebbe non problematico in caso di frazionamento istone (sezione 5) viene eseguita. In terzo luogo, è essenziale che la reazione propionylation viene eseguita rapidamente (paragrafo 6.3 - 6.7). Per fare ciò, evitare di utilizzare lo stesso mix propionylation (propionico anidride + acetonitrile) per più di 3 - 4 campioni consecutivi. Inoltre, il pH è l'aspetto più importante della tripsina digestione (sezione 7). Altrimentiintorno a 8,0 (7,5-8,5) la digestione sarà inefficace. Questo può accadere, come il campione sarà ricco di acido propionico in questa fase. NH 4 OH può essere aggiunto finché non è necessario. Inoltre, per i ricercatori che hanno familiarità con i flussi di lavoro di proteomica si sentirà normale per acidificare il campione di interrompere tripsina digestione. Questo non dovrebbe essere fatto, in quanto metterà a repentaglio la seguente reazione, vale a dire, propionylation di peptide N-Termini (paragrafo 8.1). Infine, nello stesso numero, è importante da ricordare per l'analisi dei dati che i peptidi non modificati, non sono in realtà non modificato; Tutti i residui liberi di lisina e N-termini saranno occupati da propionylation (56,026 Da). Così, eseguendo l'estrazione cromatografia ionica della massa corrispondente univoco alla sequenza peptidica porterebbe ad alcun risultato.
I limiti del metodo sono per lo più legati alla incapacità di rilevare PTM combinatorie, a causa delle brevi sequenze peptidiche, ei pregiudizi per raggiungere il vero Abundanza di una modifica, dovuta al fatto che i peptidi in diverse forme modificate possono ionizzare con diversa efficienza. Il primo problema può essere risolto mediante la combinazione di questa tecnica con una media verso il basso o approccio top-down (rivisto in 16). Questo tipo di analisi, anche se tecnicamente più impegnativo, è ideale per studiare le frequenze di coesistenza di modifiche. Inoltre, esso consente una migliore discriminazione di varianti istoni, che non sempre possono essere conseguiti con bottom-up poiché alcuni peptidi hanno la stessa sequenza in diverse varianti istoni. Il secondo problema, legato alla efficienza di ionizzazione, può essere risolto utilizzando una libreria di peptidi sintetici 31. Questo approccio garantisce una stima più accurata della relativa abbondanza di PTM istoni. Tuttavia, nella maggior parte degli esperimenti, il risultato desiderato è le variazioni relative dei trovati modifiche tra condizioni analizzati. In questo caso, tale correzione non è necessaria, dovuta al fatto che tutti i campioni hanno gli stessi biaS.
In conclusione, questo protocollo permette l'analisi di PTM istoni che può essere completato in 3 giorni utilizzando nLC accoppiato a tandem MS. I confronti con tecniche diverse da MS, cioè, utilizzando strategie di anticorpi base come discusso nell'Introduzione, non sono adatti, in quanto non possono ottenere ancora quasi questo livello di produttività. Inoltre, le tecniche di anticorpi base non consentono la scoperta di nuovi modifiche, ma si basano esclusivamente su conferma e quantificazione contrassegni previsti. Abbiamo quindi ipotizzare che la proteomica bottom-up su peptidi istoni si guadagnare popolarità in laboratori proteomica grazie ai vantaggi intuitivi a conoscere la regolamentazione dei marchi istoni, che sono protagonisti di espressione genica messa a punto e quindi influenzare la regolazione del proteoma. Inoltre, il protocollo descritto include i recenti miglioramenti nella preparazione dei campioni e software per l'analisi dei dati, che rendono l'analisi istone più banale anche per laboraTories che mai sperimentato caratterizzazione di questo tipo di peptidi hypermodified.
Divulgazioni
Gli autori dichiarano di non avere interessi finanziari concorrenti.
Riconoscimenti
Questo lavoro è stato sostenuto da un finanziamento dal NIH sovvenzioni (DP2OD007447, R01GM110174 e R01AI118891).
Materiali
| Name | Company | Catalog Number | Comments |
| Trypsin 0.25% EDTA | Invitrogen | 25200056 | For harvesting cells |
| PBS | Invitrogen | 14200075 | |
| Tris | Roche | 77-86-1 | |
| Potassium Chloride | Fisher Scientific | BP366-500 | |
| Sodium Chloride | Sigma | S9888 | |
| Magnesium Chloride hexahydrate | Sigma | M9272 | |
| Calcium Chloride, anhydrous | Sigma | C1016 | |
| Sucrose | Fisher Scientific | BP220-1 | |
| DTT | Invitrogen | 15508-013 | |
| AEBSF | EMD Millipore Corp | 101500 | |
| Microcystin | Sigma | M4194 | |
| Sodium Butyrate | Sigma | B5887 | |
| Halt Protease and Phosphatase Inhibitor Cocktail, EDTA-free (100X) | Fisher Scientific | 78445 | |
| NP-40 Alternative | CALBIOCHEM | 492016 | |
| Sulfuric Acid, ACS grade | Fisher Chemical | 7664-93-9 | |
| Trichloroacetic acid | Sigma | T6399 | |
| Acetone | Sigma | 179124 | |
| HCl | Fisher Chemical | A144-500 | |
| Bradford reagent | Biorad | 500-0006 | |
| 30% acrylamide/bis 29:1 -- 500ml | Biorad | 1610156 | |
| Coomassie | Fisher Scientific | 20278 | |
| C18 Column (5um) 2.1mm x 250mm | Grace | 218TP52 | |
| C18 Column (5um) 4.6mm x 250mm | Grace | 218TP54 | |
| HPLC grade acetonitrile | Fisher Chemical | A955-4 | |
| HPLC grade water | Fisher Scientific | W6 4 | |
| TFA | Fisher Scientific | A11650 | |
| Ammonium Bicarbonate | Sigma | A6141 | |
| ammonium hydroxide | Sigma | 338818 | |
| propionic anhydride | Sigma | 240311 | |
| Sequencing grade modified trypsin | Promega | PRV5113 | For digesting histones for MS |
| Acetic Acid | Sigma | 49199 | |
| C18 extraction disk | Empore | 2215 | |
| Formic Acid | Sigma | F0507 |
Riferimenti
- Waddington, C. H. Canalization of development and the inheritance of acquired characters. Nature. 150, 563-565 (1942).
- Sharma, S., Kelly, T. K., Jones, P. A. Epigenetics in cancer. Carcinogenesis. 31 (1), 27-36 (2010).
- Reik, W., Dean, W., Walter, J. Epigenetic reprogramming in mammalian development. Science. 293 (5532), 1089-1093 (2001).
- Kouzarides, T. Chromatin modifications and their function. Cell. 128 (4), 693-705 (2007).
- Tessarz, P., Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Bio. 15 (11), 703-708 (2014).
- Fischle, W., Wang, Y. M., Allis, C. D. Histone and chromatin cross-talk. Curr Opin Cell Biol. 15 (2), 172-183 (2003).
- Lee, J. S., Smith, E., Shilatifard, A. The language of histone crosstalk. Cell. 142 (5), 682-685 (2010).
- Simboeck, E., et al. A Phosphorylation Switch Regulates the Transcriptional Activation of Cell Cycle Regulator p21 by Histone Deacetylase Inhibitors. J Biol Chem. 285 (52), 41062-41073 (2010).
- Hirota, T., Lipp, J. J., Toh, B. H., Peters, J. M. Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature. 438 (7071), 1176-1180 (2005).
- Xhemalce, B., Kouzarides, T. A chromodomain switch mediated by histone H3 Lys 4 acetylation regulates heterochromatin assembly. Genes Dev. 24 (7), 647-652 (2010).
- Vermeulen, M., et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. 142 (6), 967-980 (2010).
- van Attikum, H., Gasser, S. M. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol. 19 (5), 207-217 (2009).
- Fernandez-Capetillo, O., et al. H2AX is required for chromatin remodeling and inactivation of sex chromosomes in male mouse meiosis. Dev Cell. 4 (4), 497-508 (2003).
- Santaguida, S., Musacchio, A. The life and miracles of kinetochores. Embo J. 28 (17), 2511-2531 (2009).
- Egelhofer, T. A., et al. An assessment of histone-modification antibody quality. Nat Struct Mol Biol. 18 (1), 91-93 (2011).
- Sidoli, S., Cheng, L., Jensen, O. N. Proteomics in chromatin biology and epigenetics: Elucidation of post-translational modifications of histone proteins by mass spectrometry. J Proteomics. 75 (12), 3419-3433 (2012).
- Plazas-Mayorca, M. D., et al. One-Pot Shotgun Quantitative Mass Spectrometry Characterization of Histones. J Proteome Res. 8 (11), 5367-5374 (2009).
- Sidoli, S., et al. Drawbacks in the use of unconventional hydrophobic anhydrides for histone derivatization in bottom-up proteomics PTM analysis. Proteomics. 15 (9), 1459-1469 (2015).
- Lin, S., Garcia, B. A. Examining histone posttranslational modification patterns by high-resolution mass spectrometry. Methods Enzymol. 512, 3-28 (2012).
- Sidoli, S., et al. SWATH Analysis for Characterization and Quantification of Histone Post-translational Modifications. Mol Cell Proteomics. , (2015).
- Krautkramer, K. A., Reiter, L., Denu, J. M., Dowell, J. A. Quantification of SAHA-Dependent Changes in Histone Modifications Using Data-Independent Acquisition Mass Spectrometry. J Proteome Res. , (2015).
- Yuan, Z. F., et al. EpiProfile Quantifies Histone Peptides With Modifications by Extracting Retention Time and Intensity in High-resolution Mass Spectra. Mol Cell Proteomics. 14 (6), 1696-1707 (2015).
- MacLean, B., et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 26 (7), 966-968 (2010).
- Yuan, Z. F., Lin, S., Molden, R. C., Garcia, B. A. Evaluation of proteomic search engines for the analysis of histone modifications. J Proteome Res. 13 (10), 4470-4478 (2014).
- Tan, Y., Xue, Y., Song, C., Grunstein, M. Acetylated histone H3K56 interacts with Oct4 to promote mouse embryonic stem cell pluripotency. Proc Natl Acad Sci U S A. 110 (28), 11493-11498 (2013).
- Vonholt, C., et al. Isolation and Characterization of Histones. Methods Enzymol. 170, 431-523 (1989).
- Zhang, K. L., et al. Identification of acetylation and methylation sites of histone H3 from chicken erythrocytes by high-accuracy matrix-assisted laser desorption ionization-time-of-flight, matrix-assisted laser desorption ionization-postsource decay, and nanoelectrospray ionization tandem mass spectrometry. Anal. Biochem. 306 (2), 259-269 (2002).
- Jufvas, A., Stralfors, P., Vener, A. V. Histone Variants and Their Post-Translational Modifications in Primary Human Fat Cells. Plos One. 6 (1), e15960 (2011).
- Bonaldi, T., Imhof, A., Regula, J. T. A combination of different mass spectroscopic techniques for the analysis of dynamic changes of histone modifications. Proteomics. 4 (5), 1382-1396 (2004).
- Zhao, X. L., et al. Comparative Proteomic Analysis of Histone Post-translational Modifications upon Ischemia/Reperfusion-Induced Retinal Injury. J Proteome Res. 13 (4), 2175-2186 (2014).
- Lin, S., et al. Stable-isotope-labeled histone peptide library for histone post-translational modification and variant quantification by mass spectrometry. Mol Cell Proteomics. 13 (9), 2450-2466 (2014).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati