
Method Article
Flux de travail complet pour l'analyse des histones modifications post-traductionnelles utilisant la spectrométrie de masse Bottom-up: De Histone Extraction à l'analyse des données
Dans cet article
Résumé
Ce protocole décrit un flux de travail entièrement intégrée pour caractériser histone modifications post-traductionnelles par spectrométrie de masse (MS). Le flux de travail comprend histone purification à partir de cultures de cellules ou de tissus, histone dérivatisation et la digestion, l'analyse de MS en utilisant la chromatographie et des instructions pour l'analyse des données de liquide nano-flux. Le protocole est conçu pour l'achèvement dans les 2 - 3 jours.
Résumé
Les nucléosomes sont l'unité structurelle la plus petite de la chromatine, composé de 147 paires de bases d'ADN enroulé autour d'un octamère d'histones. fonction Histone est médiée par une vaste modification post-traductionnelle par une myriade de protéines nucléaires. Ces modifications sont essentielles pour l'intégrité nucléaire comme ils régulent les enzymes de structure et de recruter chromatine impliqués dans la régulation des gènes, réparation de l'ADN et la condensation des chromosomes. Même si une grande partie de la communauté scientifique adopte des techniques à base d'anticorps pour caractériser histone PTM abondance, ces approches sont bas débit et des préjugés contre les protéines hypermodified, comme l'épitope peut être obstrué par des modifications à proximité. Ce protocole décrit l'utilisation d'une chromatographie liquide nano (NLC) et la spectrométrie de masse (MS) pour la quantification précise des modifications des histones. Cette méthode est conçue pour caractériser une grande variété de PTM histones et l'abondance relative de plusieurs variantes histone dans les sIngle analyse. Dans ce protocole, les histones sont dérivatisés avec de l'anhydride propionique suivie par une digestion avec de la trypsine pour produire des peptides de 5 à 20 aa en longueur. Après digestion, la N-terminales nouvellement exposé des peptides histones sont dérivatisé pour améliorer la rétention chromatographique pendant NLC-MS. Ce procédé permet la quantification relative de l'histone PTM sur quatre ordres de grandeur.
Introduction
L' épigénétique est définie comme l'étude des changements héréditaires dans l' expression des gènes qui se posent par des mécanismes autres que la modification de la séquence d'ADN sous - jacente 1. La régulation épigénétique est critique au cours du développement que l'organisme subit phénotypique dramatique change même si sa teneur en ADN ne change pas. Il y a plusieurs composants critiques nécessaires à l' entretien épigénétique appropriée, y compris les modifications des histones post-traductionnelles (PTM), des variants d'histones, des ARN non codantes, méthylation de l' ADN et les facteurs de liaison d' ADN, chacun qui affectent l' expression des gènes par le biais de différents mécanismes 2. Par exemple, alors que la méthylation d' ADN est une modification très stable qui réprime la traduction du gène 3, des variants d'histones et PTM des histones sont beaucoup plus dynamiques et peuvent influencer la chromatine dans une variété de façons 4.
PTM histones sont principalement localisés sur les queues de N-terminal, car ils sont la région la plus exposée et flexiblede la protéine. Cependant, le noyau nucléosome est également fortement modifié par rapport aux protéines moyenne 5. Même si les marques histones ont été caractérisées en détail dans la dernière décennie, de nombreux liens entre les marques d'histone connues et leur fonction sont encore peu claires. Ceci est largement dû au fait que la plupart des PTM histones ne travaillent pas seuls, mais plutôt la fonction en tandem avec d' autres PTM ( "cross-talk") pour modifier un processus spécifique, comme la transcription 6,7. Par exemple, la marque H3S10K14ac combinatoire sur le gène p21 active sa transcription, qui ne se produirait pas avec un seul des deux PTM 8. Les comprimés de protéine HP1 chromatine en reconnaissant H3K9me2 / me3 et la diffusion de la modification de nucléosomes à proximité. Cependant, HP1 ne peut pas lier H3K9me2 / 3 lorsque le S10 adjacent est phosphorylée 9. Acétylation de H3K4 inhibe la liaison de la protéine à spChp1 H3K9me2 / ME3 à 10 Schizosaccharomyces pombe. En outre, l'histone lysine demethylase PHF8 a la plus haute efficacité de liaison de nucléosome lorsque trois PTM H3K4me3, K9ac et K14ac sont présents 11. Ces exemples mettent en évidence l'importance de parvenir à une vision globale des changements de PTM histones plutôt que de se concentrer sur les modifications simples.
La présence de variants de séquence augmente également la complexité de l'analyse des histones, comme isotypes d'histones ont généralement des séquences très similaires, mais ont souvent des rôles différents dans la chromatine. Par exemple, H2A.X a une séquence C-terminale qui est plus facilement phosphorylée sur les dommages de l' ADN par rapport à H2A canonique 12, et il est nécessaire pour l' inactivation des chromosomes sexuels dans la méiose de la souris mâle 13; de même, CENP-A substitue canonique histone H3 dans centromères 14. En dépit de leurs différentes fonctions, ces variants partagent une grande partie de leur séquence d'acides aminés avec l'histone canonique respective, ce qui rend difficile d'identifier et de quantifier séparément.
techniques à base d'anticorps, tels que Western blot ont été largement adoptées pour caractériser histones. Cependant, les approches basées sur les anticorps sont limitées pour les raisons suivantes: (i) ils ne peuvent confirmer la présence d'une modification et ne peuvent pas identifier PTM inconnus; (Ii) elles sont sollicitées en raison de la présence de marques coexistantes, ce qui peut influencer l'affinité de liaison; (iii) ils ne peuvent pas identifier les marques combinatoires, comme très peu d' anticorps sont disponibles à cette fin et (iv) ils réagissent de manière croisée entre les variants d'histones très similaires ou PTM similaires (par exemple, di- et trimethylation des résidus de lysine). Egelhofer et al. décrit que plus de 25% des anticorps commerciaux échec des tests de spécificité par dot - blot ou transfert de Western, et parmi les anticorps spécifiques de plus de 20% échouent dans des expériences d' immunoprécipitation de la chromatine 15. La spectrométrie de masse (MS) est actuellement l'outil analytique le plus approprié pour étudier de nouvelles et / ou combinatoires PTM,et il a été largement mis en oeuvre pour histones (passé en revue dans 16). Cela est principalement dû à la haute sensibilité et la précision de masse de MS, et la possibilité d'effectuer des analyses à grande échelle.
La stratégie bottom-up est la stratégie de protéomique à base de MS le plus couramment utilisé pour la caractérisation des histones et leurs PTM, dans lequel la protéine intacte est enzymatiquement digéré en peptides courts (5-20 aa). Cette digestion facilite à la fois la séparation de LC et de détection MS. Messes dans la gamme de 600 - 2000 Da sont généralement plus facilement ionisés et identifiés avec une précision de masse plus élevée et la résolution que les grandes masses. SM / SM la fragmentation est aussi améliorée, sous forme de peptides courts sont généralement bien adaptés à une dissociation induite par collision (CID). Cependant, les histones constituent un défi pour ascendante MS car ils sont très riches en résidus d'acides aminés basiques, à savoir la lysine et l'arginine. Par conséquent, la digestion de trypsine conduit à la génération de peptides qui sont trop smtous pour la rétention de LC et de localisation sans ambiguïté du PTM. Pour contourner ce problème, notre protocole comprend la lysine et le peptide dérivé N-terminal chimique 17. L'utilisation d'anhydride propionique est efficace recommandée pour la dérivatisation chimique par rapport à d' autres réactifs 18. De tels blocs de dérivatisation des groupes de résidus lysine non modifiés et monométhyliques Ɛ-amino trypsine, ce qui permet d'effectuer une protéolyse uniquement à l'extrémité C-terminale des résidus d'arginine. des amines dérivatisées ne peuvent échanger des protons avec la solution et donc les peptides ne sont généralement doublement ou triplement chargé, ce qui facilite la détection et la MS MS / MS. En outre, la dérivatisation N-terminal augmente peptide hydrophobie et en phase inverse ainsi la rétention chromatographique. Ici, nous décrivons le flux de travail pour purifier les histones et les préparer pour l' analyse PTM via protéomique bottom-up (figure 1). Cette stratégie permet d'obtenir une quantification des marques d'histone simples et marques combinatoires fou PTM histones qui sont relativement proches dans la séquence d'acides aminés.
Protocole
1. Collecte de cellules provenant de la culture
- Si les cellules ont été cultivées en suspension, recueillir des cellules par centrifugation à 300 rcf pendant 5 min. Si le milieu de cellules adhérentes, aspirer et jeter. Rincer les cellules attachées avec du PBS sans Ca 2+ et Mg 2+ (appelé PBS aller de l' avant). Incuber les cellules dans trypsine ou de la trypsine-EDTA (0,025% - 0,5% selon la lignée cellulaire) avec un volume suffisant pour couvrir toute la surface des plaques à 37 ° C jusqu'à ce que les cellules se détachent (le temps varie selon les lignées cellulaires).
- Recueillir les cellules par centrifugation à 300 rcf pendant 5 min. Laver les cellules deux fois dans du PBS et de recueillir par centrifugation.
- Estimer le volume de cellules tassées environ des graduations marquées sur les 1,8 ml tubes ou 15 ml tubes coniques.
Note: Les cellules de culture peuvent être enfichable congelés dans l'azote liquide et stockés à -80 ° C indéfiniment à ce stade.
2. Isolement des noyaux de cellules intactes
- Décongeler les cellules sur la glace.
- Tampon d'isolement nucléaire Thaw (NIB, tableau 1).
- Préparer environ 5 ml de tampon NIB (tableau 1) pour chaque volume de cellules tassées de 100 pi. Pour chaque 1 ml de tampon NIB, ajouter des inhibiteurs de la protéase et des agents stabilisants comme suit: 1 pl de 1 M de DTT, 2,5 ul de 200 mM d'AEBSF, 2 pi de 2,5 uM microcystine et 2 pl de 5 M de butyrate de sodium. NIB avec des inhibiteurs sera appelé NIB partir de ce point.
Remarque: Si histone phosphorylation est étudié, comprennent l'EDTA libre protéase et inhibiteur de la phosphatase cocktail. - Retirer un cinquième volume de mémoire tampon NIB ainsi préparée et ajoute NP-40 alternatif (tableau 1) à une concentration finale de 0,2%. Les quatre cinquième volume restant sera utilisé pour les lavages.
- Wash culot cellulaire en 1h10 culot cellulaire à NIB sans NP-40 Rapport alternatif (v / v). Retirer le surnageant par centrifugation à 700 rcf pendant 5 min.
- Lyse cell culot en le plaçant sur de la glace et en ajoutant culot cellulaire à 01:10 NIB avec 0,2% de NP-40 alternatif (v / v).
- Si l'extraction à partir d'échantillons de tissus, homogénéiser en utilisant homogénéisateurs mortier ou Dounce. Les cellules cultivées peuvent être homogénéisés par pipetage doux.
- Incuber les cellules homogénéisées sur la glace pendant 5 - 10 min. Les cellules seront lyser et libérer les noyaux.
- Centrifugeuse à 1000 rcf pendant 5 - 10 min à 4 ° C. Le culot contient essentiellement cellule noyaux, tandis que le surnageant contient des composants essentiellement cytoplasmiques. Enregistrer la fraction cytoplasmique si désiré.
- Laver le culot des noyaux en remettant en suspension doucement en 1h10 (v / v) NIB sans NP-40 Alternative.
Remarque: Cette étape de lavage est uniquement pour éliminer les traces de détergents avant extraction histones des noyaux. - Centrifugeuse à 1000 rcf pendant 5 min à 4 ° C et retirer le surnageant.
- Répétez l'étape 2.10 - 2.11 au moins deux fois pour supprimer complètement NP-40 Alternative. Enlèvement de NP-40 Alternative est évidentes pipetage doux au cours de l'étape de lavage ne forme plus de bulles.
- Pour l'extraction à partir de tissus d'histone:
- Rincer les tissus frais ou décongelé congelé dans la glace froide NIB.
- Transfert tissu à une boîte de Pétri placée sur la glace avec NIB, juste assez pour maintenir le tissu humide.
- Dice en plus petits morceaux (<1 mm) avec une lame de rasoir pour augmenter la surface de contact pour l'isolement des noyaux.
- Transfert tissu haché à un homogénéisateur pré-réfrigérés et laver à NIB par pipetage de haut en bas.
- Retirer le tampon par centrifugation à 300 rcf pendant 5 min.
- Ajouter NIB contenant du NP-40 alternative aux cellules dans une des cellules: tampon rapport de 01:10 (v / v) et homogénéiser par 5 - 10 coups.
- Vérifiez la lyse des cellules et répéter l'homogénéisation au besoin. Un bon indicateur que les cellules ont été lysées est la réduction du volume de la pastille. Le culot ne doit contenir que des noyaux.
- Centrifuger à 700 rcf pendant 5 min et enregistrer granulés. Cette pastille peut être extrait 1 - 2 plus de tempss à 1:10 (v / v) de NIB contenant NP-40 alternatif; à ce stade, les histones sont extraites hors de la chromatine et le culot a considérablement réduit.
- Laver deux fois avec 2 - 3 ml de NIB sans NP40 Alternative pour éliminer les traces de détergent.
Note: point d'arrêt provisoire: L'échantillon peut être remis en suspension dans le volume minimum de NIB + 5% de glycérol, et stocké à -80 ° C.
3. Extraction et purification des histones de Nuclei
Nota: Les histones sont très riches en résidus basiques d'acides aminés, leur permettant de coopérer étroitement avec le squelette de l'acide phosphorique de l'ADN. Histones font partie des protéines les plus élémentaires dans le noyau, ce qui permet de les extraire dans de l' acide sulfurique refroidi à la glace (0,2 MH 2 SO 4) , avec une contamination minimale des protéines non histones, qui précipitent dans l' acide fort. TCA hautement concentrée (à une concentration finale de 33%) peut ensuite être utilisé pour précipiter les histones de l'acide sulfuriqueacide. TCA est stocké sous forme de 100% en bouteille brune à 4 ° C.
- Les noyaux cellulaires remettre en suspension dans 1: 5 (v / v) refroidis 0,2 MH 2 SO 4 (tableau 1) par pipetage doux.
- Incuber l'échantillon avec une rotation constante ou douce agitation pendant 2-4 heures à 4 ° C. En règle générale, pour les échantillons avec plus de 500 pi culot cellulaire, une extraction de 2 heures est suffisante pour extraire histones; une incubation plus longue peut conduire à l'extraction d'autres protéines basiques. Pour pastilles nucléaires plus petites (<200 pi), 4 extraction h fournit un meilleur rendement.
- Centrifuger à 3400 rcf à 4 ° C pendant 5 min.
- Transférer le surnageant dans un nouveau tube.
- Répétez les étapes 3.3 à 3.4 pour éliminer toute matière insoluble.
- Pour précipiter les histones, ajouter refroidi TCA à 100% (tableau 1) au surnageant recueilli (maintenant histones contenant) dans le rapport 1: 3 (v / v) afin d'obtenir une concentration de TCA final de 33%. Mélanger en inversant le tube quelques times.
Remarque: Les échantillons se trouble par addition de TCA, indiquant la présence d'histones. - Incuber le mélange sur de la glace pendant au moins 1 h. Pour les plus petites tailles de granulés de départ, les précipitations durant la nuit est recommandé.
- Centrifuger à 3400 rcf pendant 5 min. Le manteau de histones les côtés des tubes et également déposer au fond. Une pastille blanche insoluble se forme également au bas du tube, qui contient principalement des protéines non-histones et d'autres biomolécules. Retirer le surnageant par aspiration, soigneusement sans érafler les côtés ou le culot.
- En utilisant une pipette Pasteur en verre, rincer le tube avec de l' acétone glacée + 0,1% de HCl (tableau 1) de manière à recouvrir les protéines précipitées enrobant les côtés et le fond.
- Centrifuger à 3400 rcf pendant 2 min et aspirer le surnageant, soigneusement sans érafler les côtés ou le culot.
- Répétez les étapes 3.9 à 3.10 en utilisant 100% acétone glacée.
- granulés à sec avec un débit d'air ou avec un vacuum centrifuger, ou tout simplement en laissant le tube ouvert. Acétone évapore rapidement.
- Dissoudre les histones avec ddH 2 O (eau distillée deux fois) dans un volume minimum possible pour dissoudre la couche blanche complètement. Les histones sont facilement solubles dans l'eau. Pour granulés dans un tube de 1,5 ml, 100 ul ddH 2 O est généralement suffisant pour recueillir des histones.
- Centrifuger à 3400 rcf pendant 2 min et transférer le surnageant dans un nouveau tube.
4. Estimation de la concentration en protéine et de pureté
- Pour la mesure de la concentration en protéines, en utilisant BCA, Bradford dosage des protéines ou des analyses d'acides aminés (AAA). Ne pas utiliser des techniques qui adoptent l'absorbance à 280 nm, comme les histones sont pauvres en résidus d'acides aminés aromatiques.
- Vérifier la pureté des histones extraites par analyse SDS-PAGE avec un gel d'acrylamide à 15% et une coloration de Coomassie (en option).
- Si des variantes seule histone de haute pureté sont souhaitées, continuer à fractionnement HPLC-UVvariants d'histones (section 5). Sinon, passez directement à préparation des échantillons pour l'analyse histone PTM bottom-up (section 6).
5. Séparation des Histone Variantes par HPLC en phase inverse (Facultatif)
Remarque: Les variants d'histones de haute pureté peuvent être obtenus par fractionnement du mélange d'histones brut en utilisant une HPLC en phase inverse couplée à un détecteur UV. Ces histones purifiées sont utiles pour les études qui nécessitent une plus grande sensibilité et de pureté. Cependant, pour la norme de caractérisation histone PTM, cette étape peut être ignorée parce que l'analyse est suffisamment sensible et exhaustive. Fractionnement des histone intacte variantes idéalement nécessite au moins 100-300 ug de matériau de départ.
- Connecter une colonne appropriée C 18 5 um à une HPLC en fonction de la concentration d'histone de départ: avec environ 100 pg d'histones, utilisez 2,1 mm x 250 mm colonne avec un débit de 0,2 ml / min; avec environ 300 pg histones, utilisez la colonne 4,6 x 250 mm avecun débit de 0,8 ml / min. Préparer le tampon A et B en utilisant la verrerie dédiée comme suit:
- Préparer le tampon A: 5% d'acétonitrile de qualité HPLC, 0,1% de TFA dans de l'eau de qualité HPLC.
- Préparer le tampon B: 95% d'acétonitrile de qualité HPLC, 0,1% de TFA dans de l'eau de qualité HPLC.
- Connectez la colonne à un détecteur UV, et régler l'absorbance à 210-220 nm.
- Acidifier l'échantillon histone dissous dans l'eau avec 100% de TFA pour obtenir une concentration finale de 0,1 à 1% de TFA.
- Équilibrer la colonne avec 100% de tampon A pendant au moins 15 min au débit recommandé, ce qui correspond approximativement à trois volumes de colonne. Utilisez ce signal pour régler le niveau zéro absorbance du détecteur UV.
- Préparer des tubes dimensionnés de façon appropriée pour recueillir des fractions soit manuellement, soit dans un collecteur automatique d'échantillons.
- Injecter l'échantillon à une concentration d'environ 1 ug / ul ou plus. Les échantillons dissous dans des volumes plus importants pourraient modifier l'équilibre de la colonne duanneau de chargement et de plomb à la rétention inférieure.
- Exécuter le gradient programmé comme suit: de 0 à 30% B en 1 min, 30 à 60% B en 90 min, et 60 à 90% de B en 1 min.
- Recueillir des fractions ( par exemple chromatogramme représentées sur la figure 2) à des intervalles de 1 min à l' aide d' un collecteur automatique de fractions. Recueillir des fractions dans des tubes de taille appropriée pour contenir la totalité du volume.
- Sécher les échantillons fractionnés vers le bas dans un concentrateur sous vide.
Note: point d'arrêt provisoire: fractions d'histone séchées peuvent être conservés à température ambiante pendant de courtes périodes (1 - 2 jours) ou à -80 ° C congélateur pour des périodes de longue durée.
6. Chemical dérivatisation des histones Utilisation propionique Anhydride pour l'analyse bottom-up
- (Quantité recommandée: 50 - 100 ug) Dissoudre les échantillons d'histones dans 40 pl de 50 mM NH 4 HCO 3, pH 8,0. Si les échantillons étaient en pur ddH 2 O, ajouter concentré NH 4 HCO 3 pour compenser 50 mM, pH 8.0.
- Mouiller une pointe P10 de la pipette dans l'échantillon pour vérifier le pH en utilisant pH bandes indicatrices sans perte d'échantillon. NH4OH et l' acide formique peuvent être utilisés pour ajuster le pH à 8,0.
Remarque: La partie suivante du protocole (étapes 6/3 à 6/7) devrait être fait dans les lots de maximum de trois à quatre échantillons, afin de maintenir l'anhydride propionique réactif. - Utiliser la hotte de fumée pour les étapes ultérieures où l'anhydride propionique est utilisé. Préparer le réactif de propionylation frais en mélangeant l'anhydride propionique avec de l'acétonitrile dans le rapport 1: 3 (v / v). Ajouter un réactif de propionylation d'échantillonner en 1: 4 (v / v). Pour 40 histones ul, ajouter 10 ul réactif propionylation.
Note: Il est possible d'observer les débris blanc à cette étape. Cependant, celle-ci contient la plupart des sels et de l'acide propionique, et donc aucune action spécifique doit être prise. - Ajouter rapidement NH4OH pour rétablir un pH de 8,0 à la solution. Remarque: l'anhydride propionique réagir avec les amines libres des peptides produit propl'acide ionique qui diminue le pH. En général, l' addition de NH4OH à l'échantillon avec un rapport de 1: 5 (v / v) est appropriée pour rétablir un pH de 8,0, par exemple, 8 pl de NH4OH à 40 ul d'échantillon.
- Mélanger par tourbillonnement immédiatement.
- Vérifier le pH avec le même mode opératoire que dans l'étape 6.2.
Attention: lorsque le pH est supérieur à 10,0, à l'étiquetage d'autres résidus d'acides aminés ayant un pKa supérieur est possible. - Incuber les échantillons à température ambiante pendant 15 min.
- Répétez les étapes 06.03 à 06.07, effectuer strictement la réaction de pas plus de 3 ou 4 échantillons par lot de réactif propionylation.
- Des échantillons secs jusqu'à 10 - 20 pl dans un concentrateur sous vide. Ce qui n'a pas réagi évapore l' anhydride propionique, l' acétonitrile, l' acide acétique et de l' ammoniac gazeux libéré à partir de NH 4 OH. Si les échantillons sécher complètement, pas de pertes d'échantillons importants se produisent.
Remarque: Isopropanol peut être utilisé au lieu de l'acétonitrile. Cependant, l'acétonitrile a faible tension de surface et donc plusévaporation rapide. - Resuspendre ou diluer les échantillons avec ddH 2 O jusqu'à 40 pi de volume final est atteint.
- Répétez les étapes 06.02 à 06.09. Un double tour de histone propionylation assure> 95% de la réaction d'achèvement.
- Remplir une bouteille d'anhydride propionique avec un gaz d'argon, de manière à empêcher la formation d'acide acétique en contact avec l'humidité dans la bouteille.
Note: point d'arrêt provisoire: L' échantillon peut être conservé à -80 ° C reconstituée dans ddH 2 O ou séché.
7. La digestion protéolytique avec de la trypsine
- Histones Remettre en suspension dans 50 mM de NH 4 HCO 3 pour obtenir une concentration optimale de 1 ug / ul ou plus. Plus d'échantillons dilués conduisent à diminuer l'efficacité de la trypsine.
Remarque: Les histones à cette étape doivent être à pH 8,0 Si elle reste acide, puis ajouter NH 4 HCO 3 sel à l' échantillon en utilisant la pointe de la pipette. - Ajouter la trypsine à des échantillons d'histone dans un rapport 1:10 (poids / poids).
- incubate à 37 ° C pendant 6-8 heures.
- Arrêtez la digestion par congélation dans -80 ° C.
- Sécher l'échantillon vers le bas 10 - 20 ul dans un concentrateur sous vide.
Note: point d'arrêt provisoire: L'échantillon peut être conservé à -80 ° C.
8. propionylation de Histone Peptides à N-terminales
Remarque: Cette section décrit la dérivatisation du peptide N-termini généré à partir du trypsine digest. Cette procédure améliore la rétention HPLC des peptides courts (par exemple, l' acide aminé 3-8 de l' histone H3), le groupe propionyle peptide augmente le caractère hydrophobe.
- Des échantillons de remettre en suspension dans 30 pl de 100 mM de NH 4 HCO 3.
- Répétez les étapes 6/1 à 6/9.
Remarque: Il est normal que le séchage des échantillons dans le vide prend plus de temps à cette étape. - Resuspendre ou diluer les échantillons avec 50 - 100 ul ddH 2 O + soit 0,1% d' acide acétique TFA ou 0,5%. Remarque: L'acide acétique est recommandé pour les longs stockages, comme TFA facilitates oxydation de la méthionine à long terme. D'autre part, le TFA est recommandé si l'étape de basculement (section 9) est effectuée le même jour, que le TFA aide une meilleure rétention chromatographique.
Note: point d'arrêt provisoire: L'échantillon peut être conservé à -80 ° C.
9. dessalage de l'échantillon avec Stage-conseils
Note: A ce stade, il est le sel présent dans l'échantillon. Les sels empêchent l'analyse HPLC-MS, car ils ionisent pendant électrospray, supprimer le signal à partir de peptides. Les sels peuvent également former des adduits ioniques, des peptides, en réduisant l'intensité du signal pour le peptide non fixées par addition. Comme le peptide d'adduits aura une masse différente, le peptide ne sera pas correctement identifié ou quantifié.
- En utilisant une pointe de pipette P1000, perforer un disque de C 18 matériau à partir d' un solide disque d'extraction en phase. Poussez le minidisque hors de la pointe de P1000 en utilisant un capillaire de silice fondue et déposer le minidisque au fond d'une pointe P100 / 200 pipette. Veiller à ce que le dISK est bien calé au fond de la pointe (Figure 3).
Remarque: La pointe de P1000 a un assez petit pour percer le disque C 18. Il convient de couper le dernier centimètre de la pointe afin d'avoir un trou de plus grand diamètre. - Utilisez deux C 18 coups de poing dans la même pointe P100 / P200 si dessalage plus de 25 ug d'échantillon.
- Utilisez un adaptateur de centrifugeuse pour maintenir stade-conseils en place dans 1,5 ml ou 2 tubes ml de microcentrifugation. Utilisez lente (300 - 400 rcf) rotation; les solvants passent normalement par la résine en moins d'une minute.
- Rincer la résine par centrifugation avec 50 ul d'acétonitrile à 100% pour activer le matériau C 18 et d' éliminer les contaminations potentielles.
- Equilibrer disque en rinçant 80 ul de TFA à 0,1% par centrifugation lente.
- Acidifier l'échantillon à un pH de 4,0 ou moins avec de l'acide acétique. Vérifier le pH avec des bandes de pH pour minimiser la perte de l'échantillon. échantillon de charge sur le disque par centrifugation lente.
- lavageéchantillon par balayage 70 - 80 ul de TFA à 0,1% par centrifugation lente.
- Éluer l'échantillon en rinçant 70 ul de 75% d'acétonitrile et d'acide acétique à 0,5% par centrifugation lente. Collecter l'échantillon dans un tube de 1,5 ml.
- Extrait sec dans un concentrateur sous vide.
Note: point d'arrêt provisoire: L'échantillon peut être conservé à -80 ° C.
10. Analyse des histones Peptides
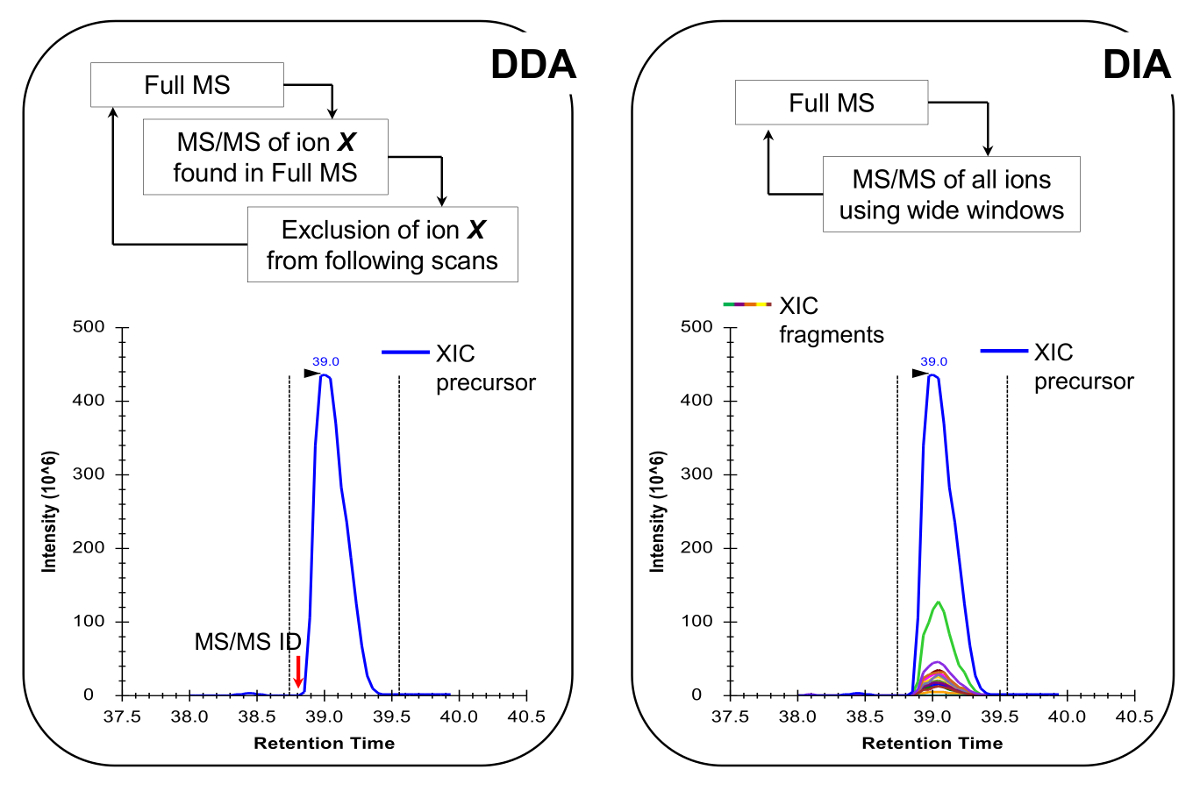
Remarque: La plate-forme NLC-MS devrait être mis en place comme cela se fait dans l'analyse traditionnelle des peptides. L'utilisation de 200-300 colonne de flux nl (75 um ID colonne analytique, C 18 particules) est recommandé, car ils sont un excellent compromis entre la sensibilité et la stabilité. La méthode d'acquisition MS peut être une combinaison d'acquisition dépendant des données (DDA) avec des scans ciblés 19 ou une acquisition (DIA) indépendante des données 20,21, les deux décrits dans Les résultats représentatifs et Figure 4.
- Préparation des tampons HPLC - A: acide formique à 0,1% dansl'eau de qualité HPLC; B: l'acide formique à 0,1% dans l'acétonitrile de qualité HPLC.
- Programmer la méthode HPLC de la manière suivante: de 0 à 30% de tampon B en 30 min, 30 à 100% de B pendant les 5 minutes suivantes et à isocratique 100% de B pendant 8 min. Si la HPLC est pas programmé pour automatisé colonne équilibration avant le chargement des échantillons, puis comprendre ce qui suit: gradient de 100 à 0% B en 1 min et le débit isocratique à 0% de B pendant 10 min. Régler la vitesse de l'analyse à 250 débit - 300 nl / min.
- Programmer la méthode de l' acquisition MS pour effectuer soit DDA combinée à des analyses ciblées 19 ou DIA 20,21 (figure 4). Assurez-vous que le cycle de service de MS permet plein MS scan tous les ~ 2 sec, afin d'avoir assez de données à travers le pic chromatographique, ce qui permet une quantification plus précise. Remarque: Avec C 18 Chromatographie, la largeur moyenne de référence de pointe est d' environ 30 secondes pour le gradient décrit ci - dessus.
- Chargez environ 1 ug d'échantillon sur la HPLC cOLONNE.
- Exécutez la méthode HPLC-MS / MS comme programmé.
Remarque: Dans le protocole, nous ne recommandons pas de détails des colonnes, des instruments MS ou MS détails des paramètres, comme toute configuration optimale qu'un protéomique laboratoire individuel développé sera adapté pour la méthode. les laboratoires de protéomique devraient utiliser leur configuration optimisée, car les peptides histones séparés en tant que peptides traditionnels.
11. Analyse des données
- Importez les fichiers bruts MS dans le logiciel pour effectuer l'intégration de la zone de pointe. Note: EpiProfile 22 est recommandée, car elle est optimisée pour les peptides histones; en utilisant la connaissance du temps de rétention d'élution chromatographique il effectue l'extraction de l'aire du pic fiable des peptides histone connus. Alternativement, Skyline 23 est un autre logiciel idéal pour le but.
- Calculer l'abondance relative d'un peptide donné en divisant la surface par la surface totale de ce peptide dans l'ensemble de ses formes modifiées. Remarque: En cas de découverte analysis Mascot est recommandé d'identifier les spectres des peptides histones modifiées. La performance de cet outil a été décrit récemment 24. Tous les autres moteurs de recherche de base de données pour la protéomique sont également utilisables, mais sur nos tests ont fourni des performances moindres.
Résultats
A titre d'exemple, nous avons analysé les histones extraites à partir de cellules souches embryonnaires humaines (hESC) avec et sans acide rétinoïque (RA), la stimulation, à partir de 200 culots cellulaires ul. La présence de RA dans la culture cellulaire conduit à la différenciation ESC. A partir du culot de cellules, environ 50 à 100 ug d'histones ont été extraits, ce qui est plus que suffisant pour effectuer de multiples injections de peptides histone LC-MS. Après dérivatisation, la digestion et dessalage, les échantillons ont été chargés sur un 75 pm x 15 cm colonne C18 (diamètre de particule de 3 um, taille de pores 300 Å) en mode série avec un système de chromatographie nano liquide à haute performance avec des puces microfluidiques couplés à un hybride linéaire piège quadripolaire - spectromètre de masse Orbitrap. acquisition MS a été réalisée en utilisant DIA. En parallèle, des échantillons ont également été analysés à l'aide d'une méthode utilisant une LDV CLUHP nano-flux couplée à un piège Orbitrap spectromètre de masse d'ions hybrides (données non présentées). Danschaque cycle, une détection complète MS Orbitrap a été réalisée avec la plage de balayage de 290 à 1400 m / z, une résolution de 60.000 (à 200 m / z) et AGC de 10 6. Ensuite, le mode d'acquisition dépend de données a été appliquée avec une dynamique exclusion de 30 sec. scans MS / MS ont été suivis sur des ions parents de plus intenses les. Ions avec un état d'une charge ont été exclues de MS / MS. Une fenêtre d'isolement de 2 m / z a été utilisé. Les ions sont fragmentés en utilisant une dissociation induite par collision (CID) avec une énergie de collision de 35%. Détection de piège ionique a été utilisé avec le mode de plage de balayage normal et la vitesse de balayage normale avec AGC de 10 4.
Les données brutes ont été analysées MS adoptant un logiciel pour l'extraction des précurseurs et des ions fragments chromatogrammes, à savoir Skyline 23 et 22 EpiProfile. EpiProfile a été optimisé pour les peptides histones, car il intègre l'extraction de la zone de pointe intelligente en raison de la connaissance antérieure du pepmarée temps de rétention. D'autre part, Skyline est optimisé pour DIA analyse, et donc les chiffres affichés DIA (figures 4 et 5A) sont des captures d' écran de ce logiciel. À partir du chromatogramme d'ions extrait, l'aire sous la courbe est récupérée, ce qui permet d'estimer l'abondance de chaque peptide. La surface du pic chromatographique a été calculé pour [M + H] +, [M + 2H] 2+ et [M + 3H] 3+ des ions du même peptide, bien que dans la plupart des cas , le [M + 2H] 2+ était la forme la plus courante. Ceci fournit l'abondance brute d'une forme modifiée donnée d'un peptide. Afin d'atteindre l'abondance relative de PTM, la somme de toutes les différentes formes modifiées d'un peptide de l'histone a été considéré comme 100%, et la région du peptide particulier a été divisée par la superficie totale de cette histone peptide sous toutes ses formes modifiées .
peptides histones sont présents dans un variété de formes isobares (Figure 5). Peptides isobariques, par exemple, K18ac et K23ac, ne peuvent être quantifiés au niveau MS / MS, où leurs ions fragments uniques sont utilisés pour déterminer le rapport des espèces isobares (figure 5A et 5B). Ce rapport est utilisé pour diviser l'aire du pic chromatographique entre les deux espèces. Lors de l'utilisation du PDD, ces formes isobares ont été inclus dans la liste des masses ciblées, parce que ces peptides doivent être sélectionnés pour la fragmentation par l'ensemble de leur élution, ce qui ne se produira pas dans une expérience DDA standard. La discrimination de l'abondance relative de l'espèce isobares est ensuite effectuée en contrôlant le profil d'élution des ions fragments. D'autre part, le type DIA d'acquisition ne nécessite pas de liste d'inclusion. Cependant, ce type de procédé d'acquisition est incompatible avec la recherche de base de données classique, et donc peut empêcher la découverte de peptides modifiés inconnus.
Lysine acétylation (+ 42,011 Da) a été victime de la trimethylation presque isobarique (+ 42,047 Da) en utilisant haute résolution MS acquisition (> 30 000). En outre, l'acétylation est plus hydrophobe que trimethylation, conduisant à élution de peptides acétylés plus tard que ceux triméthylés respectifs. La forme non modifiée du même peptide est élué plus tard encore, en raison du fait que la lysine est propionylated. En résumé, l'ordre d'hydrophobie pour un peptide avec un site modifiable est di- et Trimethylated
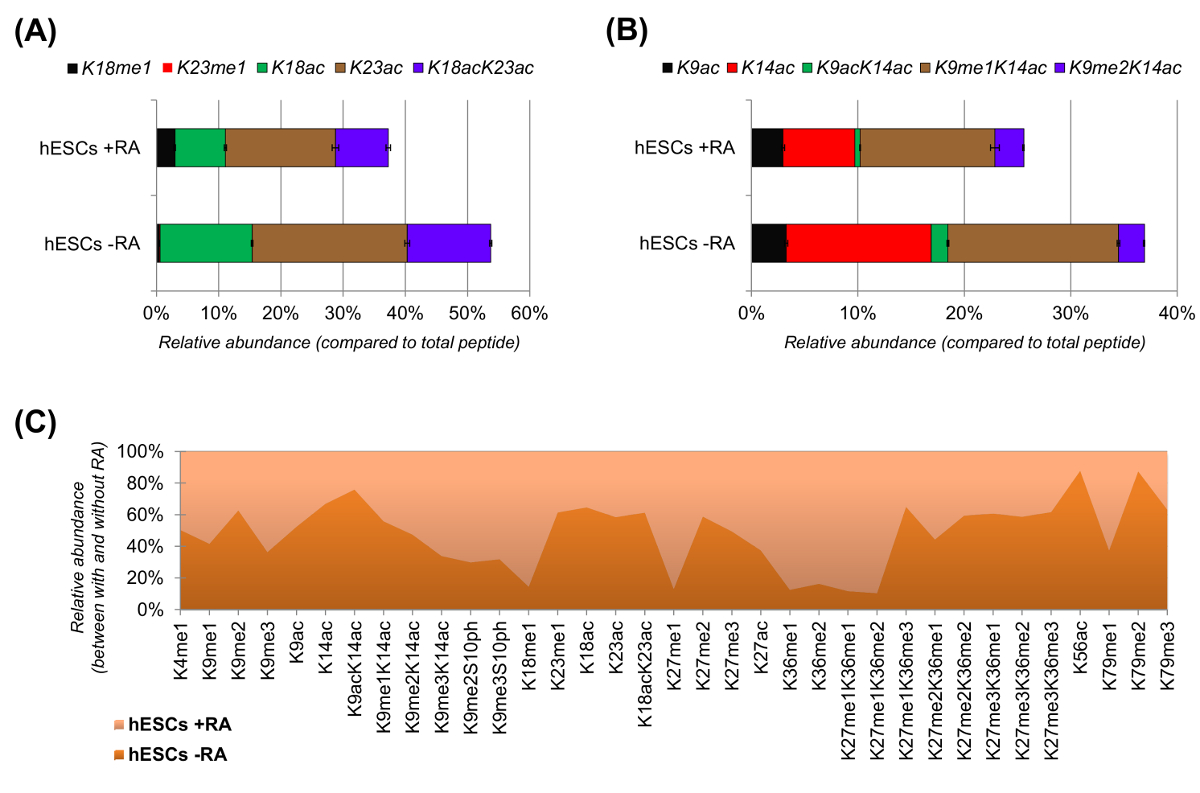
CSEh ont montré une nette diminution des peptides acétylés lorsqu'ils sont stimulés pour la différenciation (figure 6A et 6B). Ce ne fut pas surprenant que les résultats précédents ont rapporté acétylation supérieur en CES par rapport à ceux de différenciation 25,reflétant la nature généralement permissive de la chromatine pluripotentes. En se concentrant sur l' histone H3, 35 formes modifiées différentes ont été quantifiées (Figure 6C). Cependant, tous les proteoforms histones qui peuvent être étudiés avec cette approche sont plus de 200, y compris toutes les variantes des histones et des modifications de faible abondance (données non présentées). En outre, notre analyse a montré que la reproductibilité élevée peut être obtenue entre les réplicats techniques, comme en témoigne la petite taille des barres d'erreur représentant (± écart type). Pris ensemble, cette section décrit comment extraire l'abondance relative des histones modifiées peptides en utilisant des données NLC-MS.

Figure 1:. Workflow pour MS / MS Histone Analyse Bottom-up Les dix étapes pour l' analyse de l' histone sont présentés, y compris une estimation du temps nécessaire pour chaque étape. Le numéro de section est indiquée entre parenthèses comme présent dans le manuscrit. Section 5, décrivant le fractionnement des échantillons pour isoler les différentes variantes d'histones, peut être omis , sauf si il y a un besoin pour une analyse très sensible d'une variante donnée. S'il vous plaît cliquez ici pour voir une version plus grande de cette figure.
{kind=link}

Figure 2: phase inverse de débit élevé LC pour la variante Fractionnement et des histones Coomassie Gel (A) Chromatogramme LC-UV qui représente la séparation histone intacte.. des variants de l'histone H3 peuvent être distingués les uns des autres en fonction de leur temps d'élution. Fractions peuvent être collectées soit manuellement ou en utilisant un collecteur de fractions automatisé. (B) Coomassie gel de trois répétitions de la purification de l' histone.= "Https://www.jove.com/files/ftp_upload/54112/54112fig2large.jpg" target = "_ blank"> S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.

Figure 3:. Making of Stage-basculement fiche avec une pointe de pipette P1000, poinçon un disque en C 18 matériau à partir d' un disque d'extraction en phase solide (deuxième panneau). Le minidisque restera dans la pointe (panneau du milieu), de sorte qu'il peut être poussé dehors dans une pointe de pipette plus petit P100 / 200 en utilisant toute sorte de petit capillaire. Dans cet exemple, nous avons utilisé un diamètre extérieur des tubes de silice fondue de 700 pm. Le minidisque devrait être poussé vers le bas de la pointe de la pipette P100 / 200 jusqu'à ce qu'il ne peut pas aller plus loin (dernier panneau). La pointe de la scène est prête pour histone dessalage, car il a une capacité suffisante pour conserver suffisamment de matériau d'échantillon pour de nombreuses répétitions. Plus précisément, un minidisque est suffisant pour 15 - 20 pg de sample. Si plus échantillon est nécessaire, plusieurs disques peuvent être emballés sur un autre. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}

Figure 4:. Représentation schématique du PDD et DIA Méthodes Lors de l' utilisation du PDD, le cycle MS scan se caractérise par la sélection séquentielle des ions précurseurs pour MS / MS fragmentation en fonction de leur intensité et de l' état de charge. Une fois qu'un ion précurseur a été fragmenté est placé dans une liste d'exclusion pour éviter la sélection répétitive du même peptide, de sorte que la sclérose en plaques peut "dig" en signaux moins abondants. Cette méthode d'acquisition est la technique de choix en protéomique pour le mode de détection. Quantification est obtenue en intégrant le signal de balayage complet d'un ion donné à côté des MS identifiés /spectre MS. Dans DIA, toute la gamme m / z est fragmenté à chaque cycle. Cette approche est moins approprié pour le mode de détection, mais il produit un profil chromatographique de tous les ions, des précurseurs et des produits. Cela conduit à une quantification plus confiante et la discrimination des formes isobares. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}

Figure 5: Quantification de isobariques Peptides (A) Exemple de deux peptides isobares couramment abondantes dans l' analyse de l' histone.. Le chromatogramme ionique extrait (XIC) de leur masse précurseur et isotopes relatifs (ci-dessus) est identique. Cependant, le XIC des ions produits (ci-dessous) permet de discriminer les deux formes isobares. Notamment, les ions fragments seulement uniques devraient nous êtreed pour estimer l'abondance relative des deux espèces. (B) Représentation des ions de fragments uniques pour les deux peptides décrits (en rouge). (C) Liste des peptides couramment analysés dans Homo sapiens ayant au moins un équivalent isobarique. Séquence des variantes entre les peptides histones énumérés sont indiqués. S'il vous plaît cliquez ici pour voir une version plus grande de cette figure.
{kind=link}

Figure 6: Les résultats représentatifs de cellules souches embryonnaires humaines avec et sans traitement acide rétinoïque (A) la quantification relative du peptide histone H3 KQLATKAAR (aa 18-26) dans toutes ses proteoforms modifiés.. L'abondance relative a été estimée en utilisant tous les proteoforms 100% (relpourcentage ative du peptide non modifié est non montré) (B) , la quantification relative du peptide de l' histone H3 KSTGGKAPR (aa 9 -.. 17) , (C) de l' abondance relative des peptides détectés pour canonique histone H3 avec et sans traitement des cellules avec de l' acide rétinoïque. Le chiffre indique dans lequel des deux traitements les modifications données sont plus abondantes (> 50%). Dans l' ensemble, nous démontrons que l' histone H3 acétylation diminue dans la plupart des résidus de lysine sur l' induction de la différenciation cellulaire. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}
| Solution # | Composition | ||||||
| 1 | Isolation nucléaire Buffer (NIB) stock est effectué comme suit et conservé congelé en aliquotes de 100 ml à -20 ° C; décongeléNIB peut être conservé à 4 ° C pendant quelques semaines: Tris 15 mM, 60 mM KCl, 15 mM de NaCl, 5 mM de MgCl2, 1 mM de CaCl2 et 250 mM de saccharose. Le pH du tampon est ajusté à 7,5 avec du HCl. | ||||||
| 2 | Les inhibiteurs de protéase (ajouter des frais aux tampons avant utilisation): 1 M dithiothréitol (DTT) dans ddH 2 O (1,000x); MM AEBSF 200 en ddH 2 O (400x) | ||||||
| 3 | inhibiteur de la phosphatase (ajouter frais à des tampons avant utilisation): 2,5 uM Microcystine dans 100% d'éthanol (500x) | ||||||
| 4 | inhibiteur de HDAC (ajouter frais à des tampons avant utilisation): 5 butyrate de sodium M, faites par titration de l'acide 5 M butyrique en utilisant NaOH à pH 7,0 (500x) | ||||||
| 5 | NP-40 alternatif: 10% v / v dans ddH 2 O | ||||||
| 6 | 0,2 MH 2 SO 4 dans ddH 2 O | ||||||
| 7 | L' acide trichloroacétique (TCA): 100% p / v dans ddH 2 O | ||||||
| 8 | Acétone + 0,1% d'acide chlorhydrique (HCl): 0,1% v / v de HCl dans de l'acétone | ||||||
Tableau 1. Solutions.
Discussion
Le protocole décrit ici est optimisé tenant compte des coûts, le temps et la performance. D'autres préparations sont possibles, mais ils ont des limites, en particulier dans le cas de couplage avec l'analyse MS. Par exemple, le protocole d'extraction riche en sel peut être utilisé pour purifier histones 26 au lieu de la précipitation de TCA (section 3). protocole haut-sel est intrinsèquement plus doux, car il ne pas utiliser un acide fort. Ceci préserve PTM labiles aux acides et augmente le rendement des histones extraites, comme précipitation au TCA coprécipités de nombreuses autres protéines de liaison à la chromatine. Cependant, l'extraction riche en sel conduit à des échantillons contenant du sel trop concentré pour HPLC-MS / MS. Dans une autre préparation, histone digestion peut être effectuée sans propionylation (section 6-8), par exemple en réduisant le temps d'incubation de la trypsine et le rapport enzyme / substrat 27 ou en utilisant argc que la digestion enzymatique 28-30. Cependant, dérivatisation avec l'anhydride propionique est recommandé, comme iT conduit à la génération de peptides hydrophobes, qui sont mieux conservées au cours de la chromatographie liquide.
Pour dérivatisation chimique, une variété d'anhydrides d'acides organiques ont été évalués et leurs mérites globalement discuté 18. Néanmoins, l'anhydride propionique avéré le meilleur compromis entre l'efficacité, les produits secondaires minimisés et l'amélioration de l'hydrophobie de peptide. Potentiellement, l'anhydride propionique peut être acheté sous forme isotopiquement marquée; cela permet une analyse de multiplexage en raison de la possibilité de mélanger plusieurs échantillons et de discriminer les au niveau MS sur la base des différentes masses imparties de l'étiquette lourde. Cependant, cette analyse conduit à une complexité accrue du chromatogramme CL-SM et réduit la quantité d'échantillon qui peut être injecté pour chaque condition unique.
À cet égard, certains aspects critiques du protocole devraient être mis en évidence. Ce qui suit doit être utilisé comme checklist pour trouver des erreurs dans l'exécution de la procédure en cas on obtient des résultats négatifs. Tout d'abord, après la précipitation des noyaux le culot doit être soigneusement lavé avec NIB sans NP-40 Alternative (article 2.10) jusqu'à l'élimination complète du détergent (visible par l'absence de bulles pendant le mélange). Ne pas le faire pourrait compromettre l'extraction des histone avec des acides. Deuxièmement, après précipitation histone avec TCA (section 3.9) lavages du culot avec de l'acétone est crucial. Présence d'acide concentré nuirait à l'étape suivante si propionylation et la digestion (section 6.1) sont directement effectués. Il serait pas problématique en cas histone fractionnement est effectué (section 5). Troisièmement, il est essentiel que la réaction de propionylation est effectuée rapidement (section 06.03 au 06.07). Pour ce faire, évitez d'utiliser le même mélange de propionylation (anhydride propionique + acétonitrile) pendant plus de 3 - 4 échantillons consécutifs. En outre, le pH est l'aspect le plus important de la digestion par la trypsine (section 7). Si nonautour de 8,0 (7,5 à 8,5), la digestion sera inefficace. Cela peut se produire, comme l'échantillon sera riche en acide propionique à cette étape. NH 4 OH peut être ajouté jusqu'à ce que nécessaire. En outre, pour les chercheurs familiers avec les flux de travail de la protéomique, il se sentira normale pour acidifier l'échantillon de mettre fin à la trypsine digestion. Cela ne devrait pas être fait, car il mettra en péril la réaction suivante, à savoir, propionylation du peptide N-terminales (section 8.1). Enfin, dans le même numéro, il est important de se rappeler pour l'analyse des données que les peptides non modifiés ne sont pas réellement non modifié; tous les résidus de lysine libre et N-terminales seront occupés par propionylation (56,026 Da). Ainsi, la réalisation d'une chromatographie d'extraction d'ions de la masse correspondant de manière unique à la séquence peptidique conduirait à aucun résultat.
Les limitations de la méthode sont principalement liés à l'incapacité de détecter PTM combinatoires, en raison des séquences peptidiques courtes et les biais dans la réalisation de la vraie abundanse d'une modification, due au fait que les peptides de différentes formes modifiées peuvent ioniser avec des rendements différents. Le premier problème peut être résolu en combinant cette technique avec une baisse moyenne ou une approche top-down (revue dans 16). Ce type d'analyse, même si cela est techniquement plus difficile, est idéal pour étudier les fréquences de co-existence de modifications. En outre, il permet une meilleure discrimination des variants d'histones, qui ne peuvent pas toujours être obtenus avec bottom-up car certains peptides ont la même séquence dans différentes variantes d'histones. La deuxième question, liée à l'efficacité d'ionisation, peut être résolu en utilisant une bibliothèque de peptides synthétiques 31. Cette approche permet une estimation plus précise de l'abondance relative des PTM histones. Cependant, dans la plupart des expériences, le résultat souhaité est l'évolution relative des modifications données entre les conditions analysées. Dans ce cas, une telle correction est pas nécessaire, en raison du fait que tous les échantillons ont les mêmes bias.
En conclusion, ce protocole permet l'analyse des PTM histones qui peut être complété en 3 jours en utilisant nCL couplé tandem MS. Les comparaisons avec d' autres techniques que MS, à savoir, en utilisant des stratégies d'anticorps à base tel que discuté dans l'introduction, ne sont pas adaptés, car ils ne peuvent pas atteindre même près de ce niveau de débit. En outre, des techniques à base d'anticorps ne permettent pas la découverte de nouvelles modifications, mais elles sont basées exclusivement sur la confirmation et la quantification des marques prévues. Nous pensons donc que la protéomique bottom-up sur les peptides histones va gagner en popularité dans les laboratoires de protéomique en raison des avantages intuitifs à connaître la réglementation des marques des histones, qui sont les protagonistes de l'expression des gènes d'accord et donc affecter la régulation du protéome. En outre, le protocole décrit comprend des améliorations récentes dans la préparation des échantillons et des logiciels d'analyse de données, ce qui rend l'analyse des histone plus trivial aussi pour laboratories qui n'a jamais connu la caractérisation de ce type de peptides hypermodified.
Déclarations de divulgation
Les auteurs déclarent qu'ils ont aucun intérêt financier concurrents.
Remerciements
Ce travail a été soutenu financièrement par des subventions des NIH (DP2OD007447, R01GM110174 et R01AI118891).
matériels
| Name | Company | Catalog Number | Comments |
| Trypsin 0.25% EDTA | Invitrogen | 25200056 | For harvesting cells |
| PBS | Invitrogen | 14200075 | |
| Tris | Roche | 77-86-1 | |
| Potassium Chloride | Fisher Scientific | BP366-500 | |
| Sodium Chloride | Sigma | S9888 | |
| Magnesium Chloride hexahydrate | Sigma | M9272 | |
| Calcium Chloride, anhydrous | Sigma | C1016 | |
| Sucrose | Fisher Scientific | BP220-1 | |
| DTT | Invitrogen | 15508-013 | |
| AEBSF | EMD Millipore Corp | 101500 | |
| Microcystin | Sigma | M4194 | |
| Sodium Butyrate | Sigma | B5887 | |
| Halt Protease and Phosphatase Inhibitor Cocktail, EDTA-free (100x) | Fisher Scientific | 78445 | |
| NP- 40 Alternative | CALBIOCHEM | 492016 | |
| Sulfuric Acid, ACS grade | Fisher Chemical | 7664-93-9 | |
| Trichloroacetic acid | Sigma | T6399 | |
| Acetone | Sigma | 179124 | |
| HCl | Fisher Chemical | A144-500 | |
| Bradford reagent | Biorad | 500-0006 | |
| 30% acrylamide/bis 29:1 — 500 ml | Biorad | 1610156 | |
| Coomassie | Fisher Scientific | 20278 | |
| C18 Column (5 µm) 2.1 mm x 250 mm | Grace | 218TP52 | |
| C18 Column (5 µm) 4.6 mm x 250 mm | Grace | 218TP54 | |
| HPLC grade acetonitrile | Fisher Chemical | A955-4 | |
| HPLC grade water | Fisher Scientific | W6 4 | |
| TFA | Fisher Scientific | A11650 | |
| Ammonium Bicarbonate | Sigma | A6141 | |
| ammonium hydroxide | Sigma | 338818 | |
| propionic anhydride | Sigma | 240311 | |
| Sequencing grade modified trypsin | Promega | PRV5113 | For digesting histones for MS |
| Acetic Acid | Sigma | 49199 | |
| C18 extraction disk | Empore | 2215 | |
| Formic Acid | Sigma | F0507 |
Références
- Waddington, C. H. Canalization of development and the inheritance of acquired characters. Nature. 150, 563-565 (1942).
- Sharma, S., Kelly, T. K., Jones, P. A. Epigenetics in cancer. Carcinogenesis. 31 (1), 27-36 (2010).
- Reik, W., Dean, W., Walter, J. Epigenetic reprogramming in mammalian development. Science. 293 (5532), 1089-1093 (2001).
- Kouzarides, T. Chromatin modifications and their function. Cell. 128 (4), 693-705 (2007).
- Tessarz, P., Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Bio. 15 (11), 703-708 (2014).
- Fischle, W., Wang, Y. M., Allis, C. D. Histone and chromatin cross-talk. Curr Opin Cell Biol. 15 (2), 172-183 (2003).
- Lee, J. S., Smith, E., Shilatifard, A. The language of histone crosstalk. Cell. 142 (5), 682-685 (2010).
- Simboeck, E., et al. A Phosphorylation Switch Regulates the Transcriptional Activation of Cell Cycle Regulator p21 by Histone Deacetylase Inhibitors. J Biol Chem. 285 (52), 41062-41073 (2010).
- Hirota, T., Lipp, J. J., Toh, B. H., Peters, J. M. Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature. 438 (7071), 1176-1180 (2005).
- Xhemalce, B., Kouzarides, T. A chromodomain switch mediated by histone H3 Lys 4 acetylation regulates heterochromatin assembly. Genes Dev. 24 (7), 647-652 (2010).
- Vermeulen, M., et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. 142 (6), 967-980 (2010).
- van Attikum, H., Gasser, S. M. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol. 19 (5), 207-217 (2009).
- Fernandez-Capetillo, O., et al. H2AX is required for chromatin remodeling and inactivation of sex chromosomes in male mouse meiosis. Dev Cell. 4 (4), 497-508 (2003).
- Santaguida, S., Musacchio, A. The life and miracles of kinetochores. Embo J. 28 (17), 2511-2531 (2009).
- Egelhofer, T. A., et al. An assessment of histone-modification antibody quality. Nat Struct Mol Biol. 18 (1), 91-93 (2011).
- Sidoli, S., Cheng, L., Jensen, O. N. Proteomics in chromatin biology and epigenetics: Elucidation of post-translational modifications of histone proteins by mass spectrometry. J Proteomics. 75 (12), 3419-3433 (2012).
- Plazas-Mayorca, M. D., et al. One-Pot Shotgun Quantitative Mass Spectrometry Characterization of Histones. J Proteome Res. 8 (11), 5367-5374 (2009).
- Sidoli, S., et al. Drawbacks in the use of unconventional hydrophobic anhydrides for histone derivatization in bottom-up proteomics PTM analysis. Proteomics. 15 (9), 1459-1469 (2015).
- Lin, S., Garcia, B. A. Examining histone posttranslational modification patterns by high-resolution mass spectrometry. Methods Enzymol. 512, 3-28 (2012).
- Sidoli, S., et al. SWATH Analysis for Characterization and Quantification of Histone Post-translational Modifications. Mol Cell Proteomics. , (2015).
- Krautkramer, K. A., Reiter, L., Denu, J. M., Dowell, J. A. Quantification of SAHA-Dependent Changes in Histone Modifications Using Data-Independent Acquisition Mass Spectrometry. J Proteome Res. , (2015).
- Yuan, Z. F., et al. EpiProfile Quantifies Histone Peptides With Modifications by Extracting Retention Time and Intensity in High-resolution Mass Spectra. Mol Cell Proteomics. 14 (6), 1696-1707 (2015).
- MacLean, B., et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 26 (7), 966-968 (2010).
- Yuan, Z. F., Lin, S., Molden, R. C., Garcia, B. A. Evaluation of proteomic search engines for the analysis of histone modifications. J Proteome Res. 13 (10), 4470-4478 (2014).
- Tan, Y., Xue, Y., Song, C., Grunstein, M. Acetylated histone H3K56 interacts with Oct4 to promote mouse embryonic stem cell pluripotency. Proc Natl Acad Sci U S A. 110 (28), 11493-11498 (2013).
- Vonholt, C., et al. Isolation and Characterization of Histones. Methods Enzymol. 170, 431-523 (1989).
- Zhang, K. L., et al. Identification of acetylation and methylation sites of histone H3 from chicken erythrocytes by high-accuracy matrix-assisted laser desorption ionization-time-of-flight, matrix-assisted laser desorption ionization-postsource decay, and nanoelectrospray ionization tandem mass spectrometry. Anal. Biochem. 306 (2), 259-269 (2002).
- Jufvas, A., Stralfors, P., Vener, A. V. Histone Variants and Their Post-Translational Modifications in Primary Human Fat Cells. Plos One. 6 (1), e15960(2011).
- Bonaldi, T., Imhof, A., Regula, J. T. A combination of different mass spectroscopic techniques for the analysis of dynamic changes of histone modifications. Proteomics. 4 (5), 1382-1396 (2004).
- Zhao, X. L., et al. Comparative Proteomic Analysis of Histone Post-translational Modifications upon Ischemia/Reperfusion-Induced Retinal Injury. J Proteome Res. 13 (4), 2175-2186 (2014).
- Lin, S., et al. Stable-isotope-labeled histone peptide library for histone post-translational modification and variant quantification by mass spectrometry. Mol Cell Proteomics. 13 (9), 2450-2466 (2014).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.