
Method Article
Fluxo de trabalho completo para Análise de histona modificações pós-traducionais Usando Bottom-up Espectrometria de Massa: De histona Extração de Análise de Dados
Neste Artigo
Resumo
Este protocolo descreve um fluxo de trabalho totalmente integrado para a caracterização de histonas modificações pós-traducionais, utilizando espectrometria de massa (MS). O fluxo de trabalho inclui a purificação histona de culturas de células ou tecidos, derivação histona e digestão, análise de MS utilizando cromatografia líquida de nano-fluxo e instruções para a análise de dados. O protocolo é projetado para a conclusão dentro de 2 - 3 dias.
Resumo
Nucleossomas são a unidade estrutural menor da cromatina, composto de 147 pares de bases de ADN envolvida em torno de um octâmero de proteínas histona. Histona função é mediada por extensa modificação pós-tradução por uma miríade de proteínas nucleares. Essas modificações são essenciais para a integridade nuclear como eles regulam estrutura e recrutar enzimas cromatina envolvidos na regulação do gene, a reparação do ADN e condensação cromossomo. Mesmo que uma grande parte da comunidade científica adota técnicas baseadas em anticorpos para caracterizar histona PTM abundância, essas abordagens são de baixo rendimento e tendenciosa contra proteínas hypermodified, como o epitopo pode ser obstruída por modificações nas proximidades. Este protocolo descreve o uso de cromatograf ia líquida nano (NLC) e espectrometria de massa (MS) para a quantificação precisa de modificações de histonas. Este método foi concebido para caracterizar uma grande variedade de PTMs de histonas e a abundância relativa de várias variantes dentro da histona sanalisa ingle. Neste protocolo, histonas são derivatizados com anidrido propiónico seguido de digestão com tripsina a gerar péptidos de 5 - 20 aa de comprimento. Após a digestão, os terminais N recentemente exposto da histona péptidos são derivatizados para melhorar a retenção cromatográfica durante NLC-MS. Este método permite a quantificação relativa de PTMs histona abrangendo quatro ordens de grandeza.
Introdução
Epigenética é definida como o estudo de alterações hereditárias na expressão do gene que surgem por outros que não altera a sequência de ADN subjacente uma mecanismos. regulação epigenética é fundamental durante o desenvolvimento como o organismo sofre fenotípica dramática muda, embora o seu conteúdo de DNA não muda. Existem vários componentes críticos necessários para a manutenção epigenética adequada, incluindo histonas modificações pós-traducionais (PTMs), variantes de histona, RNAs não-codificantes, a metilação do DNA e dos factores de ligação de ADN, cada um dos quais afectam a expressão do gene através de diferentes mecanismos 2. Por exemplo, enquanto que a metilação do ADN é uma modificação altamente estável que reprime a tradução do gene 3, variantes de histona e PTMs histonas são muito mais dinâmico e podem influenciar a cromatina numa variedade de maneiras 4.
PTMs histonas estão principalmente localizadas nas caudas N-terminais, como são a região mais exposta e flexívelda proteína. No entanto, o núcleo nucleossomo também é fortemente modificada em comparação com proteínas média 5. Mesmo que as marcas de histonas têm sido amplamente caracterizada, na última década, muitas ligações entre marcas histonas conhecidos e sua função ainda não são claras. Isto é principalmente devido ao fato de que a maioria dos PTMs histonas não trabalham sozinhos, mas sim a função em conjunto com outra PTMs ( "cross-talk") para alterar um processo específico, como a transcrição 6,7. Por exemplo, a marca H3S10K14ac combinatória no gene p21 activa a sua transcrição, o que não ocorreria com apenas um dos dois PTMs 8. Os compactos HP1 proteína cromatina, reconhecendo H3K9me2 / me3 e difundir a modificação nucleossomos nas proximidades. No entanto, HP1 não pode vincular H3K9me2 / 3 quando o S10 adjacente é fosforilada 9. Acetilação da H3K4 inibe a ligação da proteína para spChp1 H3K9me2 / me3 em Schizosaccharomyces pombe 10. Além disso, a histona d lisinaemethylase PHF8 tem a maior eficiência de ligação nucleossomo quando três PTMs H3K4me3, K9ac e K14ac estão presentes 11. Estes exemplos destacam a importância de alcançar uma visão global da evolução da PTM histonas, em vez de se concentrar em modificações individuais.
A presença de variantes de sequência também aumenta a complexidade da análise das histonas, como isotipos de histonas têm geralmente sequências muito semelhantes, mas muitas vezes têm papéis diferentes na cromatina. Por exemplo, H2A.x tem uma sequência C-terminal que é mais facilmente fosforilada após danos no DNA em comparação com H2A canônica 12, e é necessário para inativação de cromossomos sexuais em meiose ratinho macho 13; Da mesma forma, CENP-A substitui histona H3 canônica em centrômeros 14. Apesar das suas diferentes funções, estas variantes compartilham uma grande porção da sua sequência de aminoácidos com o respectivo histona canónica, tornando-o difícil de identificar e quantificar-los separadamente.
técnicas à base de anticorpos, tais como Western blotting foram amplamente adoptada para caracterizar histonas. No entanto, as abordagens baseadas em anticorpos são limitados pelas seguintes razões: (i) só se pode confirmar a presença de uma modificação e pode não identificar PTMs desconhecidos; (II) que são inclinadas devido à presença de sinais co-existentes, o que pode influenciar a afinidade de ligação; (iii) que não pode identificar marcas combinatória, uma vez que apenas muito poucos anticorpos estão disponíveis para tal fim e (iv) reagir de forma cruzada entre as variantes de histona altamente semelhantes ou PTMs semelhantes (por exemplo, di- e trimethylation de resíduos de lisina). Egelhofer et ai. descrito que mais de 25% dos anticorpos comerciais não testes de especificidade por dot blot ou Western blot e anticorpos específicos entre mais do que 20% falham em experiências de imunoprecipitação da cromatina 15. Espectrometria de massa (MS) está actualmente a ferramenta analítica mais apropriada para estudar novos e / ou combinatórios PTMs,e tem sido amplamente implementadas por proteínas histona (revisto em 16). Isto é principalmente devido à alta sensibilidade e precisão massa de MS, e a possibilidade de realizar análises em grande escala.
A estratégia de baixo para cima é o mais vulgarmente utilizado estratégia proteómica à base de MS para a caracterização de histona e seus PTMs, em que a proteína intacta é enzimaticamente digeridas em péptidos curtos (5-20 aa). Esta digestão facilita tanto a separação por LC e detecção por MS. Massas na faixa de 600 - 2000 Da são comumente mais facilmente ionizado e identificados com maior precisão em massa e resolução do que massas maiores. MS / MS de fragmentação é também melhorada, como péptidos curtos são geralmente bem adequada para a dissociação induzida por colisão (CID). No entanto, as histonas apresentam um desafio para-baixo para cima MS como eles são altamente enriquecida em resíduos de aminoácidos básicos, ou seja, a lisina e a arginina. Portanto, a digestão com tripsina conduz à geração de péptidos que são demasiado SMtudo para a retenção de LC e localização inequívoca do PTMs. Para contornar este problema, o nosso protocolo inclui lisina e peptídeo N-terminal química derivação 17. A utilização de anidrido propiónico é recomendado para a derivatização química eficiente em comparação com outros reagentes 18. Tais blocos de derivatização dos grupos ɛ-amino de resíduos de lisina não modificados e monometilo, permitindo tripsina para realizar a proteólise apenas no C-terminal dos resíduos de arginina. aminas derivatizados não pode trocar protões com a solução e, assim, os péptidos são geralmente só duplamente ou triplamente carregados, facilitando a detecção de MS e MS / MS. Além disso, a derivatização N-terminal aumenta a hidrofobicidade de péptidos e retenção cromatográfica de fase reversa assim. Aqui, descrevemos o fluxo de trabalho para purificar histonas e prepará-los para análise PTM via proteômica de baixo para cima (Figura 1). Esta estratégia atinge quantificação de histona marcas individuais e marcas de f combinatóriasou PTMs histonas, que são relativamente perto na sequência de aminoácidos.
Protocolo
1. Recolha de Células de Cultura
- Se as células foram cultivadas em suspensão, recolher as células por centrifugação a 300 rcf durante 5 min. Se o meio de células aderentes, aspirado e descarte. Lavar as células unidas com PBS sem Ca 2+ e Mg 2+ (referido como PBS daqui para frente). Incubar as células em qualquer tripsina ou tripsina-EDTA (0,025% - 0,5%, dependendo da linha celular) com um volume suficiente para cobrir a superfície das placas a 37 ° C até que as células de separar (tempo varia para diferentes linhas de células).
- Recolher as células por centrifugação a 300 rcf durante 5 min. Lave as células duas mais vezes em PBS e recolher por centrifugação.
- Estimar o volume globular aproximadamente desde as graduações marcadas em 1,8 ml tubos ou tubos de 15 ml cônico.
Nota: As células de cultura podem ser congeladas em azoto líquido e armazenado a -80 ° C indefinidamente nesta fase.
2. Isolamento de núcleos a partir de células intactas
- células descongelamento em gelo.
- Tampão de isolamento nuclear Thaw (NIB, Tabela 1).
- Prepare a aproximadamente 5 ml de tampão NIB (Tabela 1) para cada volume de células empacotadas 100 ul. Para cada 1 ml de tampão NIB, adicionar inibidores da protease e agentes de estabilização como se segue: 1 ul de DTT 1 M, 2,5 ul de AEBSF 200 mM, 2 ul de 2,5 uM e microcistina 2 ul de butirato de sódio 5 M. NIB com inibidores será referido como NIB a partir deste ponto em diante.
Nota: Se a fosforilação da histona é estudado, incluem EDTA protease livre e coquetel de inibidores de fosfatase. - Remover um quinto do volume do tampão NIB assim preparada e adicionar NP-40 Alternativa (Tabela 1) para uma concentração final de 0,2%. O volume de quatro quinto restante será utilizado para lavagens.
- Lavar sedimento celular em 01:10 sedimento celular para o NIB sem NP-40 rácio Alternativa (v / v). Remover o sobrenadante por centrifugação a 700 rcf durante 5 min.
- Lyse o cell sedimento, colocando-o em gelo e adicionando 01:10 sedimento celular para o NIB com 0,2% de NP-40 Alternativa (v / v).
- Se a extração de amostras de tecidos, homogeneizar utilizando homogeneizadores almofariz e pilão ou Dounce. células cultivadas podem ser homogeneizadas por pipetagem suave.
- Incubar as células homogeneizadas em gelo durante 5 - 10 min. As células irá lisar e libertar os núcleos.
- Centrifuga-se a 1000 RCF durante 5 - 10 min a 4 ° C. O sedimento contém principalmente núcleos celulares, enquanto que o sobrenadante contém principalmente componentes citoplasmáticos. Salve a fração citoplasmática, se desejar.
- Lavar o sedimento de núcleos por ressuspensão-o suavemente em 01:10 (v / v) NIB sem NP-40 alternativo.
Nota: Esta etapa de lavagem é apenas para remover vestígios de detergentes antes da extração de histonas dos núcleos. - Centrifuga-se a 1000 RCF durante 5 minutos a 4 ° C e remover o sobrenadante.
- Repita o passo 2.10 - 2.11 pelo menos duas vezes para remover completamente o NP-40 Alternativa. Remoção de NP-40 Alternativa é evidente as pipetagem suave durante a etapa de lavagem não forma bolhas.
- Para a extracção a partir de tecidos de histona:
- Lavar o tecido fresco congelado ou descongelado no NIB gelada.
- Transferir o tecido a uma placa de petri colocada em gelo com PONTA, apenas o suficiente para manter o tecido molhado.
- Dice em pedaços menores (<1 mm) com lâmina de barbear para aumentar a superfície de contato para o isolamento de núcleos.
- Transferir o tecido picada para um homogeneizador pré-refrigerados e lave em NIB pipetando cima e para baixo.
- Remover o tampão por centrifugação a 300 rcf durante 5 min.
- Adicionar NIB contendo NP-40 Alternativa para as células de uma células: tampão proporção de 1:10 (v / v), e homogeneizar por 5 - 10 pancadas.
- Verifique para a lise celular e repetir homogeneização, conforme necessário. Um bom indicador de que as células foram lisadas, é a redução do volume de sedimento. O sedimento deveria conter apenas núcleos.
- Centrifugar a 700 rcf durante 5 min e salvar pellet. Este sedimento pode ser extraída 1-2 mais tempos em 01:10 (v / v) de NIB contendo NP-40 Alternativa; Nesta fase, as histonas são extraídos para fora da cromatina e a pelete foi encolhida consideravelmente.
- Lavar duas vezes com 2 - 3 ml de NIB sem NP40 alternativo para remover os vestígios de detergente.
Nota: ponto de paragem Provisório: A amostra pode ser ressuspenso num volume mínimo de NIB + 5% de glicerol e armazenadas a -80 ° C.
3. Extracção e purificação de histonas de Núcleos
Nota: As histonas são muito ricas em resíduos de aminoácidos básicos, o que lhes permite interagir fortemente com a espinha dorsal do ácido fosfórico de ADN. As histonas estão entre as proteínas mais básicas no núcleo, permitindo-lhes assim serem extraídos em ácido sulfúrico arrefecido em gelo (0,2 H 2 SO 4) com contaminação mínima de proteínas não-histonas, que precipitam no ácido forte. TCA altamente concentrado (para uma concentração final de 33%) pode então ser usado para precipitar histonas do sulfúricoácido. TCA é armazenado como 100% no frasco de vidro castanho, a 4 ° C.
- Ressuspender em núcleos de células 1: 5 (v / v) arrefecida 0,2 H 2 SO 4 (Tabela 1) por pipetagem suave.
- Incubar a amostra com rotação constante ou agitação suave durante 2-4 h a 4 ° C. Tipicamente, para amostras com mais de 500 ul de sedimento celular, uma extracção de 2 horas é suficiente para extrair histonas; mais tempo de incubação pode resultar na extracção de outras proteínas básicas. Para sedimentos nucleares pequenos (<200 uL), 4 h de extracção fornece um rendimento melhor.
- Centrifuga-se a 3400 RCF a 4 ° C durante 5 min.
- Transferir o sobrenadante para um novo tubo.
- Repita os passos de 3,3-3,4 para remover qualquer material insolúvel.
- Para precipitar as histonas, adicionar refrigerados 100% de TCA (Tabela 1) ao sobrenadante recolhido (agora contendo as histonas) na proporção de 1: 3 (v / v), a fim de se obter uma concentração final de TCA de 33%. Misture invertendo o tubo um pouco times.
Nota: As amostras vai ficar turva por adição de TCA, o que indica a presença de histonas. - Incubar a mistura em gelo durante pelo menos 1 h. Para tamanhos menores de pelotas de partida, a precipitação durante a noite é recomendado.
- Centrifugar a 3400 rcf durante 5 min. O histonas revestir os lados das câmaras de ar e também depositar na parte inferior. Um sedimento branco insolúvel também forma a parte inferior do tubo, o qual contém principalmente proteínas não-histonas e outras biomoléculas. Remover o sobrenadante por aspiração, sem cuidadosamente raspando os lados ou do sedimento.
- Usando uma pipeta de Pasteur de vidro, enxaguar o tubo com gelo-acetona fria + 0,1% de HCl (Tabela 1) de modo a cobrir as proteínas precipitadas revestimento dos lados e fundo.
- Centrifuga-se a 3400 RCF durante 2 min e o sobrenadante aspirado, cuidadosamente sem raspar os lados ou do sedimento.
- Repita os passos 3,9-3,10 usando 100% de acetona gelada.
- grânulo seco com o fluxo de ar ou com um Vacuhum centrifugar, ou apenas deixando o tubo aberto. Acetona evapora-se rapidamente.
- Dissolver as histonas com DDH 2 O (água bidestilada) em volume mínimo possível dissolver a camada branca completamente. As histonas são facilmente solúveis em água. Para as pelotas em um tubo de microcentrífuga de 1,5 ml, 100 ul ddH2O é geralmente suficiente para recolher histonas.
- Centrifuga-se a 3400 RCF durante 2 min e transferir o sobrenadante para um novo tubo.
4. Estimativa da concentração de proteína e Pureza
- Para medir a concentração de proteína, utilizar BCA, ensaio de proteína de Bradford, ou a análise de aminoácidos (AAA). Não utilizar técnicas que adotam a absorvância a 280 nm, como histonas são pobres em resíduos de aminoácidos aromáticos.
- Verifique a pureza das histonas extraídos por análise de SDS-PAGE com um gel de acrilamida a 15% e coloração com Coomassie (opcional).
- Se alta pureza variantes única histonas são desejados, continuar a fracionamento HPLC-UV devariantes histonas (Seção 5). Se não, saltar directamente para preparação da amostra para análise histona PTM bottom-up (secção 6).
5. Separação de Histona Variantes por HPLC de fase reversa (Opcional)
Nota: variantes de histona de elevada pureza pode ser obtida por fraccionamento da mistura de histona bruto utilizando HPLC de fase inversa acoplado a um detector de UV. Estas histonas purificadas são úteis para estudos que requerem maior sensibilidade e pureza. No entanto, para a caracterização da histona PTM padrão, esta etapa pode ser ignorada, porque a análise é suficientemente sensível e exaustiva. Fraccionamento de histona intacta variantes idealmente requer, pelo menos, 100-300 ug de material de partida.
- Conectar uma coluna apropriada C 18 5 um e uma HPLC em função da concentração de histona de partida: com cerca de 100 ug de histonas, UTILIZADA 2.1 mm x 250 mm com um caudal de 0,2 ml / min; com cerca de 300 ug histonas, usar coluna 4,6 x 250 mm comum caudal de 0,8 ml / min. Preparar tampão A e B utilizando material de vidro, do seguinte modo:
- Preparar tampão A: 5% de acetonitrilo de grau HPLC, 0,1% de TFA em água de grau HPLC.
- Prepare Tampão B: 95% de acetonitrilo de grau HPLC, 0,1% de TFA em água de grau HPLC.
- Ligar a coluna a um detector de UV, e definir a absorvência a 210-220 nm.
- Acidifica-se a amostra de histona dissolvida em água com 100% de TFA para obter uma concentração final de 0,1-1% de TFA.
- Equilibrar a coluna com 100% de tampão A durante pelo menos 15 min no caudal recomendado, o que corresponde, aproximadamente, a três volumes de coluna. Usar este sinal para definir o nível zero de absorvância do detector de UV.
- Prepare tubos dimensionados adequadamente para recolher fracções manualmente ou em um coletor automático de amostras.
- Injectar amostra a uma concentração de aproximadamente 1 ug / ul ou superior. As amostras dissolvidas em volumes maiores pode alterar o equilíbrio da coluna duCarregando ringue e levar à retenção inferior.
- Executar o gradiente, programado como se segue: 0-30% de B em 1 min, de 30 a 60% de B em 90 min, e 60 a 90% de B em 1 min.
- Recolha fracções (cromatograma exemplo mostrado na Figura 2) em intervalos de 1 min, utilizando um colector de fracções automático. Recolha fracções em tubos com as dimensões apropriadas para conter a totalidade do volume.
- Seque as amostras fracionadas em um concentrador de vácuo.
Nota: ponto de parada Intercalar: frações de histonas secas podem ser armazenadas à temperatura ambiente durante curtos períodos (1 - 2 dias) ou em -80 ° C congelador para períodos de longo prazo.
6. Chemical Derivatização de histonas Usando Propionic anidrido de Análise Bottom-up
- (Quantidade recomendada: 50 - 100 ug) Dissolve-se amostras de histonas em 40 ul de 50 mM NH 4 HCO 3, pH 8,0. Se as amostras foram em puro ddH2O, adicionar concentrada de NH 4 HCO 3 a tornar-se 50 mM, pH 8.0.
- Molhe uma ponta P10 pipeta na amostra para verificar o pH utilizando papel indicador de pH sem perdas amostrais. NH 4 OH e ácido fórmico pode ser usado para ajustar o pH a 8,0.
Nota: A seguinte parte do protocolo (passos 6,3-6,7) deve ser realizado em lotes de máximo de três a quatro amostras, a fim de manter o anidrido propiónico reactivo. - Use exaustor para as etapas subsequentes se anidrido propiônico é usado. Prepare propionylation reagente fresco por mistura de anidrido propiónico com acetonitrilo na proporção de 1: 3 (v / v). Adiciona-se reagente propionylation a amostra em 1: 4 (v / v). Por 40 histonas ml, adicionar 10 reagente propionylation ul.
Nota: É possível observar detritos branco nesta fase. No entanto, este contém principalmente sais e ácido propiónico, e, assim, nenhuma acção específica deve ser tomado. - Rapidamente adicionar NH4OH, para re-estabelecer a pH 8,0 para a solução. Nota: anidrido propiónico reagir com as aminas livres dos péptidos produz propácido iónico que diminui o pH. Geralmente, a adição de NH4OH, para a amostra com um rácio de 1: 5 (v / v) é apropriado para restabelecer o pH 8.0; por exemplo, 8 mL de NH 4 OH a 40 ul de amostra.
- Misturar imediatamente por vórtice.
- Verificar o pH com o mesmo procedimento como no passo 6.2.
Atenção: Quando o pH é superior a 10,0, a rotulagem de outros resíduos de aminoácidos, com maior pKa é possível. - Incubar as amostras à temperatura ambiente durante 15 min.
- Repita os passos 6,3-6,7, realizando estritamente a reação para não mais do que 3 ou 4 amostras por lote de reagente propionylation.
- As amostras secas para 10 - 20 mL num concentrador de vácuo. Este evapora-se anidrido propiónico que não reagiu, acetonitrilo, ácido acético e gás amoníaco libertado a partir de NH 4 OH. Se as amostras de secar completamente, sem perdas amostra significativa ocorrer.
Nota: isopropanol podem ser utilizados em vez de acetonitrilo. No entanto, tem de acetonitrilo inferior a tensão superficial e, portanto, maisevaporação rápida. - Volte a suspender ou diluir as amostras com ddH2O até 40 l de volume final seja alcançado.
- Repita os passos 6,2-6,9. A rodada dupla de propionylation histona garante> 95% de conclusão da reacção.
- Encha garrafa anidrido propiónico com gás árgon, de modo a prevenir a formação de ácido acético em contacto com a humidade na garrafa.
Nota: ponto de paragem Provisório: A amostra pode ser armazenada a -80 ° C reconstituído em ddH2O ou secas.
7. Digestão proteolítica com tripsina
- Histonas Ressuspender em 50 mM NH 4 HCO 3 para atingir uma concentração óptima de 1 ng / ul ou superior. Mais amostras diluídas levam a uma diminuição da eficiência de tripsina.
Nota: As histonas nesta etapa precisa estar no pH 8,0 Se ainda ácido, em seguida, adicione NH 4 HCO 3 sal para provar usando ponta da pipeta. - Adicionar tripsina às amostras de histona a uma razão de 1:10 (p / p).
- Incubate a 37 ° C durante 6-8 h.
- Parar a digestão por congelação em -80 ° C.
- Seca-se a amostra para 10 - 20 mL num concentrador de vácuo.
Nota: ponto de paragem Provisório: A amostra pode ser armazenada a -80 ° C.
8. Propionylation de Histona Os péptidos no terminal N
Nota: Esta secção descreve a derivatização de péptido de terminal N gerado a partir da tripsina Digest. Este procedimento melhora a retenção de HPLC dos péptidos mais curtos (por exemplo, aminoácidos 3-8 de histona H3), como do grupo propionilo aumenta a hidrofobicidade dos péptidos.
- Suspenda as amostras em 30 ul de 100 mM NH 4 HCO 3.
- Repita os passos 6,1-6,9.
Nota: É normal que a secagem de amostras em vácuo leva mais tempo a esta etapa. - Ressuspender ou diluir as amostras com 50 - 100 ul de ddH2O + quer TFA a 0,1% ou 0,5% de ácido acético. Nota: O ácido acético é recomendado por longos armazenamentos, como TFA facilitates de oxidação de metionina, a longo prazo. Por outro lado, TFA é recomendado se stage-depósito (secção 9) é realizada no mesmo dia, como TFA auxilia uma melhor retenção cromatográfica.
Nota: ponto de paragem Provisório: A amostra pode ser armazenada a -80 ° C.
9. Dessalinização da amostra com estágio dicas
Nota: Nesta fase, existe sal presente na amostra. Sais impedir a análise HPLC-MS porque ionizar durante a electropulverização, a supressão do sinal a partir de péptidos. Os sais também podem formar aductos iónicos em péptidos, reduzindo a intensidade do sinal para o péptido não-aduzido. Como o péptido aduzido terá uma massa diferente, o péptido não será devidamente identificados ou quantificados.
- Usando uma ponteira P1000, perfurar um disco de C 18 material de um disco de extração em fase sólida. Empurrar o minidisk para fora da ponta P1000 usando um capilar de sílica fundida e depositar o minidisk para o fundo de uma ponta de pipeta P100 / 200. Certifique-se de que o dISK está firmemente entalada na parte inferior da ponta (Figura 3).
Nota: A ponta P1000 tem um pequeno furo para perfurar o disco C 18. É apropriado para cortar o último centímetro da ponta, a fim de ter um furo com o diâmetro maior. - Use dois C 18 socos na mesma ponta P100 / P200 se dessalinização mais de 25 mg de amostra.
- Use um adaptador de centrífuga para manter estágio-dicas em prática em 1,5 ml ou 2 tubos ml de microcentrífuga. Use lento - rotação (300 400 rcf); os dissolventes passam normalmente através da resina em menos de um minuto.
- Lavar a resina por fiação com 50 ul de 100% de acetonitrilo para activar o material 18 C e remover potenciais contaminações.
- Equilibrar disco através de lavagem 80 mL de 0,1% de TFA por centrifugação lenta.
- Acidifica-se a amostra a pH 4,0 ou inferior, com ácido acético. Verificar o pH com tiras de pH para minimizar a perda de amostra. amostra de carga para o disco por centrifugação lenta.
- lavaramostra por lavagem 70 - 80 ul de 0,1% de TFA por centrifugação lenta.
- Eluir amostra por lavagem de 70 mL acetonitrilo a 75% e 0,5% de ácido acético por centrifugação lenta. Recolher a amostra para um tubo de 1,5 ml.
- amostra seca num concentrador de vácuo.
Nota: ponto de paragem Provisório: A amostra pode ser armazenada a -80 ° C.
10. Análise de Peptídeos histona
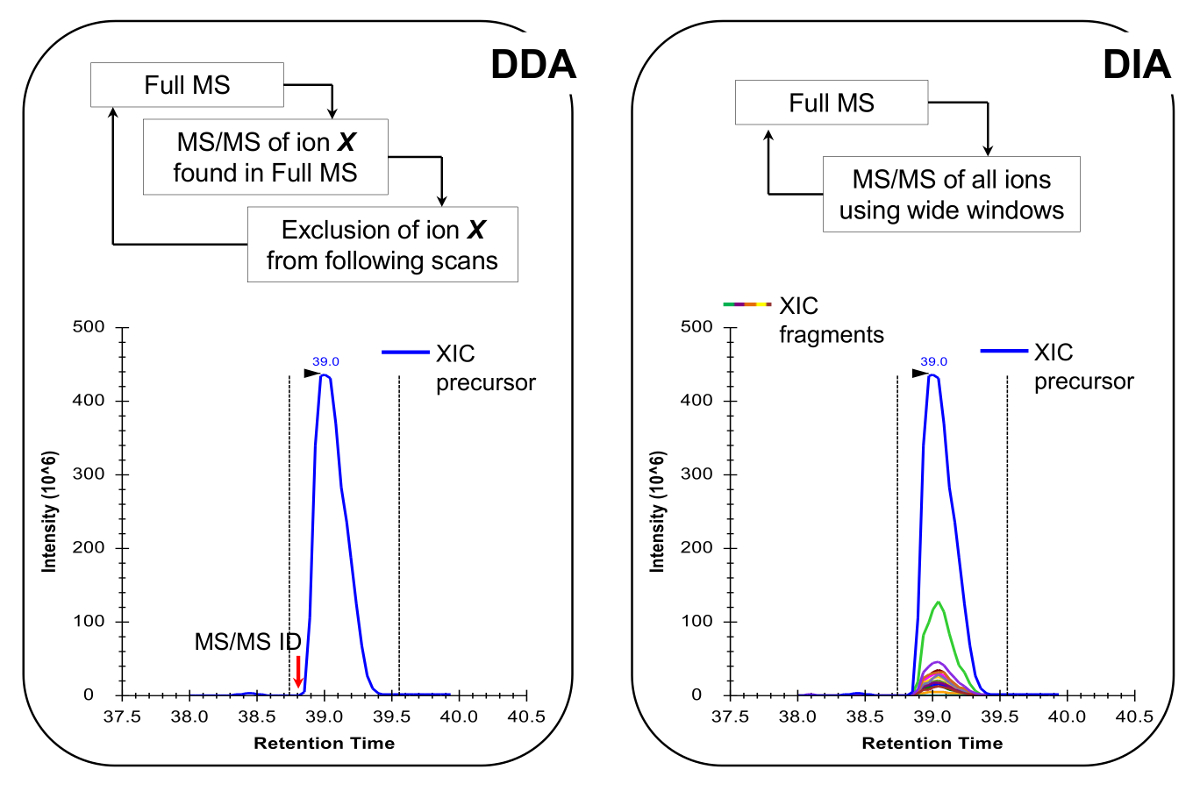
Nota: A plataforma NLC-MS deve ser configurado como feito na análise peptídeo tradicional. O uso de 200-300 coluna de fluxo nl (75 mm ID coluna analítica, C 18 partículas) é recomendado, pois eles são um excelente compromisso entre a sensibilidade e estabilidade. O método de aquisição MS pode ser uma combinação de aquisição dependente de dados (DDA) com verificações direcionadas 19 ou a aquisição de dados independente (DIA) 20,21, ambos descritos em resultados representativos e Figura 4.
- Preparar tampões de HPLC - A: 0,1% de ácido fórmico emágua HPLC-grade; B: ido fmico 0,1% em acetonitrilo de grau HPLC.
- Programar o método de HPLC como segue: a partir de 0 a 30% de tampão B em 30 min, de 30 a 100% de B durante os próximos 5 min e à isocrático 100% de B durante 8 min. Se a HPLC não está programado para equilibração em coluna automatizada antes do carregamento da amostra, em seguida, incluir o seguinte: gradiente de 100 a 0% B em 1 min e fluxo isocrático a 0% de B durante 10 min. Definir a taxa de fluxo da análise de 250-300 nl / min.
- Programar o método de aquisição MS para executar qualquer DDA combinado com verificações direcionadas 19 ou DIA 20,21 (Figura 4). Certifique-se de que o ciclo de trabalho MS permite uma verificação completa do MS cada ~ 2 segundos, a fim de ter pontos de dados suficientes para todo o pico cromatográfico, o que permite a quantificação mais precisa. Nota: Com C 18 cromatografia, a largura média da linha de base do pico é de cerca de 30 seg para o gradiente acima descrito.
- Carregar cerca de 1 ^ g de amostra na coluna de HPLC Column.
- Executar o método de HPLC-MS / MS como programado.
Nota: No protocolo não recomendamos especificidades de colunas, instrumentos MS ou detalhes dos parâmetros de MS, como qualquer configuração ideal que um laboratório de proteômica indivíduo desenvolvido será adequado para o método. laboratórios Proteomics deve usar a sua configuração otimizada, uma vez que os péptidos histonas separar como peptídeos tradicionais.
Análise 11. Dados
- Importar os arquivos de matérias-MS em software para realizar a integração da área dos picos. Nota: EpiProfile 22 é recomendado, pois é otimizado para peptídeos histonas; usando o conhecimento do tempo de retenção cromatograf ico de eluição realiza a extracção de péptidos conhecidos histona fiável área do pico. Alternativamente, Horizonte 23 é um outro software ideal para a finalidade.
- Calcular a abundância relativa de um dado péptido dividindo a sua área de superfície total do referido péptido em todas as suas formas modificadas. Nota: Em caso de Analy descobertasis Mascot é recomendado para identificar os espectros de péptidos modificados histonas. O desempenho desta ferramenta tem sido descrito recentemente 24. Todos os outros motores de busca de banco de dados para proteômica também são utilizáveis, mas em nossos testes que proporcionou desempenho inferior.
Resultados
Como exemplo, foram analisadas as histonas extraídos a partir de células estaminais embrionárias humanas (hESCs) com e sem ácido retinóico estimulação (RA), começando com 200 ul de sedimentos celulares. Presença de RA em cultura de células leva à diferenciação de ESC. A partir do sedimento de células, cerca de 50 - 100 ug de histonas foram extraídas, o que é mais que suficiente para efectuar injecções múltiplas de LC-MS de péptidos de histonas. Após derivatização, a digestão, e dessalinização, as amostras foram carregadas num 75 uM cm x 15 cm coluna C 18 (diâmetro de partícula de 3 um, tamanho de poro de 300 Â) no modo em série com um sistema de cromatografia em nano líquida de alta eficiência com chips de microfluidos acoplados aos uma armadilha de quadrupolo linear híbrida - espectrômetro de massa Orbitrap. aquisição MS foi realizada utilizando DIA. Em paralelo, as amostras foram também analisadas com um método utilizando uma DDA UHPLC nano-fluxo acoplado a um espectrómetro de massa de armadilha de ião híbrido-Orbitrap (dados não mostrados). Dentrocada ciclo, uma detecção completa MS Orbitrap foi realizada com a gama de varrimento de 290 a 1400 m / z, a resolução de 60.000 (a 200 m / z) e de AGC de 10 6. Em seguida, o modo de aquisição dependente dos dados foi aplicada com uma dinâmica exclusão de 30 seg. scans MS / MS foram seguidos em iões parentais desde os mais intensos. Os iões com um estado de carga de um foram excluídos da MS / MS. Utilizou-se uma janela de isolamento de 2 m / z. Os iões foram fragmentados utilizando induzida por colisão de dissociação (CID) com uma energia de colisão de 35%. Detecção ion trap foi utilizado com o modo de faixa de varredura normal e taxa de varredura normal com AGC, de 10 4.
Os dados em bruto MS foram analisados adotando software para a extração de precursores e de iões de fragmento de cromatogramas, ou seja, Skyline 23 e EpiProfile 22. EpiProfile foi optimizado para peptídeos histonas, uma vez que integra a extração área do pico inteligente devido ao conhecimento prévio de peptempo de retenção maré. Por outro lado, Skyline é otimizado para análises DIA, e, assim, os números DIA apresentados (Figuras 4 e 5A) são screenshots deste software. A partir do cromatograma iónico extraiu-se, a área sob a curva é recuperado, e este é utilizado para calcular a abundância de cada péptido. A área do pico cromatográfico foi calculado para [M + H] +, [M + 2H] 2+, e [M + 3H] 3+ da mesma péptido, embora na maioria dos casos, o [M + 2H] 2+ foi a forma predominante. Isto proporciona a abundância em bruto de uma dada forma modificada de um péptido. A fim de alcançar a abundância relativa de PTMs, a soma de todas as formas modificadas diferentes de um péptido de histona foi considerada como 100%, e a região do peptídeo particular, foi dividida pela área total para que o péptido de histona em todas as suas formas modificadas .
péptidos de histonas estão presentes numa VAriety de formas isobáricas (Figura 5). Péptidos isobáricas, por exemplo, K18ac e K23ac, só podem ser quantificados no nível do MS / MS, em que os seus iões do fragmento original são utilizados para determinar a proporção das espécies isobárica (Figura 5A e 5B). Esta relação é utilizada para dividir a área do pico cromatográfico entre as duas espécies. Quando utilizando DDA, estas formas isobáricas foram incluídos numa lista de massas específicas, porque estes péptidos devem ser seleccionados para a fragmentação através de toda a sua eluição, o que não ocorreria numa experiência de DDA padrão. A discriminação da abundância relativa da espécie isobáricas é então realizada através da monitorização do perfil de eluição dos iões do fragmento. Por outro lado, DIA tipo de aquisição não requer qualquer lista de inclusão. No entanto, este tipo de método de aquisição não é compatível com a pesquisa na base de dados tradicionais, e portanto pode impedir a descoberta dos péptidos modificados desconhecidos.
Lisina acetilação (+ 42,011 Da) foi discriminado do trimethylation quase isobaric (+ 42,047 Da) usando alta resolução de aquisição MS (> 30.000). Além disso, a acetilação é mais hidrofóbica do que trimethylation, levando a eluição de péptidos acetilados mais tarde do que os respectivos queridos trimethylated. A forma não modificada do mesmo péptido elui mesmo mais tarde, devido ao fato de que a lisina é propionylated. Em resumo, a ordem de hidrofobicidade para um péptido com um local modificável é di- e trimetilada
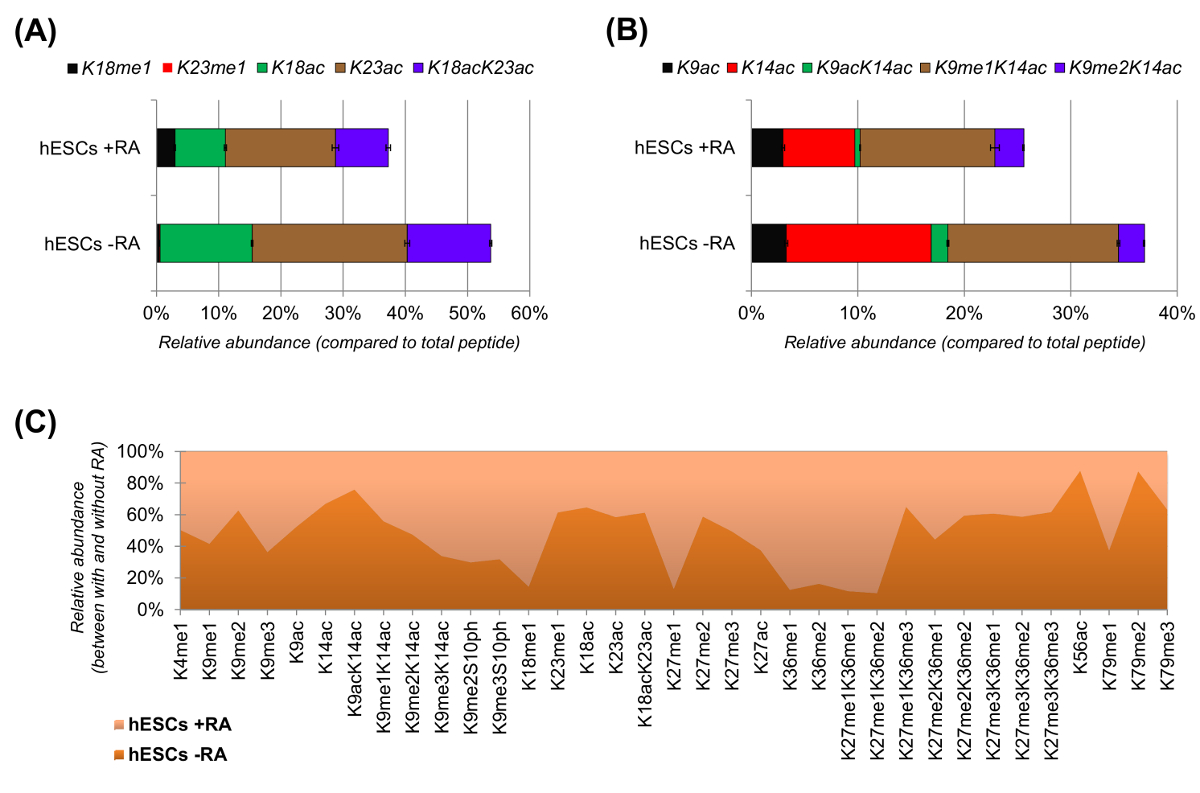
hESCs mostrou uma clara redução de peptídeos acetilados quando estimulados para a diferenciação (Figura 6A e 6B). Este não foi surpreendente, pois os resultados anteriores relataram maior acetilação na CES, em comparação com diferenciando os 25,refletindo a natureza geralmente permissiva da cromatina pluripotentes. Ao se concentrar em histona H3, 35 formas modificadas diferentes foram quantificados (Figura 6C). No entanto, todos os proteoforms histona que podem ser investigadas com esta abordagem são mais do que 200, incluindo todas as variantes e modificações das histonas de baixa abundância (dados não mostrados). Além disso, a análise mostrou que a elevada reprodutibilidade pode ser obtido entre repetições técnicas, como evidenciado pelo pequeno tamanho das barras de erro representam (± desvio padrão). No seu conjunto, esta seção descreve como extrair a abundância relativa de peptídeos histonas modificadas usando dados NLC-MS.

Figura 1:. Fluxo de trabalho para o MS / MS Análise Histona de baixo para cima Os dez passos para a análise das histonas são mostradas, incluindo uma estimativa do tempo necessário para cada passo. O número da seção é dado entre parênteses como presente no manuscrito. Seção 5, descrevendo fracionamento da amostra para isolar as diversas variantes de histonas, pode ser omitida a menos que haja uma necessidade de análise altamente sensível de uma determinada variante. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: Fase Inversa High Flow LC para histona Variant Fracionamento e Coomassie Gel (A) cromatograma LC-UV representando separação histona intacta.. variantes de histona H3 podem ser discriminados a partir de um outro de acordo com o seu tempo de eluição. As frações podem ser recolhidos manualmente ou utilizando um colector de fracções automático. (B) gel Coomassie de três réplicas de purificação de histona.= "Https://www.jove.com/files/ftp_upload/54112/54112fig2large.jpg" target = "_ blank"> Clique aqui para ver uma versão maior desta figura.

Figura 3:. Making of Stage-tombamento ficha com uma ponteira P1000, perfurar um disco feito de C 18 material de um disco de extração em fase sólida (segundo painel). O minidisk vai ficar na ponta (painel do meio), de modo que pode ser empurrado para fora para uma ponta de pipeta menor P100 / 200 usando qualquer tipo de pequenos capilares. Neste exemplo, foi utilizado um diâmetro exterior tubo de sílica fundida 700 mm. O minidisco deve ser empurrada para o fundo do P100 / 200 ponteira até que ele não pode ir (último painel) ainda mais. A ponta de fase está pronto para dessalinização de histona, uma vez que tem capacidade suficiente para reter o material de amostra suficiente para numerosas repetições. Especificamente, um minidisk é suficiente para 15 - 20? G de SAmple. Se mais de amostra é necessária, vários discos pode ser embalado em um outro. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4:. Representação esquemática do DDA e DIA Métodos Ao utilizar DDA, o ciclo de varredura MS é caracterizada pela seleção seqüencial de íons precursores para MS / MS fragmentação de acordo com a sua intensidade e estado de carga. Uma vez que um ião precursor foi fragmentado é colocado em uma lista de exclusão para evitar a selecção repetitiva do mesmo péptido, de modo que a MS pode "escavar" em sinais menos abundantes. Este método de aquisição é a técnica de escolha em proteômica para o modo de descoberta. A quantificação é conseguida através da integração do sinal de varrimento completo de um determinado ião junto aos identificados MS /espectro de MS. No DIA, todo o intervalo de razões m / z é fragmentada em cada ciclo de verificação. Esta abordagem é menos adequado para o modo de descoberta, mas produz um perfil cromatográfico de todos os iões, os precursores e os produtos. Isto leva a quantificação mais confiante e discriminação de formas isobáricos. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 5: Quantificação de isobáricas péptidos (A) Exemplo de dois péptidos isobáricas geralmente abundantes em análise de histona.. O cromatograma de iões extraído (XIC) da sua massa precursor e isótopos relativos (acima) é idêntica. No entanto, o XIC dos iões dos produtos (a seguir) permite a discriminação das duas formas isobáricas. Notavelmente, iões fragmentados exclusivos somente deve ser-nosEd para estimar a abundância relativa das duas espécies. (B) Representação dos iões do fragmento original para os dois péptidos descritos (realçado a vermelho). (C) Lista dos péptidos vulgarmente analisados em Homo sapiens possuindo, pelo menos, um equivalente isobárica. Variantes de sequência entre os peptídeos histonas listadas são indicadas. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 6: Os resultados representativos de células estaminais embrionárias humanas com e sem tratamento com ácido retinóico (A) A quantificação relativa do peptídeo histona H3 KQLATKAAR (aa 18-26) em todas as suas proteoforms modificados.. A abundância relativa foi estimada utilizando todos os proteoforms como 100% (o relpercentagem operatória do péptido não modificado não é mostrado) (B) A quantificação relativa do péptido de histona H3 KSTGGKAPR (AA 9 -.. 17) (C) relativa abundância de péptidos detectada histona H3 canónica com e sem tratamento de células com ácido retinóico. A figura indica em qual dos dois tratamentos dadas as modificações são mais abundante (> 50%). No geral, nós demonstramos que a histona H3 acetilação diminui na maioria dos resíduos de lisina sobre a indução de diferenciação celular. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
| solução # | Composição | ||||||
| 1 | O isolamento nuclear Buffer (NIB) estoque é feita como se segue e armazenado congelado como alíquotas de 100 ml à temperatura de -20 ° C; descongeladoNIB podem ser armazenadas a 4 ° C durante algumas semanas: Tris 15 mM, KCl 60 mM, NaCl 15 mM, MgCl 2 5 mM, CaCl 2 1 mM, e 250 mM de sacarose. O pH do tampão é ajustado para 7,5 com HCl. | ||||||
| 2 | Os inibidores de protease (adicionar fresco para tampões antes do uso): 1 M ditiotreitol (DTT) em ddH2O (1.000X); AEBSF 200 mM em ddH2O (400x) | ||||||
| 3 | inibidor de fosfatase (adicionar fresco para buffers antes de usar): 2,5 mM microcistina em 100% de etanol (500x) | ||||||
| 4 | inibidor HDAC (adicionar fresco para buffers antes da utilização): 5 butirato de sódio M, feitas por titulação do ácido 5 M butírico usando NaOH para pH 7,0 (500x) | ||||||
| 5 | NP-40 Alternativa: 10% v / v em ddH2O | ||||||
| 6 | 0,2 MH 2 SO 4 em ddH2O | ||||||
| 7 | Ácido tricloroacético (TCA): 100% w / v em ddH2O | ||||||
| 8 | Acetona + 0,1% de ácido clorídrico (HCl): 0,1% v / v de HCI em acetona | ||||||
Tabela 1. Solutions.
Discussão
O protocolo aqui descrito é otimizada considerando custos, tempo e desempenho. Outras preparações são possíveis, mas eles têm limitações, especialmente no caso de acoplamento com análise por MS. Por exemplo, o protocolo de extracção de alto teor salino pode ser utilizada para purificar histonas 26, em vez da precipitação com TCA (secção 3). protocolo de alto teor salino é intrinsecamente mais suave, uma vez que não utiliza um ácido forte. Isso preserva PTMs lábeis a ácidos e aumenta o rendimento das histonas extraídos, como precipitação TCA co-precipita muitas outras proteínas de ligação da cromatina. No entanto, a extracção de alto teor salino leva a amostras que contêm o sal muito concentrada por HPLC-MS / MS. Numa preparação alternativa, a digestão de histona pode ser realizada sem propionylation (secção 6-8), por exemplo, reduzindo o tempo de incubação com tripsina e a proporção de enzima / substrato 27 ou usando argc com enzima de digestão 28-30. No entanto, derivatização com anidrido propiônico é recomendado, pois iT conduz à geração de peptídeos mais hidrofóbicos, que são melhor retidos durante a cromatografia líquida.
Para a derivatização química, uma variedade de anidridos de ácidos orgânicos foram avaliados e os seus méritos exaustivamente discutidos 18. No entanto, anidrido propiônico provou o melhor compromisso entre eficiência, produtos colaterais minimizados e melhoria da hidrofobia peptídeo. Potencialmente, anidrido propiónico pode ser adquirida na forma isotopicamente marcada; esta permite a análise de multiplexagem, devido à possibilidade de múltiplas amostras de mistura e discriminar-las ao nível de MS baseado nos diferentes massas transmitidos a partir do rótulo pesado. No entanto, esta análise conduz a um aumento na complexidade do cromatograma LC-MS e reduz a quantidade de amostra que pode ser injectado para cada condição única.
A este respeito, deve-se destacar alguns aspectos críticos do protocolo. O seguinte deve ser usada como checklist para encontrar erros na realização do procedimento no caso são obtidos resultados negativos. Em primeiro lugar, após precipitação núcleos o sedimento deve ser cuidadosamente lavado com NIB sem NP-40 Alternativa (secção 2.10) até remoção completa do detergente (notável pela falta de bolhas durante a mistura). Não fazer isso poderia comprometer a extração de histona com ácidos. Em segundo lugar, após precipitação histona com TCA (Ponto 3.9) lavagens do sedimento com acetona é crucial. Presença de ácido concentrado prejudicaria o seguinte passo se propionylation e digestão (secção 6.1) são realizadas diretamente. Não seria problemático em fracionamento caso histona é realizada (seção 5). Em terceiro lugar, é essencial que a reacção é realizada rapidamente propionylation (secção de 6,3-6,7). Para fazer isso, evite usar a mesma mistura propionylation (anidrido propiônico + acetonitrilo) por mais de 3 - 4 amostras consecutivas. Além disso, o pH é o aspecto mais importante da digestão de tripsina (secção 7). Se nãocerca de 8,0 (7,5-8,5) a digestão será ineficaz. Isto pode acontecer, como a amostra será rica em ácido propiónico neste passo. NH4OH pode ser adicionado até que seja necessário. Além disso, para os investigadores familiares com fluxos de trabalho proteômica ele vai se sentir normal para acidificar a amostra para terminar a digestão de tripsina. Isso não deve ser feito, uma vez que irá comprometer a seguinte reacção, ou seja, propionylation do peptídeo N-terminais (seção 8.1). Finalmente, na mesma edição, é importante lembrar-se de análise de dados que os peptídeos não modificados não são, na verdade, não modificado; todos os resíduos de lisina livre e N-terminais serão ocupados por propionylation (56,026 Da). Assim, a realização de cromatografia iónica extracção da massa correspondente exclusivamente à sequência peptídica conduziria a nenhum resultado.
As limitações do método são principalmente relacionado com a incapacidade de detectar PTMs combinatórias, devido às sequências peptídicas curtas, e os desvios na realização do verdadeiro Abundança de uma modificação, devido ao fato de que os péptidos em diferentes formas modificadas podem ionizar com eficiências diferentes. O primeiro problema pode ser resolvido por combinar esta técnica com uma meia-baixo ou abordagem de cima para baixo (revisto em 16). Este tipo de análise, mesmo se tecnicamente mais desafiador, é ideal para estudar freqüências co-existência de modificações. Além disso, ele permite uma melhor discriminação de variantes de histona, que nem sempre podem ser alcançados com baixo para cima uma vez que alguns péptidos têm a mesma sequência em diferentes variantes de histona. O segundo problema, relacionado com a eficiência de ionização, pode ser resolvido usando uma biblioteca de péptidos sintéticos 31. Esta abordagem assegura uma estimativa mais precisa da abundância relativa de PTMs histonas. No entanto, na maioria dos experimentos, o resultado desejado é que as alterações relativas de dados modificações entre as condições analisadas. Neste caso, essa correcção não é necessário, devido ao facto de todas as amostras têm os mesmos BIAs.
Em conclusão, este protocolo permite a análise das histonas PTMs que pode ser completada em 3 dias utilizando NLC acoplado a MS em tandem. Comparações com outros do que MS técnicas, ou seja, o uso de estratégias de anticorpos baseada como discutido na introdução, não são adequados, como eles não podem conseguir ainda quase esse nível de taxa de transferência. Além disso, técnicas de anticorpo com base não permitem a descoberta de novas modificações, mas que se baseiam exclusivamente em confirmar e quantificar marcas previstas. Nós, portanto, especular que proteômica bottom-up em peptídeos histonas vai ganhar popularidade em laboratórios proteômica devido às vantagens intuitivas em saber o regulamento de marcas de histonas, que são protagonistas na expressão gênica de sintonia e, portanto, afetam a regulação do proteoma. Além disso, o protocolo descrito inclui melhorias recentes na preparação de amostras e software para análise de dados, que fazem análise de histona mais trivial também para laboratórios que nunca experimentaram caracterização desse tipo de péptidos hypermodified.
Divulgações
Os autores declaram que não têm interesses financeiros concorrentes.
Agradecimentos
Este trabalho foi apoiado pelo financiamento do NIH subvenções (DP2OD007447, R01GM110174 e R01AI118891).
Materiais
| Name | Company | Catalog Number | Comments |
| Trypsin 0.25% EDTA | Invitrogen | 25200056 | For harvesting cells |
| PBS | Invitrogen | 14200075 | |
| Tris | Roche | 77-86-1 | |
| Potassium Chloride | Fisher Scientific | BP366-500 | |
| Sodium Chloride | Sigma | S9888 | |
| Magnesium Chloride hexahydrate | Sigma | M9272 | |
| Calcium Chloride, anhydrous | Sigma | C1016 | |
| Sucrose | Fisher Scientific | BP220-1 | |
| DTT | Invitrogen | 15508-013 | |
| AEBSF | EMD Millipore Corp | 101500 | |
| Microcystin | Sigma | M4194 | |
| Sodium Butyrate | Sigma | B5887 | |
| Halt Protease and Phosphatase Inhibitor Cocktail, EDTA-free (100x) | Fisher Scientific | 78445 | |
| NP- 40 Alternative | CALBIOCHEM | 492016 | |
| Sulfuric Acid, ACS grade | Fisher Chemical | 7664-93-9 | |
| Trichloroacetic acid | Sigma | T6399 | |
| Acetone | Sigma | 179124 | |
| HCl | Fisher Chemical | A144-500 | |
| Bradford reagent | Biorad | 500-0006 | |
| 30% acrylamide/bis 29:1 — 500 ml | Biorad | 1610156 | |
| Coomassie | Fisher Scientific | 20278 | |
| C18 Column (5 µm) 2.1 mm x 250 mm | Grace | 218TP52 | |
| C18 Column (5 µm) 4.6 mm x 250 mm | Grace | 218TP54 | |
| HPLC grade acetonitrile | Fisher Chemical | A955-4 | |
| HPLC grade water | Fisher Scientific | W6 4 | |
| TFA | Fisher Scientific | A11650 | |
| Ammonium Bicarbonate | Sigma | A6141 | |
| ammonium hydroxide | Sigma | 338818 | |
| propionic anhydride | Sigma | 240311 | |
| Sequencing grade modified trypsin | Promega | PRV5113 | For digesting histones for MS |
| Acetic Acid | Sigma | 49199 | |
| C18 extraction disk | Empore | 2215 | |
| Formic Acid | Sigma | F0507 |
Referências
- Waddington, C. H. Canalization of development and the inheritance of acquired characters. Nature. 150, 563-565 (1942).
- Sharma, S., Kelly, T. K., Jones, P. A. Epigenetics in cancer. Carcinogenesis. 31 (1), 27-36 (2010).
- Reik, W., Dean, W., Walter, J. Epigenetic reprogramming in mammalian development. Science. 293 (5532), 1089-1093 (2001).
- Kouzarides, T. Chromatin modifications and their function. Cell. 128 (4), 693-705 (2007).
- Tessarz, P., Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Bio. 15 (11), 703-708 (2014).
- Fischle, W., Wang, Y. M., Allis, C. D. Histone and chromatin cross-talk. Curr Opin Cell Biol. 15 (2), 172-183 (2003).
- Lee, J. S., Smith, E., Shilatifard, A. The language of histone crosstalk. Cell. 142 (5), 682-685 (2010).
- Simboeck, E., et al. A Phosphorylation Switch Regulates the Transcriptional Activation of Cell Cycle Regulator p21 by Histone Deacetylase Inhibitors. J Biol Chem. 285 (52), 41062-41073 (2010).
- Hirota, T., Lipp, J. J., Toh, B. H., Peters, J. M. Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature. 438 (7071), 1176-1180 (2005).
- Xhemalce, B., Kouzarides, T. A chromodomain switch mediated by histone H3 Lys 4 acetylation regulates heterochromatin assembly. Genes Dev. 24 (7), 647-652 (2010).
- Vermeulen, M., et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. 142 (6), 967-980 (2010).
- van Attikum, H., Gasser, S. M. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol. 19 (5), 207-217 (2009).
- Fernandez-Capetillo, O., et al. H2AX is required for chromatin remodeling and inactivation of sex chromosomes in male mouse meiosis. Dev Cell. 4 (4), 497-508 (2003).
- Santaguida, S., Musacchio, A. The life and miracles of kinetochores. Embo J. 28 (17), 2511-2531 (2009).
- Egelhofer, T. A., et al. An assessment of histone-modification antibody quality. Nat Struct Mol Biol. 18 (1), 91-93 (2011).
- Sidoli, S., Cheng, L., Jensen, O. N. Proteomics in chromatin biology and epigenetics: Elucidation of post-translational modifications of histone proteins by mass spectrometry. J Proteomics. 75 (12), 3419-3433 (2012).
- Plazas-Mayorca, M. D., et al. One-Pot Shotgun Quantitative Mass Spectrometry Characterization of Histones. J Proteome Res. 8 (11), 5367-5374 (2009).
- Sidoli, S., et al. Drawbacks in the use of unconventional hydrophobic anhydrides for histone derivatization in bottom-up proteomics PTM analysis. Proteomics. 15 (9), 1459-1469 (2015).
- Lin, S., Garcia, B. A. Examining histone posttranslational modification patterns by high-resolution mass spectrometry. Methods Enzymol. 512, 3-28 (2012).
- Sidoli, S., et al. SWATH Analysis for Characterization and Quantification of Histone Post-translational Modifications. Mol Cell Proteomics. , (2015).
- Krautkramer, K. A., Reiter, L., Denu, J. M., Dowell, J. A. Quantification of SAHA-Dependent Changes in Histone Modifications Using Data-Independent Acquisition Mass Spectrometry. J Proteome Res. , (2015).
- Yuan, Z. F., et al. EpiProfile Quantifies Histone Peptides With Modifications by Extracting Retention Time and Intensity in High-resolution Mass Spectra. Mol Cell Proteomics. 14 (6), 1696-1707 (2015).
- MacLean, B., et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 26 (7), 966-968 (2010).
- Yuan, Z. F., Lin, S., Molden, R. C., Garcia, B. A. Evaluation of proteomic search engines for the analysis of histone modifications. J Proteome Res. 13 (10), 4470-4478 (2014).
- Tan, Y., Xue, Y., Song, C., Grunstein, M. Acetylated histone H3K56 interacts with Oct4 to promote mouse embryonic stem cell pluripotency. Proc Natl Acad Sci U S A. 110 (28), 11493-11498 (2013).
- Vonholt, C., et al. Isolation and Characterization of Histones. Methods Enzymol. 170, 431-523 (1989).
- Zhang, K. L., et al. Identification of acetylation and methylation sites of histone H3 from chicken erythrocytes by high-accuracy matrix-assisted laser desorption ionization-time-of-flight, matrix-assisted laser desorption ionization-postsource decay, and nanoelectrospray ionization tandem mass spectrometry. Anal. Biochem. 306 (2), 259-269 (2002).
- Jufvas, A., Stralfors, P., Vener, A. V. Histone Variants and Their Post-Translational Modifications in Primary Human Fat Cells. Plos One. 6 (1), e15960(2011).
- Bonaldi, T., Imhof, A., Regula, J. T. A combination of different mass spectroscopic techniques for the analysis of dynamic changes of histone modifications. Proteomics. 4 (5), 1382-1396 (2004).
- Zhao, X. L., et al. Comparative Proteomic Analysis of Histone Post-translational Modifications upon Ischemia/Reperfusion-Induced Retinal Injury. J Proteome Res. 13 (4), 2175-2186 (2014).
- Lin, S., et al. Stable-isotope-labeled histone peptide library for histone post-translational modification and variant quantification by mass spectrometry. Mol Cell Proteomics. 13 (9), 2450-2466 (2014).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados