
Method Article
Komplette Workflow zur Analyse von Histon-post-translationale Modifikationen Mit Bottom-up-Massenspektrometrie: Von Histone Extraktion in Datenanalyse
In diesem Artikel
Zusammenfassung
Dieses Protokoll beschreibt einen voll integrierten Workflow für die Charakterisierung von Histon-post-translationale Modifikationen mittels Massenspektrometrie (MS). Der Workflow umfasst Histon-Reinigung aus Zellkulturen oder Geweben, Histon-Derivatisierung und Verdauung, MS-Analyse unter Verwendung von Nano-Flow-Flüssigkeitschromatographie und Anweisungen für die Datenanalyse. 3 Tage - Das Protokoll wird innerhalb von 2 für die Fertigstellung konzipiert.
Zusammenfassung
Nukleosomen sind die kleinsten Struktureinheit des Chromatins, bestehend aus 147 Basenpaaren um eine Oktamers von Histon-Proteine gewickelt DNA. Histon-Funktion wird durch umfangreiche posttranslationale Modifikation durch eine Vielzahl von Kernproteine vermittelt. Diese Änderungen sind von entscheidender Bedeutung für die nukleare Integrität, wie sie in der Genregulation, DNA-Reparatur und Chromosomenkondensation beteiligt Chromatinstruktur und rekrutieren Enzyme regulieren. Obwohl ein großer Teil der wissenschaftlichen Gemeinschaft Antikörper-basierte Techniken nimmt Histon PTM Überfluß zu charakterisieren, sind diese Ansätze geringen Durchsatz und voreingenommen gegen hyper Proteine, wie das Epitop, das durch in der Nähe Modifikationen behindert werden könnten. Dieses Protokoll beschreibt die Verwendung von Nano-Flüssigchromatographie (nLC) und Massenspektrometrie (MS) für eine genaue Quantifizierung von Histon-Modifikationen. Dieses Verfahren ist für eine große Vielzahl von Histon-PTMs und die relative Häufigkeit mehrerer Histon zu charakterisieren Varianten innerhalb single analysiert. In diesem Protokoll werden Histone mit Propionsäureanhydrid durch Verdau gefolgt derivatisierten Peptiden mit Trypsin von 5 zu erzeugen - 20 aa lang. Nach dem Verdau werden die neu freigelegten N-Termini der Peptide Histon derivatisierten chromatographischen Retention während NLC-MS zu verbessern. Dieses Verfahren ermöglicht die relative Quantifizierung von Histon PTMs über vier Größenordnungen.
Einleitung
Epigenetics ist als das Studium der erblichen Veränderungen in der Genexpression definiert , die durch Mechanismen entstehen andere als die zugrundeliegende DNA - Sequenz 1 zu verändern. Epigenetische Regulation ist von entscheidender Bedeutung bei der Entwicklung als der Organismus dramatische phänotypische Veränderungen erfährt, obwohl seine DNA-Gehalt ändert sich nicht. Es gibt mehrere kritische Komponenten für die richtige epigenetische Wartung erforderlich, einschließlich Histon post-translationale Modifikationen (PTMs), Histon - Varianten, nicht-kodierenden RNAs, DNA - Methylierung und DNA - Bindungsfaktoren, von denen jede 2 - Genexpression durch verschiedene Mechanismen beeinflussen. Während zum Beispiel ist die DNA - Methylierung eine hoch stabile Modifikation , die Gen - Übersetzung 3, Histonvarianten und Histon PTMs beeinflussen 4 sind viel dynamischer und Chromatin in vielfältiger Weise reprimiert.
Histon-PTMs sind meist an den N-terminalen Enden lokalisiert, da sie die am meisten ausgesetzt und flexibel sind regionsdes Proteins. Allerdings ist auch die Nukleosomen Kern stark verändert im Vergleich zu durchschnittlichen Proteine 5. Auch wenn Histonmarkierungen haben in den letzten zehn Jahren viele Verbindungen zwischen den bekannten Histonmarkierungen und deren Funktion noch unklar sind ausführlich charakterisiert worden. Dies ist vor allem auf die Tatsache zurückzuführen, dass die meisten Histon PTM allein nicht funktionieren, sondern Funktion zusammen mit anderen PTM ( "cross-talk") einen bestimmten Prozess wie Transkription 6,7 zu verändern. Zum Beispiel aktiviert die kombinatorische Markierung H3S10K14ac auf dem Gen p21 seine Transkription, die mit nur einem der beiden nicht PTMs 8 auftreten würden. Die HP1 Protein Compacts Chromatin durch die Anerkennung H3K9me2 / me3 und die Änderung in der Nähe Nukleosomen zu verbreiten. Allerdings kann HP1 nicht binden H3K9me2 / 3 , wenn die benachbarte S10 phosphoryliert ist 9. Acetylierung von H3K4 hemmt die Bindung des Proteins an spChp1 H3K9me2 / me3 in Schizosaccharomyces pombe 10. Weiterhin d das Histon Lysinemethylase PHF8 hat die höchste Nukleosomen Bindungseffizienz , wenn drei PTM H3K4me3, K9ac und K14ac 11 vorhanden sind. Diese Beispiele unterstreichen die Bedeutung der einen globalen Überblick über Histon PTM Veränderungen zu erreichen, anstatt sich auf einzelne Modifikationen.
Das Vorhandensein von Sequenzvarianten erhöht auch die Komplexität der Histon-Analyse, wie Histon-Isotypen im allgemeinen stark ähnliche Sequenzen haben, aber haben oft unterschiedliche Rollen in Chromatin. Zum Beispiel hat H2A.X eine C-terminale Sequenz , die auf DNA - Schädigung im Vergleich zur kanonischen H2A 12, und es ist erforderlich für die Inaktivierung von Geschlechtschromosomen in männlichen Maus Meiose leichter 13 phosphoryliert wird; In ähnlicher Weise ersetzt CENP-A kanonische Histon H3 in Romeren 14. Trotz ihrer unterschiedlichen Funktionen teilen sich diese Varianten, einen großen Teil ihrer Aminosäuresequenz mit der jeweiligen kanonischen Histon, was es schwierig macht, sie getrennt zu identifizieren und zu quantifizieren.
Antikörper-basierte Techniken wie Western-Blotting wurden ausgiebig angenommen Histone zu charakterisieren. Jedoch sind Antikörper-basierte Ansätze begrenzt aus den folgenden Gründen: (i) sie können nur das Vorhandensein einer Änderung zu bestätigen und kann nicht unbekannt PTMs identifizieren; (Ii) sie aufgrund des Vorhandenseins von koexistierenden Markierungen vorgespannt sind, die Bindungsaffinität beeinflussen können; (iii) sie können nicht kombinatorischen Markierungen identifizieren, da nur sehr wenige Antikörper für diesen Zweck verfügbar sind , und (iv) sie kreuzreagieren zwischen sehr ähnlichen Histonvarianten oder ähnliche PTMs (beispielsweise Di- und Trimethylierung von Lysinresten). Egelhofer et al. durch Dot - Blot oder Western mehr als 25% der kommerziellen Antikörper - Spezifität Tests fehlschlagen Blot, und unter spezifischen Antikörpern mehr als 20% scheitern in Chromatinimmunpräzipitation Experimente 15 beschrieben ist. Die Massenspektrometrie (MS) ist derzeit der am besten geeignete Analysewerkzeug neuartige und / oder kombinatorischen PTM zu studieren,und es wurde für Histon - Proteine ( beschrieben in 16) weitgehend umgesetzt. Dies ist vor allem aufgrund der hohen Empfindlichkeit und Massengenauigkeit von MS, und die Möglichkeit zur Durchführung groß angelegte Analysen.
Der Bottom-up-Strategie ist die am häufigsten verwendeten MS-basierte Proteomik Strategie für Histon-Charakterisierung und deren PTMs, wobei das intakte Protein enzymatisch in kleine Peptide gespalten (5 bis 20 aa). Diese Verdauung erleichtert sowohl LC-Trennung und MS-Detektion. Messen im Bereich von 600 - 2.000 Da häufig sind leichter ionisiert und mit höherer Massengenauigkeit und Auflösung als größere Massen identifiziert. MS / MS-Fragmentierung wird auch verbessert, wie auch kurze Peptide im allgemeinen gut geeignet für kollisionsinduzierte Dissoziation (CID). Histone stellen jedoch eine Herausforderung für die Bottom-up-MS, wie sie in basischen Aminosäureresten hoch angereichert sind, nämlich Lysin und Arginin. Daher führt Trypsin-Verdau zur Generierung von Peptiden, die zu sm sindalle für LC Bindung und eine eindeutige Lokalisierung des PTM. Um dieses Problem zu umgehen, unser Protokoll enthält Lysin und Peptid - N-terminale chemische Derivatisierung 17. Die Verwendung von Propionsäureanhydrid wird für eine effiziente chemische Derivatisierung zu empfehlen , da im Vergleich zu anderen Reagenzien 18. Solche Derivatisierung blockiert die ɛ -Aminogruppen von unmodifizierten und Monomethylether Lysinresten, Trypsin die Proteolyse durchzuführen erlaubt nur an der C-terminalen Argininreste. Derivatisierten Amine können nicht Protonen mit der Lösung auszutauschen, und somit werden die Peptide im Allgemeinen nur zweifach oder dreifach geladenen, erleichtert MS und MS / MS-Detektion. Darüber hinaus erhöht N-terminale Peptid Derivatisierung Hydrophobizität und somit Umkehrphasen-chromatographische Retention. Hier beschreiben wir den Workflow Histone zu reinigen und bereiten sie für die PTM - Analyse über Bottom-up Proteomik (Abbildung 1). Diese Strategie erreicht Quantifizierung einzelner Histonmarkierungen und kombinatorische Markierungen foder Histon PTM, die relativ nahe in der Aminosäuresequenz sind.
Protokoll
1. Sammlung von Zellen, die aus Kultur
- Wenn Zellen in Suspension gezüchtet wurden, zu sammeln Zellen durch Zentrifugation bei 300 RCF für 5 min. Wenn Haft-, absaugen und verwerfen Zellmedium. Spülen Sie die anhaftenden Zellen mit PBS ohne Ca 2+ und Mg 2+ (bezeichnet als PBS geht nach vorn). Inkubieren der Zellen in entweder Trypsin oder Trypsin-EDTA (0,025% - 0,5% auf der Zelllinie abhängig) mit genügend Volumen, um die Oberfläche der Platten bei 37 ° C zu decken, bis die Zellen abzulösen (Zeit variiert für verschiedene Zelllinien).
- Sammle die Zellen durch Zentrifugation bei 300 RCF für 5 min. Wash-Zellen zwei Mal in PBS und sammeln durch Zentrifugation.
- Schätzen Sie die Hämatokrit etwa von den markierten Abstufungen auf den 1,8-ml-Röhrchen oder 15 ml konischen Röhrchen.
Anmerkung: Die Zellen aus der Kultur kann in flüssigem Stickstoff schockgefroren und bei -80 ° C unbegrenzt in diesem Stadium.
2. Isolierung von Nuclei von intakten Zellen
- Thaw Zellen auf Eis.
- Thaw Kernisolationspuffer (NIB, Tabelle 1).
- Bereiten ca. 5 ml NIB - Puffer (Tabelle 1) für jede 100 & mgr; l gepackte Zellvolumen. Zu jedem 1 ml NIB-Puffer, Proteaseinhibitoren und Stabilisierungsmittel hinzuzufügen, wie folgt: 1 ul 1 M DTT, 2,5 ul 200 mM AEBSF, 2 ul 2,5 uM Microcystin und 2 ul 5 M Natriumbutyrat. NIB mit Inhibitoren wird vorwärts als NIB von diesem Punkt bezeichnet.
Hinweis: Wenn Histon-Phosphorylierung untersucht wird, sind freie EDTA-Protease und Phosphatase-Inhibitor-Cocktail. - Entfernen eines fünften Band von NIB Puffer so hergestellten und fügen NP-40 Alternative (Tabelle 1) bis zu einer Endkonzentration von 0,2%. Die restlichen vier Fünftel Volumen wird für Waschungen verwendet werden.
- Waschzellpellet in 01.10 Zellpellet NIB ohne NP-40 Alternative Verhältnis (v / v). Entfernen Stand durch Zentrifugation bei 700 rcf für 5 min.
- Lyse der cell Pellet indem sie auf Eis gestellt und das Hinzufügen von 01.10 Zellpellet NIB mit 0,2% NP-40 Alternative (v / v).
- Wenn aus Gewebeproben zu extrahieren, zu homogenisieren Homogenisatoren mit Mörser und Stößel oder Dounce. Kultivierte Zellen kann durch vorsichtiges Pipettieren homogenisiert werden.
- Inkubieren homogenisierten Zellen auf Eis für 5 - 10 min. Die Zellen werden lysiert und die Kerne freizusetzen.
- Zentrifuge bei 1000 rcf 5 - 10 min bei 4 ° C. Das Pellet enthält meist Kerne Zelle, während der Überstand meist Cytoplasma-Komponenten enthält. Speichern Sie die Zytoplasmafraktion, falls gewünscht.
- Waschen Sie das Pellet-Kerne durch vorsichtiges Resuspendieren in 01.10 (v / v) NIB ohne NP-40 Alternative.
Hinweis: Dieser Waschschritt nur Spuren von Reinigungsmitteln zu entfernen, ist vor der Histone aus Kernen zu extrahieren. - Zentrifuge bei 1000 rcf für 5 min bei 4 ° C und entfernt den Überstand.
- Wiederholen Sie Schritt 2,10-2,11 mindestens zweimal vollständig NP-40 Alternative entfernen. Entfernung von NP-40 Alternative ist offensichtlich eins vorsichtiges Pipettieren während der Waschschritt nicht mehr Blasen bildet.
- Für Histon-Extraktion aus Gewebe:
- Spülen Sie frische oder gefrorene Gewebe in eiskaltem NIB aufgetaut.
- Übertragen Gewebe in eine Petrischale auf Eis gelegt mit NIB, gerade genug, um das Gewebe feucht zu halten.
- Dice in kleinste Stücke (<1 mm) mit Rasierklinge den Oberflächenkontakt für Kerne Isolation zu erhöhen.
- Übertragen zerkleinerte Gewebe auf ein vorgekühltes Homogenisator und waschen in NIB durch Pipettieren von oben und unten.
- Entfernen Puffer von 300 rcf für 5 Minuten zentrifugiert.
- Hinzufügen NIB enthält NP-40 Alternative zu den Zellen in einem Zellen: Puffer-Verhältnis von 1:10 (v / v) und homogenisieren von von 5 bis 10 Hüben.
- Überprüfen Sie für Zell-Lyse und wiederholen Sie die Homogenisierung nach Bedarf. Ein guter Indikator dafür, dass Zellen lysiert worden sind, ist die Reduktion des Pelletvolumens. Das Pellet sollte nur Kerne enthalten.
- Zentrifuge bei 700 rcf für 5 Minuten und speichern Pellet. 2 mehr Zeit - Dieses Pellet kann 1 extrahiert werdens in 01.10 (v / v) NP-40 NIB Alternative enthält; in diesem Stadium werden die Histone aus Chromatin extrahiert und das Pellet beträchtlich geschrumpft.
- Wasche zweimal mit 2 - 3 ml NIB ohne NP40 Alternative Spuren des Detergens zu entfernen.
Hinweis: Zwischenhaltepunkt: Probe kann in einem minimalen Volumen von NIB + 5% Glycerin und gelagert bei -80 ° C erneut suspendiert werden.
3. Extraktion und Reinigung von Histone aus Nuclei
Anmerkung: Histone sind sehr reich an basischen Aminosäureresten, so dass sie mit der Phosphorsäure Rückgrat der DNA zu interagieren eng. Histone sind unter den basischen Proteinen in den Zellkern, so dass sie in eiskaltem Schwefelsäure (0,2 MH 2 SO 4) mit minimaler Kontamination durch Nicht-Histon - Proteine, die in starken Säure auszufällen extrahiert werden. Hochkonzentrierte TCA (bis zu einer Endkonzentration von 33%) kann dann verwendet werden Histone aus der Schwefelsäure auszufällenAcid. TCA wird bei 4 ° C als 100% in braunen Flasche aufbewahrt.
- Resuspendieren Zellkerne in 1: 5 (v / v) gekühlt 0,2 MH 2 SO 4 (Tabelle 1) durch vorsichtiges Pipettieren.
- Inkubieren Sie die Probe mit konstanter Rotation oder leichtem Schütteln für 2 - 4 Stunden bei 4 ° C. Typischerweise wird für Proben mit mehr als 500 & mgr; l Zellpellet, a 2 hr Extraktion reicht Histone zu extrahieren; längere Inkubationszeit kann bei der Extraktion von anderen basischen Proteinen führen. Für kleinere Kernpellets (<200 & mgr; l), 4 h Extraktion sorgt für eine bessere Ausbeute.
- Zentrifuge bei 3400 rcf bei 4 ° C für 5 min.
- Den Überstand in ein neues Röhrchen.
- Wiederholen Sie die Schritte 3,3-3,4 um unlösliche Material zu entfernen.
- Um die Histone, fügen gekühltes 100% TCA (Tabelle 1) zu dem Überstand gesammelt (jetzt enthaltend Histone) im Verhältnis von 1 präzipitieren: 3 (v / v), um eine endgültige Konzentration von TCA 33% zu erhalten. Mischen Sie durch das Rohr ein paar T Umkehrenimes.
Anmerkung: Die Proben werden trüb bei Zugabe von TCA, was die Anwesenheit von Histonen. - Inkubieren Sie die Mischung auf Eis für mindestens 1 Stunde. Für kleinere Ausgangsgranulatgrößen, wird über Nacht Fällung empfohlen.
- Zentrifuge bei 3.400 rcf für 5 min. Die Histone coat die Seiten der Rohre und auch am Boden abzulagern. Ein weißes unlösliches Pellet bildet auch ganz am Boden des Röhrchens, die meist nicht-Histon-Proteine und andere Biomoleküle enthält. Überstand entfernen durch Absaugen, vorsichtig, ohne die Seiten oder das Pellet Schaben.
- Durch die Verwendung einer Pasteur - Glaspipette, spülen Sie das Rohr mit eiskaltem Aceton + 0,1% HCl (Tabelle 1) , um die gefällten Proteine Beschichtung der Seiten und den Boden zu bedecken.
- Zentrifuge bei 3.400 rcf für 2 min und absaugen Überstand vorsichtig, ohne die Seiten Schaben oder das Pellet.
- Wiederholen Sie die Schritte 3,9-3,10 mit 100% eiskaltem Aceton.
- Trocknen des Pellets mit einem Luftstrom oder mit einem vacuum Zentrifuge, oder einfach nur durch das Rohr offen zu lassen. Aceton schnell verdunstet.
- Lösen Sie den Histonen mit ddH 2 O (doppelt destilliertes Wasser) in einem minimalen Volumen möglich , die weiße Schicht vollständig aufzulösen. Histone sind in Wasser leicht löslich. Für Pellets in einem 1,5 ml Mikrozentrifugenröhrchen, 100 & mgr; l ddH 2 O ist in der Regel genug Histone zu sammeln.
- Zentrifuge bei 3400 rcf für 2 min und den Überstand in ein neues Röhrchen.
4. Abschätzung der Proteinkonzentration und Reinheit
- Für die Proteinkonzentration zu messen, verwenden BCA, Bradford-Protein-Assay oder Aminosäureanalyse (AAA). verwenden Techniken nicht, dass die Absorption bei 280 nm annehmen, wie Histone in aromatischen Aminosäurereste sind schlecht.
- Überprüfen, um die Reinheit des extrahierten Histone durch SDS-PAGE-Analyse mit einer 15% igen Acrylamidgel und Coomassie-Färbung (optional).
- Wenn hoher Reinheit einzelne Histonvarianten gewünscht werden, weiterhin HPLC-UV FraktionierungHistonvarianten (Abschnitt 5). Wenn nicht, gehen Sie direkt Vorbereitung für die Bottom-up-Histon-PTM-Analyse (Abschnitt 6) zu probieren.
5. Trennung von Histonvarianten durch Umkehrphasen-HPLC (Optional)
Hinweis: Hohe Reinheit Histon-Varianten können durch Fraktionieren des Roh-Histon-Gemisch unter Verwendung Umkehrphasen-HPLC erhalten werden, gekoppelt mit einem UV-Detektor. Diese gereinigten Histone sind nützlich für Studien, die eine höhere Empfindlichkeit und Reinheit erfordern. Jedoch für Standard Histon PTM Charakterisierung kann dieser Schritt übersprungen werden, weil die Analyse ausreichend empfindlich und erschöpfend ist. Die Fraktionierung von intakten Histonvarianten erfordert idealerweise mindestens 100 bis 300 & mgr; g Ausgangsmaterial.
- Verbinden eines geeigneten C 18 5 & mgr; m - Säule mit einem HPLC je nach Ausgangs Histon - Konzentration: mit etwa 100 ug Histone, verwenden 2.1 mm x 250 mm Säule mit einer Fließgeschwindigkeit von 0,2 ml / min; mit etwa 300 ug Histone, verwenden 4,6 x 250 mm Säule miteiner Fließgeschwindigkeit von 0,8 ml / min. Bereiten Puffer A und B spezielle Glaswaren wie folgt verwendet:
- Bereiten Sie Puffer A: 5% HPLC-Acetonitril, 0,1% TFA in HPLC-Wasser.
- Bereiten Puffer B: 95% HPLC-Acetonitril, 0,1% TFA in HPLC-Wasser.
- Verbinden Sie die Spalte mit einem UV-Detektor, und stellen Sie die Absorption auf 210 bis 220 nm.
- Säuert das Histon Probe in Wasser gelöst, mit 100% iger TFA zu einer Endkonzentration von 0,1-1% TFA zu erreichen.
- Äquilibrierung der Säule mit 100% Puffer A für mindestens 15 min bei der empfohlenen Durchflussrate, die Volumina in etwa drei Spalte entspricht. Verwenden Sie dieses Signal die Null-Absorption Niveau des UV-Detektors einzustellen.
- Bereiten Sie entsprechend dimensionierten Rohren Fraktionen zu sammeln, entweder manuell oder in einem automatischen Probensammler.
- Injizieren Probe bei einer Konzentration von etwa 1 & mgr; g / & mgr; l oder höher. Die Proben in größeren Mengen aufgelöst könnte die Einstellung des Gleichgewichts der Spalte du ändernRing Laden und führen zu geringeren Retention.
- Führen des Gradienten, wie folgt programmiert: 0-30% B in 1 min, 30 bis 60% B in 90 min, und 60 bis 90% B in 1 min.
- Sammle Fraktionen (Beispiel - Chromatogramm in Figur 2 gezeigt) in 1 min - Intervallen unter Verwendung eines automatischen Fraktionssammlers. Sammle Fraktionen in geeigneter Größe Röhren das gesamte Volumen zu enthalten.
- Trocknen Sie nach unten fraktionierten Proben in einem Vakuum-Konzentrator.
Hinweis: Zwischenhaltepunkt: Getrocknete Histon-Fraktionen, die bei Raumtemperatur gelagert für kurze Zeiträume werden können (1 - 2 Tage) oder in -80 ° C Gefrierschrank für langfristige Zeiträume.
6. Chemische Derivatisierung von Histone Mit Propionsäureanhydrid für Bottom-up-Analyse
- Löse Histon - Proben in 40 ul 50 mM NH 4 HCO 3, pH 8,0 (empfohlene Menge: 50 - 100 ug). Wenn Proben in reinem ddH 2 O waren, fügen Sie konzentrierte NH 4 HCO 3 bis 50 mM bilden, pH 8.0.
- mit pH-Indikatorstreifen ohne Probenverluste Befeuchten Sie ein P10 Pipettenspitze in die Probe, die den pH-Wert zu überprüfen. NH 4 OH und Ameisensäure kann den pH - Wert auf 8,0 einzustellen verwendet werden.
Hinweis: Der folgende Teil des Protokolls (Schritte 6,3-6,7) sollten in den Reihen von maximal drei bis vier Proben durchgeführt werden, um reaktive Propionsäureanhydrid zu halten. - Verwenden Sie Dunstabzug für die weiteren Schritte in dem Propionsäureanhydrid verwendet wird. Bereiten frischen Propionylierung Reagenz durch Mischen Propionsäureanhydrid mit Acetonitril im Verhältnis 1: 3 (v / v). Hinzufügen Propionylierung Reagenz in Probe 1: 4 (v / v). Für 40 ul Histone, mit 10 & mgr; l Propionylierung Reagenz.
Hinweis: Es ist möglich, weiße Ablagerungen in diesem Schritt zu beobachten. Jedoch enthält diese meist Salze und Propionsäure, und somit keine spezielle Maßnahmen ergriffen werden. - Zur Wiederherstellung der pH - Wert 8,0 zu der Lösung schnell NH 4 OH hinzuzufügen. Hinweis: Propionsäureanhydrid mit den freien Aminen der Peptide reagieren produziert propionische Säure, die pH-Wert sinkt. Üblicherweise Zugabe NH 4 OH zu der Probe mit einem Verhältnis von 1: 5 (v / v) geeignet ist zur Wiederherstellung der pH 8,0; beispielsweise 8 & mgr; l NH 4 OH bis 40 & mgr; l Probe.
- Mischen Sie sofort durch Verwirbelung.
- Überprüfen pH mit demselben Verfahren wie in Schritt 6.2.
Vorsicht: Beim pH größer als 10,0, Kennzeichnung von anderen Aminosäureresten mit höheren pKa möglich. - Inkubieren Proben bei Raumtemperatur für 15 min.
- Wiederholen Sie die Schritte 6,3-6,7, streng die Reaktion nicht pro Charge von Propionylierung Reagenz mehr als 3 oder 4 Proben durchführen.
- Trockenproben bis zu 10 - 20 & mgr; l in einem Vakuum-Konzentrator. Dieses verdampft nicht umgesetztes Propionsäureanhydrid, Acetonitril, Essigsäure und Ammoniakgas aus NH 4 OH gelöst. Wenn Proben vollständig austrocknen, treten keine signifikanten Probenverluste.
Hinweis: Isopropanol anstelle von Acetonitril verwendet werden. Jedoch hat Acetonitril geringere Oberflächenspannung und somit mehrschnelle Verdunstung. - Resuspendieren oder verdünnten Proben mit ddH 2 O bis 40 ul Endvolumen erreicht wird.
- Wiederholen Sie die Schritte 6,2-6,9. Eine doppelte Runde von Histon Propionylierung sorgt für> 95% der Beendigung der Reaktion.
- Füllen Propionsäureanhydrid Flasche mit Argongas, um die Bildung von Essigsäure in Kontakt mit Feuchtigkeit in der Flasche zu verhindern.
Hinweis: Zwischenhaltepunkt: Probe kann in ddH 2 O oder getrocknet Rekonstitution bei -80 ° C gelagert werden.
7. proteolytischen Verdau mit Trypsin
- Resuspendieren Histone in 50 mM NH 4 HCO 3 eine optimale Konzentration von 1 & mgr; g / & mgr; l oder höher zu erreichen. Weitere verdünnten Proben führen Trypsin Effizienz zu senken.
Hinweis: Die Histone bei diesem Schritt müssen bei pH 8,0, wenn sie immer noch sauer, dann NH hinzufügen 4 HCO 3 Salz unter Verwendung von Pipettenspitze zu probieren. - Hinzufügen Trypsin zu Histon Proben im Verhältnis 1:10 (wt / wt).
- Incubate bei 37 ° C für 6 bis 8 Stunden.
- Stoppen Sie die Verdauung von in -80 ° C einfrieren.
- Trocknen Sie die Probe auf 10 - 20 & mgr; l in einem Vakuum-Konzentrator.
Hinweis: Zwischenhaltepunkt: Probe kann bei -80 ° C gelagert werden.
8. Propionylierung von Histon Peptides an N-Termini
Hinweis: Dieser Abschnitt beschreibt die Derivatisierung von Peptid N-Termini aus dem Trypsin erzeugt verdauen. Solches Verfahren verbessert HPLC Retentions der kürzesten Peptide (beispielsweise Aminosäure 3 bis 8 von Histon H3) als Propionylgruppe erhöht Peptid Hydrophobizität.
- Resuspendieren Proben in 30 ul 100 mM NH 4 HCO 3.
- Wiederholen Sie die Schritte 6,1-6,9.
Anmerkung: Es ist normal, dass die Trocknung der Proben in Vakuum eine längere Zeit in diesem Schritt stattfindet. - Resuspendieren oder verdünnten Proben mit 50 bis 100 & mgr; l ddH 2 O + entweder 0,1% TFA oder 0,5% Essigsäure. Anmerkung: Essigsäure wird für lange Lagerungen empfohlen, da TFA facilitatES Methioninoxidation auf lange Sicht. Auf der anderen Seite wird TFA zu empfehlen, wenn bühnen Kipp (Abschnitt 9) am selben Tag durchgeführt, wie TFA eine bessere chromatographische Retention unterstützt.
Hinweis: Zwischenhaltepunkt: Probe kann bei -80 ° C gelagert werden.
9. Beispiel Entsalzung mit Stufe-Tipps
Anmerkung: In diesem Stadium gibt es Salz in der Probe vorliegt. Salze behindern HPLC-MS-Analyse, da sie während des Elektrosprüh-Ionisierung, um das Signal von Peptiden unterdrücken. Salze können auch ionische Addukte an Peptiden, bilden die Signalintensität für das nicht-adduzierten Peptid zu reduzieren. Da die adducirt Peptid eine unterschiedliche Masse haben, wird das Peptid nicht richtig quantifiziert identifiziert oder werden.
- Eine P1000 Pipettenspitze, Punsch eine Scheibe aus C 18 Material aus einer Festphasenextraktion Platte Durch die Verwendung. Schieben die Minidisk aus dem P1000 Spitze durch eine Quarzglaskapillare unter Verwendung von und die Minidisk auf dem Boden eines P100 / 200 Pipettenspitze abzulagern. Stellen Sie sicher, dass der dISK ist an der Unterseite der Spitze (3) fest verkeilt.
Hinweis: Die P1000 Spitze hat eine ziemlich kleines Loch die C 18 Platte zu stanzen. Es empfiehlt sich, die letzten Zentimeter von der Spitze zu schneiden, um ein Loch mit größerem Durchmesser zu haben. - Verwenden Sie zwei C 18 Schläge in der gleichen P100 / P200 Tipp , wenn Entsalzung über 25 & mgr; g Probe.
- Verwenden Sie eine Zentrifuge Adapter Bühne Spitzen an Ort und Stelle in 1,5 ml oder 2 ml Mikrozentrifugenröhrchen zu halten. Verwenden Sie langsam (300-400 RCF) Rotation; die Lösungsmittel passieren normalerweise durch das Harz in weniger als einer Minute.
- Spülen des Harzes durch mit 50 & mgr; l 100% Acetonitril Verspinnen der C 18 Material zu aktivieren und potentiellen Verunreinigungen zu entfernen.
- Äquilibrieren Scheibe um 80 & mgr; l von 0,1% TFA Spülung durch langsame Zentrifugation.
- Man säuert die Probe auf pH-Wert 4,0 oder weniger mit Essigsäure. Überprüfen Sie den pH-Wert mit pH-Streifen Probenverlust zu minimieren. Legen Sie die Probe auf die Platte durch langsame Zentrifugation.
- WaschenProbe um 70 Spülung - durch langsame Zentrifugation 80 ul 0,1% TFA.
- Eluieren Probe durch Spülen 70 & mgr; l 75% Acetonitril und 0,5% Essigsäure durch langsame Zentrifugation. Sammeln Sie die Probe in einem 1,5-ml-Röhrchen.
- Trockene Probe in einem Vakuum-Konzentrator.
Hinweis: Zwischenhaltepunkt: Probe kann bei -80 ° C gelagert werden.
10. Die Analyse der Histone Peptides
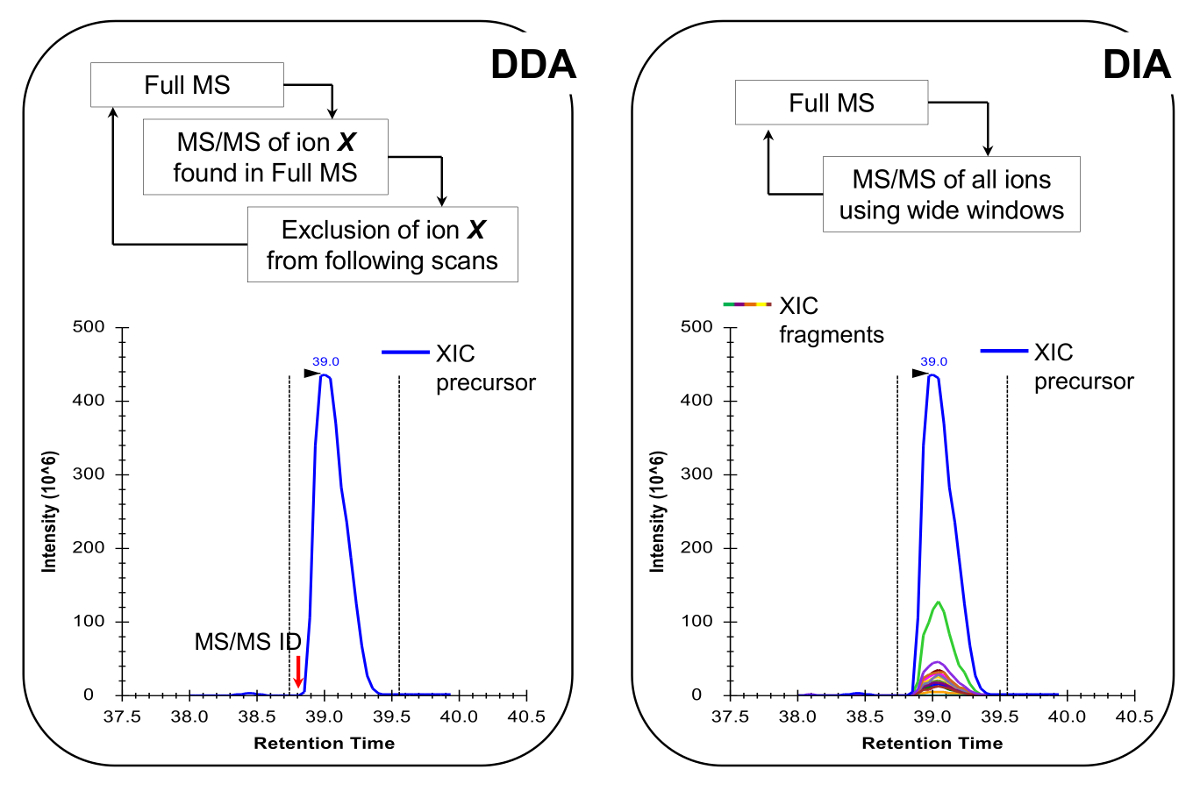
Hinweis: Die NLC-MS-Plattform in der traditionellen Peptidanalyse aufgebaut wie getan werden sollte. Die Verwendung von 200 bis 300 nl Stromkolonne (75 & mgr; m ID analytischen Säule, C 18 Partikel) wird empfohlen, da sie ein ausgezeichneter Kompromiss zwischen Empfindlichkeit und Stabilität sind. Die MS - Erfassungsverfahren kann entweder eine Kombination von datenabhängigen Erwerb (DDA) mit gezielten Scans 19 oder einer Datenunabhängigen Erwerb (DIA) 20,21, sowohl in Repräsentative Ergebnisse und Abbildung 4 beschrieben.
- Bereiten HPLC Puffer - A: 0,1% Ameisensäure inHPLC-Wasser; B: 0,1% Ameisensäure in HPLC-Qualität Acetonitril.
- Programmieren der HPLC-Methode wie folgt: 0-30% Puffer B in 30 min, 30-100% B für die nächsten 5 min und bei isokratischen 100% B für 8 min. Wenn der HPLC nicht für automatisierte Äquilibrierung der Säule vor dem Probenauftrag programmiert wird, schließen dann die folgende: Gradient 100-0% B in 1 min und isokratischen Strom bei 0% B für 10 min. Stellen Sie die Strömungsrate der Analyse von 250 bis 300 nl / min.
- Programmieren Sie die MS Erwerbsmethode zur Durchführung entweder DDA kombiniert mit gezielten Scans 19 oder DIA 20,21 (Abbildung 4). Stellen Sie sicher, dass die MS Tastverhältnis eine volle MS ermöglicht es jedem ~ 2 sec zu scannen, um genügend Datenpunkte haben, um über den chromatographischen Peaks, die eine genauere Quantifizierung ermöglicht. Anmerkung: Bei der C 18 - Chromatographie, die durchschnittliche Basispeakbreite beträgt etwa 30 sec für den Gradienten oben beschrieben.
- Legen Sie etwa 1 ug Probe auf die HPLC-cPALTE.
- Führen Sie die HPLC-MS / MS-Verfahren wie programmiert.
Hinweis: Im Protokoll wir nicht empfehlen Besonderheiten der Spalten, MS Instrumente oder MS-Parameter Details, wie jede optimale Setup, das eine individuelle Proteomics Labor entwickelt werden für das Verfahren geeignet sein. Proteomics Laboratorien sollten ihre optimierte Setup verwenden, da Histon-Peptide als traditionelle Peptide trennen.
11. Datenanalyse
- Importieren Sie die MS Raw-Dateien in die Software zu Peakflächenintegration durchführen. Anmerkung: EpiProfile 22 wird empfohlen, da es für die Histon - Peptide optimiert; durch die Retentionszeit Kenntnis von chromatographischen Elution unter Verwendung führt es zuverlässig Peakfläche Extraktion von bekannten Histon-Peptide. Alternativ ist 23 Skyline eine weitere ideale Software für den Zweck.
- Berechnen der relativen Häufigkeit eines gegebenen Peptids durch seine Fläche durch die Gesamtfläche dieses Peptids in all ihren modifizierten Formen geteilt wird. Hinweis: Bei der Entdeckung Analysis Mascot wird empfohlen Spektren von modifizierten Histone Peptide zu identifizieren. Die Leistung dieses Werkzeug wurde kürzlich 24 beschrieben. Alle anderen Datenbank-Suchmaschinen für die Proteomik sind auch brauchbar, aber bei unseren Tests, sofern sie niedriger Leistung.
Ergebnisse
Als Beispiel haben wir analysiert, aus humanen embryonalen Stammzellen (hES) mit und ohne Retinoesäure (RA) Stimulation, beginnend mit 200 & mgr; l Zellpellets extrahiert Histone. Das Vorhandensein von RA in der Zellkultur führt zu ESC Differenzierung. Aus dem Zellpellet, etwa 50 bis 100 & mgr; g von Histonen extrahiert wurden, die mehr als ausreichend ist, um mehrere LC-MS Injektionen von Histon-Peptiden durchzuführen. Nach Derivatisierung, Verdauung und Entsalzen, wurden die Proben auf eine Säule 75 um x 15 cm C 18 geladen (Teilchendurchmesser 3 um, Porengröße 300 Å) im seriellen Modus mit einem flüssigen nano Chromatographiesystem Hochleistungs mit Mikrofluidik - Chips gekoppelt ist ein Hybrid-Linearfalle Quadrupol - Orbitrap-Massenspektrometer. MS Erwerb wurde unter Verwendung von DIA durchgeführt. Parallel Proben auch auf ein Hybridionenfalle-Orbitrap-Massenspektrometer mit einem DDA-Verfahren unter Verwendung einer Nanoströmungs UHPLC gekoppelt analysiert wurden (Daten nicht gezeigt). Imjeder Zyklus eine vollständige MS Orbitrap Detektion wurde mit dem Scanbereich von 290 bis 1400 m / z, einer Auflösung von 60.000 (bei 200 m / z) und AGC von 10 6. Dann werden die Daten abhängig Erfassungsmodus mit einer dynamischen angewendet wurde durchgeführt , Ausschluss von 30 sec. MS / MS-Scans wurden von den intensivsten denen auf Elternionen gefolgt. Ionen mit einem Ladezustand von einer wurden von MS / MS ausgeschlossen. Ein Isolationsfenster von 2 m / z verwendet wurde. Ionen wurden unter Verwendung von stoßinduzierte Dissoziation (CID) mit Kollisionsenergie von 35% fragmentiert. Ionenfallen - Erkennung wurde mit normalen Scan Range - Modus und normalen Abtastrate mit AGC von 10 4 verwendet.
Roh MS Daten wurden Annahme Software für die Extraktion von Vorläufer- und Fragment Ionenchromatogramme analysiert, nämlich Skyline 23 und EpiProfile 22. EpiProfile wurde für Histon-Peptide optimiert, wie es intelligente Spitzenbereich Extraktion aufgrund Vorkenntnisse von pep integrierttide Retentionszeit. Auf der anderen Seite wird Skyline optimiert für DIA - Analysen und damit zeigten die DIA Figuren (4 und 5A) sind Screenshots von dieser Software. Aus dem extrahierten Ionenchromatogramm wird die Fläche unter der Kurve wiedergewonnen, und das verwendet wird, um die Fülle von jedem Peptid zu bestimmen. Die Fläche des chromatographischen Peak für die [M + H] berechnet wurde , +, [M + 2H] 2+ und [M + 3H] 3+ -Ionen des gleichen Peptids, obwohl in den meisten Fällen die [M + 2H] 2+ war die vorherrschende Form. Dies liefert den rohen Abundanz eines bestimmten modifizierten Form eines Peptids. Um die relative Häufigkeit von PTMs, die Summe aller verschiedenen modifizierten Formen eines Histon-Peptid zu erreichen, wurde als 100% betrachtet, und die Fläche des jeweiligen Peptids wurde durch die Gesamtfläche für die Histon-Peptid in all ihren modifizierten Formen geteilt .
Histone Peptide sind in einer vafalt von isobar Formen (Abbildung 5). Isobaren Peptide, zB K18ac und K23ac, kann nur an der MS / MS quantifiziert werden, wo ihre einzigartige Fragmentionen verwendet werden , das Verhältnis der isobaren Spezies (5A und 5B) zu bestimmen. Dieses Verhältnis wird verwendet, um die Fläche des chromatographischen Peaks zwischen den beiden Arten zu unterteilen. Wenn DDA verwendet, diese isobar Formen wurden in einer Liste von gezielten Massen enthalten, da diese Peptide für die Fragmentierung durch ihre gesamte Elution ausgewählt werden müssen, die nicht in einem Standard-DDA Experiment auftreten würde. Die Unterscheidung der relativen Abundanz der isobaren Spezies wird dann durch die Überwachung der Elutionsprofil der Fragmentionen durchgeführt. Auf der anderen Seite, DIA Art der Erfassung erfordert keine Aufnahmeliste. Allerdings ist diese Art der Erwerbsmethode nicht kompatibel mit herkömmlichen Datenbanksuche, und somit könnte die Entdeckung unbekannter modifizierten Peptide verhindern.
Lysine Acetylierung (+ 42,011 Da) wurde aus dem fast isobar Trimethylierung (+ 42.047 Da) durch die Verwendung hochauflösende MS Erfassung (> 30.000) unterschieden. Darüber hinaus ist die Acetylierung hydrophober als Trimethylierung, der Elution von acetylierten Peptide führen später als die jeweiligen trimethyliertem diejenigen. Die unmodifizierten Form des gleichen Peptid eluierte später sogar aufgrund der Tatsache, dass das Lysin propionyliert wird. Zusammenfassend ist die Reihenfolge der Hydrophobizität für ein Peptid mit einem modifizierbaren Seite Di- und trimethylierte
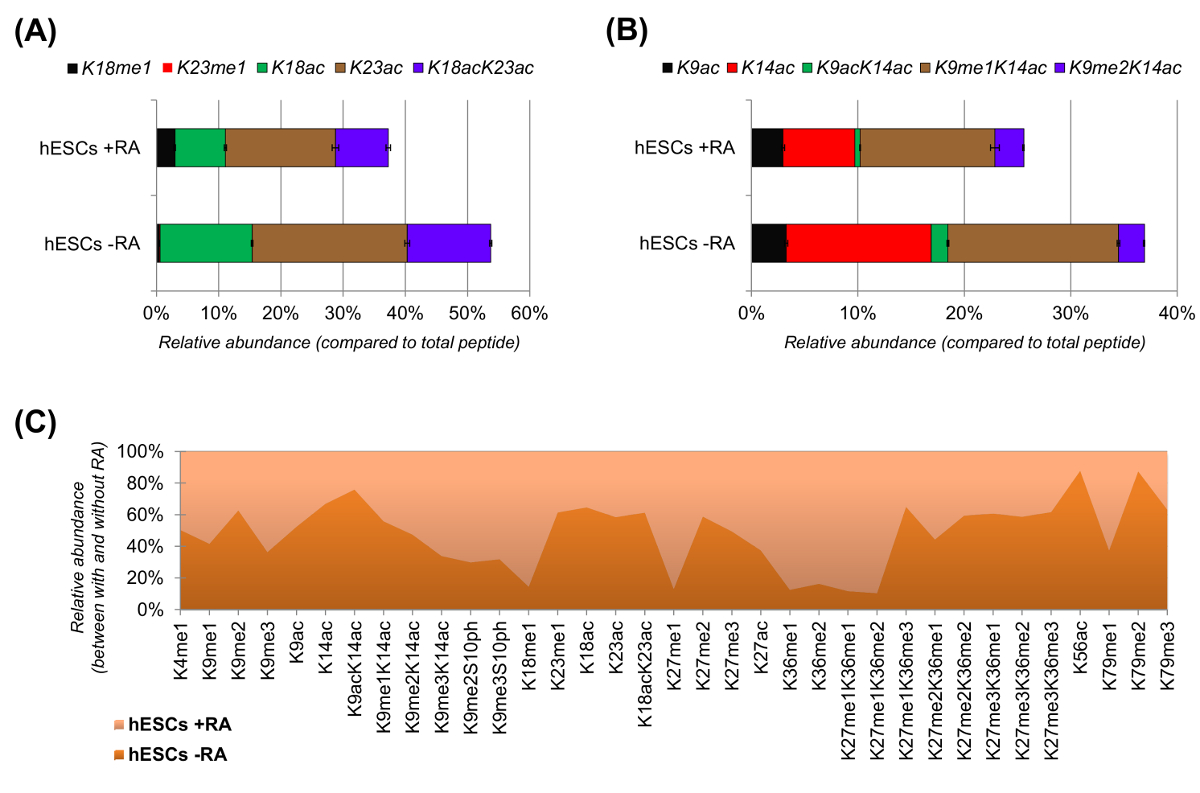
hESCs zeigte eine deutliche Reduktion der acetylierten Peptide , wenn für die Differenzierung (6A und 6B) stimuliert. Dies war nicht überraschend, da frühere Ergebnisse höhere Acetylierung in WSR berichtet , im Vergleich zu differirenden 25,was die allgemein permissive Natur des pluripotenten Chromatin. Durch die Fokussierung auf Histon H3 wurden 35 verschiedene modifizierte Formen quantifiziert (6C). Jedoch alle Histon proteoforms, die mit diesem Ansatz untersucht werden können, sind mehr als 200, einschließlich aller Histonvarianten und geringer Häufigkeit Modifikationen (Daten nicht gezeigt). Darüber hinaus zeigte unsere Analyse, dass eine hohe Reproduzierbarkeit zwischen technische Replikate erhalten werden, wie durch die geringe Größe der Fehlerbalken (entspricht ± Standardabweichung) belegt. Verwendung von NLC-MS-Daten Zusammengefasst beschreibt dieser Abschnitt, wie die relative Häufigkeit von Histon-modifizierten Peptide zu extrahieren.

Abb . 1: Workflow für den Bottom-up - MS / MS - Histon - Analyse Die zehn Schritte für Histon - Analyse gezeigt werden , einschließlich einer Schätzung der Zeit für jeden Schritt erforderlich. Die Abschnittsnummer ist in Klammern, wie sie in der Handschrift gegeben. Abschnitt 5 beschreibt Probenfraktionierung die verschiedenen Histonvarianten zu isolieren, kann verzichtet werden , wenn es eine Notwendigkeit für hochempfindliche Analyse einer bestimmten Variante ist. Bitte hier klicken , um eine größere Version dieser Figur zu sehen.
{kind=link}

Abbildung 2: Reversed-Phase High Flow LC für Histone Variant Fractionation und Coomassie - Gel (A) LC-UV - Chromatogramm intakt Histon Trennung darstellt.. Histon-H3-Varianten können voneinander entsprechend ihrer Elutionszeit unterschieden werden. Die Fraktionen können entweder manuell erfasst werden oder eine automatische Fraktionssammler verwendet wird . (B) Coomassie - Gel von drei Replikaten von Histon - Reinigung.= "Https://www.jove.com/files/ftp_upload/54112/54112fig2large.jpg" target = "_ blank"> Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.

Abbildung 3:. Herstellung von Stage-Kipp - Stecker mit einem P1000 Pipettenspitze, Punsch eine Scheibe aus C 18 Material aus einer Festphasenextraktion Scheibe (zweite Platte). Die Minidisk wird in der Spitze (Mitte) halten, so dass es in eine kleinere P100 / 200 Pipettenspitze, jede Art von kleinen Kapillare herausgeschoben werden kann. In diesem Beispiel verwendeten wir ein 700 um Außendurchmesser Quarzglasrohr. Die Minidisk- sollte auf der Unterseite des P100 / 200 Pipettenspitze geschoben werden, bis es nicht weiter (letzte Panel) gehen kann. Die Bühne Spitze ist bereit für die Histon-Entsalzung, da es eine ausreichende Kapazität hat genügend Probenmaterial für zahlreiche Wiederholungen zu behalten. Insbesondere ist ein Minidisk- genug für 15 bis 20 & mgr; g sample. Wenn mehr Probe erforderlich ist, können mehrere Platten aufeinander gepackt werden. Bitte hier klicken , um eine größere Version dieser Figur zu sehen.
{kind=link}

Abbildung 4:. Schematische Darstellung der DDA und DIA Methoden Wenn DDA verwendet, die MS - Scan - Zyklus wird durch sequentielle Auswahl von Vorläuferionen für MS / MS - Fragmentierung nach ihrer Intensität und Ladezustand charakterisiert. Sobald ein Vorläufer-Ionen fragmentiert worden ist in einer Ausschlussliste gesetzt, um sich wiederholende Auswahl des gleichen Peptids zu vermeiden, so dass die MS kann "graben" in weniger reichlich Signale. Diese Akquisition Methode ist die Methode der Wahl in der Proteomik für Discovery-Modus. Die Quantifizierung erfolgt durch die Integration des Scan-Signal eines bestimmten Ionen neben den identifizierten MS erreicht /MS-Spektrum. In DIA wird die gesamte m / z-Bereich bei jedem Zyklus fragmentiert. Dieser Ansatz ist weniger geeignet für die Discovery-Modus, aber es erzeugt eine chromatographische Profil aller Ionen, Vorprodukte und Produkte. Dies führt zu mehr Vertrauen Quantifizierung und Diskriminierung von isobar Formen. Bitte hier klicken , um eine größere Version dieser Figur zu sehen.
{kind=link}

Abbildung 5: Quantifizierung der Isobaric Peptide (A) Beispiel für zwei isobaren Peptide häufig reichlich in Histon - Analyse.. Das extrahierte Ionenchromatogramms (XIC) ihrer Vorläufermasse und relative Isotope (oben) ist identisch. Jedoch erlaubt die XIC der Produkt-Ionen (unten) für die Unterscheidung der zwei isobaren Formen. Bemerkenswert ist, nur einzigartige Fragmentionen sollten uns seined die relative Häufigkeit der beiden Arten. (B) Darstellung der einzigartigen Fragmentionen für die beiden beschriebenen Peptide (rot markiert). (C) Liste der häufigsten analysierten Peptide in Homo sapiens mindestens ein isobar Äquivalent zu schätzen. Sequenzvarianten zwischen den genannten Histon - Peptide werden angezeigt. Bitte hier klicken , um eine größere Version dieser Figur zu sehen.
{kind=link}

Abbildung 6: Repräsentative Ergebnisse von humanen embryonalen Stammzellen mit und ohne Retinsäurebehandlung (A) Relative Quantifizierung des Histon H3 - Peptid KQLATKAAR (aa 18 - 26) in all seinen modifizierten proteoforms.. Die relative Häufigkeit wurde mit allen proteoforms als 100% geschätzt (reltiven Anteil des unmodifizierten Peptids nicht dargestellt) (B) Relative Quantifizierung der Histon H3 - Peptid KSTGGKAPR (aa . 9 -. 17) , (C) die relative Häufigkeit erkannter Peptide für die kanonische Histon H3 mit und ohne Zellbehandlung mit Retinsäure. Die Figur zeigt, in welchem der beiden Behandlungen die angegebenen Änderungen mehr reichlich vorhanden sind (> 50%). Insgesamt zeigen wir , dass Histon H3 Acetylierung verringert sich in den meisten der Lysinreste nach Induktion der Zelldifferenzierung. Bitte hier klicken , um eine größere Version dieser Figur zu sehen.
{kind=link}
| Lösung # | Zusammensetzung | ||||||
| 1 | Nuclear Isolation Buffer (NIB) Lager wird wie folgt hergestellt und gefroren als 100 ml Aliquots bei -20 ° C gelagert; aufgetaut15 mM Tris, 60 mM KCl, 15 mM NaCl, 5 mM MgCl 2, 1 mM CaCl 2 und 250 mM Saccharose: NIB kann bei 4 ° C für einige Wochen gelagert werden. Der pH-Wert des Puffers wird auf 7,5 mit HCl eingestellt. | ||||||
| 2 | Protease - Inhibitoren (fügen frisch Puffer vor der Verwendung): 1 M Dithiothreitol (DTT) in ddH 2 O (1,000x); 200 mM AEBSF in ddH 2 O (400x) | ||||||
| 3 | Phosphatase-Inhibitor (fügen frisch Puffer vor der Verwendung): 2,5 & mgr; M Microcystin in 100% Ethanol (500x) | ||||||
| 4 | HDAC-Inhibitor (fügen frisch Puffer vor der Verwendung): 5 M Natriumbutyrat, hergestellt durch Titration von 5 M Buttersäure mit NaOH auf pH 7,0 (500x) | ||||||
| 5 | NP-40 Alternative: 10% v / v in ddH 2 O | ||||||
| 6 | 0,2 MH 2 SO 4 in ddH 2 O | ||||||
| 7 | Trichloressigsäure (TCA): 100% w / v in ddH 2 O | ||||||
| 8 | Aceton + 0,1% ige Salzsäure (HCl): 0,1% v / v HCl in Aceton | ||||||
Tabelle 1. Lösungen.
Diskussion
Das hier beschriebene Protokoll ist optimiert Kosten, Zeit und Leistung berücksichtigen. Andere Zubereitungen sind möglich, aber sie haben ihre Grenzen, insbesondere bei der Kupplung mit MS-Analyse. Zum Beispiel kann das Hochsalzextraktionsprotokoll verwendet werden Histone 26 anstelle von TCA - Fällung (Abschnitt 3) zu reinigen. Hochsalz Protokoll ist intrinsisch milder, da es nicht starken Säure nicht verwendet. Auf diese Weise bleibt säurelabile PTM und erhöht die Ausbeute der extrahierten Histone, als TCA-Fällung Co-Ausscheidungen viele andere Chromatin-Bindungsproteine. Jedoch führt Hochsalzextraktion Proben für HPLC-MS / MS zu konzentriert Salz enthält. Bei einer alternativen Herstellung können Histone Verdau ohne Propionylierung (Abschnitt 6 - 8) durchgeführt werden, beispielsweise durch Trypsin Inkubationszeit verringert und die Enzym / Substrat - Verhältnis 27 oder Verwendung ArgC als Verdauungsenzym 28-30. Jedoch Derivatisierung mit Propionsäureanhydrid wird empfohlen, da it führt zur Erzeugung von hydrophoberen Peptide, die besser in der Flüssigchromatographie zurückgehalten werden.
Für chemische Derivatisierung wurde eine Vielzahl von organischen Säureanhydriden wurden bewertet und ihre Vorteile diskutiert umfassend 18. Nichtsdestoweniger erwies Propionsäureanhydrid zu den besten Kompromiß zwischen Effizienz, minimiert Nebenprodukten und verbesserter Peptid Hydrophobizität. Potenziell können Propionsäureanhydrid in der isotopisch markierte Form erworben werden; Dies ermöglicht zum Multiplexen Analyse aufgrund der Möglichkeit, mehrere Proben zu mischen und sie auf der Ebene MS auf der Basis der unterschiedlichen Massen von der schweren Etikett verliehen diskriminieren. Jedoch führt diese Analyse zu einer erhöhten Komplexität des LC-MS-Chromatogramm und verringert die Menge der Probe, die für jede einzelne Bedingung eingespritzt werden kann.
In dieser Hinsicht sollten einige wichtige Aspekte des Protokolls markiert. Folgendes sollte als ch verwendet werdenecklist Fehler zu finden, in das Verfahren im Falle negativer Ergebnisse der Durchführung erhalten werden. Zuerst wird nach der Keimfällung sollte das Pellet vorsichtig mit NIB ohne NP-40 Alternative (Abschnitt 2.10) bis zur vollständigen Entfernung des Detergens (erkennbar durch das Fehlen von Blasen während des Mischens) gewaschen werden. Sie dies nicht, so würde Histon-Extraktion mit Säuren beeinträchtigen. Zweitens, nach dem Histon-Fällung mit TCA (Abschnitt 3.9) Waschungen des Pellets mit Aceton ist von entscheidender Bedeutung. Das Vorhandensein von konzentrierter Säure würde nichts schaden, den folgenden Schritt, wenn Propionylierung und Verdauung (Abschnitt 6.1) direkt durchgeführt werden. Es wäre nicht problematisch bei Histon-Fraktionierung durchgeführt wird (Teil 5). Drittens ist es wichtig, dass die Propionylierung Reaktion schnell durchgeführt wird (siehe Abschnitt 6,3-6,7). 4 aufeinander folgende Proben - mit dem gleichen Propionylierung-Mix (Propionsäureanhydrid + Acetonitril) für mehr als 3 Um dies zu tun, zu vermeiden. Darüber hinaus ist pH der wichtigste Aspekt von Trypsinverdaus (Kapitel 7). Wenn nichtrund 8,0 (7,5-8,5) wird die Verdauung unwirksam. Dies kann passieren, wenn die Probe in diesem Schritt in Propionsäure reich sein wird. NH 4 OH kann , bis Bedarf hinzugefügt werden. Auch für die Forscher vertraut mit Proteomik Workflows wird es normal fühlen, die Probe zu säuern zu Trypsinverdaus beenden. Dies sollte nicht durchgeführt werden, da es die folgende Reaktion gefährden, das heißt Propionylierung von Peptid N-Termini (Abschnitt 8.1). Schließlich wird in der gleichen Ausgabe, ist es wichtig für die Datenanalyse, die nicht-modifizierten Peptide sind eigentlich nicht unmodifizierte zu erinnern; alle freien Lysinreste und N-Termini durch Propionylierung (56,026 Da) belegt werden. Somit, keine Ergebnisse Durchführung Extrahieren Ionenchromatographie der Masse eindeutig der Peptidsequenz entsprechende führen würde.
Die Einschränkungen des Verfahrens sind vor allem im Zusammenhang mit der Unfähigkeit, die kombinatorische PTMs des Erfassens, aufgrund der kurzen Peptidsequenzen, und die Vorspannungen in den wahren abun ErreichungTanz einer Modifikation, aufgrund der Tatsache, dass Peptide in verschiedenen modifizierten Formen mit unterschiedlichen Effizienzen ionisieren können. Die erste Ausgabe kann durch die Kombination dieser Technik mit einem mittleren oder Top-down - Ansatz ( zusammengefasst in 16) gelöst werden. Diese Art der Analyse, auch wenn technisch anspruchsvoller, ist ideal für die Untersuchung Koexistenz Frequenzen von Modifikationen. Darüber hinaus erlaubt es eine bessere Unterscheidung von Histon-Varianten, die nicht immer von unten nach oben erzielt werden kann, da einige Peptide, die die gleiche Sequenz in verschiedenen Histon-Varianten haben. Das zweite Problem, bezogen auf die Ionisationseffizienz kann 31 unter Verwendung einer Bibliothek von synthetischen Peptiden , gelöst werden. Dieser Ansatz gewährleistet eine genauere Abschätzung der relativen Häufigkeit von Histon-PTMs. Jedoch in den meisten Experimenten wird das gewünschte Ergebnis die relativen Veränderungen der gegebenen Änderungen zwischen analysierten Bedingungen. In diesem Fall ist eine solche Korrektur nicht erforderlich ist, aufgrund der Tatsache, dass alle Proben die gleichen bia habens.
Schließlich ermöglicht dieses Protokoll für die Analyse von Histon-PTMs, die in 3 Tagen abgeschlossen werden kann nLC gekoppelt Tandem-MS verwendet wird. Vergleiche mit anderen Techniken als MS, dh Antikörpern basierende Strategien , wie in der Einleitung erwähnt, sind nicht geeignet, da sie nicht einmal annähernd dieses Niveau der Durchsatz erreichen können. Darüber hinaus Antikörper basierte Techniken erlauben nicht für die Entdeckung von neuen Modifikationen, aber sie sind auf der Bestätigung und Quantifizierung von vorhergesagten Marken, die ausschließlich auf Basis. Wir spekulieren, so dass die Bottom-up-Proteomik auf Histon-Peptide werden auf die intuitive Vorteile aufgrund der Popularität in der Proteomik Labors gewinnen in die Regulation der Histonmarkierungen zu wissen, die Protagonisten in Tuning-Genexpression und damit die Regulierung des Proteoms auswirken. Darüber hinaus enthält das beschriebene Protokoll jüngsten Verbesserungen in der Probenvorbereitung und die Software für die Datenanalyse, die auch für labora Histon-Analyse trivialere machenTorys, die nie Charakterisierung dieser Art von hyper Peptide erfahren.
Offenlegungen
Die Autoren erklären, dass sie keine finanziellen Interessen haben.
Danksagungen
Diese Arbeit wurde durch die Finanzierung von NIH Zuschüsse (DP2OD007447, R01GM110174 und R01AI118891) unterstützt.
Materialien
| Name | Company | Catalog Number | Comments |
| Trypsin 0.25% EDTA | Invitrogen | 25200056 | For harvesting cells |
| PBS | Invitrogen | 14200075 | |
| Tris | Roche | 77-86-1 | |
| Potassium Chloride | Fisher Scientific | BP366-500 | |
| Sodium Chloride | Sigma | S9888 | |
| Magnesium Chloride hexahydrate | Sigma | M9272 | |
| Calcium Chloride, anhydrous | Sigma | C1016 | |
| Sucrose | Fisher Scientific | BP220-1 | |
| DTT | Invitrogen | 15508-013 | |
| AEBSF | EMD Millipore Corp | 101500 | |
| Microcystin | Sigma | M4194 | |
| Sodium Butyrate | Sigma | B5887 | |
| Halt Protease and Phosphatase Inhibitor Cocktail, EDTA-free (100x) | Fisher Scientific | 78445 | |
| NP- 40 Alternative | CALBIOCHEM | 492016 | |
| Sulfuric Acid, ACS grade | Fisher Chemical | 7664-93-9 | |
| Trichloroacetic acid | Sigma | T6399 | |
| Acetone | Sigma | 179124 | |
| HCl | Fisher Chemical | A144-500 | |
| Bradford reagent | Biorad | 500-0006 | |
| 30% acrylamide/bis 29:1 — 500 ml | Biorad | 1610156 | |
| Coomassie | Fisher Scientific | 20278 | |
| C18 Column (5 µm) 2.1 mm x 250 mm | Grace | 218TP52 | |
| C18 Column (5 µm) 4.6 mm x 250 mm | Grace | 218TP54 | |
| HPLC grade acetonitrile | Fisher Chemical | A955-4 | |
| HPLC grade water | Fisher Scientific | W6 4 | |
| TFA | Fisher Scientific | A11650 | |
| Ammonium Bicarbonate | Sigma | A6141 | |
| ammonium hydroxide | Sigma | 338818 | |
| propionic anhydride | Sigma | 240311 | |
| Sequencing grade modified trypsin | Promega | PRV5113 | For digesting histones for MS |
| Acetic Acid | Sigma | 49199 | |
| C18 extraction disk | Empore | 2215 | |
| Formic Acid | Sigma | F0507 |
Referenzen
- Waddington, C. H. Canalization of development and the inheritance of acquired characters. Nature. 150, 563-565 (1942).
- Sharma, S., Kelly, T. K., Jones, P. A. Epigenetics in cancer. Carcinogenesis. 31 (1), 27-36 (2010).
- Reik, W., Dean, W., Walter, J. Epigenetic reprogramming in mammalian development. Science. 293 (5532), 1089-1093 (2001).
- Kouzarides, T. Chromatin modifications and their function. Cell. 128 (4), 693-705 (2007).
- Tessarz, P., Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Bio. 15 (11), 703-708 (2014).
- Fischle, W., Wang, Y. M., Allis, C. D. Histone and chromatin cross-talk. Curr Opin Cell Biol. 15 (2), 172-183 (2003).
- Lee, J. S., Smith, E., Shilatifard, A. The language of histone crosstalk. Cell. 142 (5), 682-685 (2010).
- Simboeck, E., et al. A Phosphorylation Switch Regulates the Transcriptional Activation of Cell Cycle Regulator p21 by Histone Deacetylase Inhibitors. J Biol Chem. 285 (52), 41062-41073 (2010).
- Hirota, T., Lipp, J. J., Toh, B. H., Peters, J. M. Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature. 438 (7071), 1176-1180 (2005).
- Xhemalce, B., Kouzarides, T. A chromodomain switch mediated by histone H3 Lys 4 acetylation regulates heterochromatin assembly. Genes Dev. 24 (7), 647-652 (2010).
- Vermeulen, M., et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. 142 (6), 967-980 (2010).
- van Attikum, H., Gasser, S. M. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol. 19 (5), 207-217 (2009).
- Fernandez-Capetillo, O., et al. H2AX is required for chromatin remodeling and inactivation of sex chromosomes in male mouse meiosis. Dev Cell. 4 (4), 497-508 (2003).
- Santaguida, S., Musacchio, A. The life and miracles of kinetochores. Embo J. 28 (17), 2511-2531 (2009).
- Egelhofer, T. A., et al. An assessment of histone-modification antibody quality. Nat Struct Mol Biol. 18 (1), 91-93 (2011).
- Sidoli, S., Cheng, L., Jensen, O. N. Proteomics in chromatin biology and epigenetics: Elucidation of post-translational modifications of histone proteins by mass spectrometry. J Proteomics. 75 (12), 3419-3433 (2012).
- Plazas-Mayorca, M. D., et al. One-Pot Shotgun Quantitative Mass Spectrometry Characterization of Histones. J Proteome Res. 8 (11), 5367-5374 (2009).
- Sidoli, S., et al. Drawbacks in the use of unconventional hydrophobic anhydrides for histone derivatization in bottom-up proteomics PTM analysis. Proteomics. 15 (9), 1459-1469 (2015).
- Lin, S., Garcia, B. A. Examining histone posttranslational modification patterns by high-resolution mass spectrometry. Methods Enzymol. 512, 3-28 (2012).
- Sidoli, S., et al. SWATH Analysis for Characterization and Quantification of Histone Post-translational Modifications. Mol Cell Proteomics. , (2015).
- Krautkramer, K. A., Reiter, L., Denu, J. M., Dowell, J. A. Quantification of SAHA-Dependent Changes in Histone Modifications Using Data-Independent Acquisition Mass Spectrometry. J Proteome Res. , (2015).
- Yuan, Z. F., et al. EpiProfile Quantifies Histone Peptides With Modifications by Extracting Retention Time and Intensity in High-resolution Mass Spectra. Mol Cell Proteomics. 14 (6), 1696-1707 (2015).
- MacLean, B., et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 26 (7), 966-968 (2010).
- Yuan, Z. F., Lin, S., Molden, R. C., Garcia, B. A. Evaluation of proteomic search engines for the analysis of histone modifications. J Proteome Res. 13 (10), 4470-4478 (2014).
- Tan, Y., Xue, Y., Song, C., Grunstein, M. Acetylated histone H3K56 interacts with Oct4 to promote mouse embryonic stem cell pluripotency. Proc Natl Acad Sci U S A. 110 (28), 11493-11498 (2013).
- Vonholt, C., et al. Isolation and Characterization of Histones. Methods Enzymol. 170, 431-523 (1989).
- Zhang, K. L., et al. Identification of acetylation and methylation sites of histone H3 from chicken erythrocytes by high-accuracy matrix-assisted laser desorption ionization-time-of-flight, matrix-assisted laser desorption ionization-postsource decay, and nanoelectrospray ionization tandem mass spectrometry. Anal. Biochem. 306 (2), 259-269 (2002).
- Jufvas, A., Stralfors, P., Vener, A. V. Histone Variants and Their Post-Translational Modifications in Primary Human Fat Cells. Plos One. 6 (1), e15960(2011).
- Bonaldi, T., Imhof, A., Regula, J. T. A combination of different mass spectroscopic techniques for the analysis of dynamic changes of histone modifications. Proteomics. 4 (5), 1382-1396 (2004).
- Zhao, X. L., et al. Comparative Proteomic Analysis of Histone Post-translational Modifications upon Ischemia/Reperfusion-Induced Retinal Injury. J Proteome Res. 13 (4), 2175-2186 (2014).
- Lin, S., et al. Stable-isotope-labeled histone peptide library for histone post-translational modification and variant quantification by mass spectrometry. Mol Cell Proteomics. 13 (9), 2450-2466 (2014).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten