
Method Article
Complete Workflow for Analysis of Histone Post-translational Modifications Using Bottom-up Mass Spectrometry: From Histone Extraction to Data Analysis
In This Article
Summary
This protocol outlines a fully integrated workflow for characterizing histone post-translational modifications using mass spectrometry (MS). The workflow includes histone purification from cell cultures or tissues, histone derivatization and digestion, MS analysis using nano-flow liquid chromatography and instructions for data analysis. The protocol is designed for completion within 2 - 3 days.
Abstract
Nucleosomes are the smallest structural unit of chromatin, composed of 147 base pairs of DNA wrapped around an octamer of histone proteins. Histone function is mediated by extensive post-translational modification by a myriad of nuclear proteins. These modifications are critical for nuclear integrity as they regulate chromatin structure and recruit enzymes involved in gene regulation, DNA repair and chromosome condensation. Even though a large part of the scientific community adopts antibody-based techniques to characterize histone PTM abundance, these approaches are low throughput and biased against hypermodified proteins, as the epitope might be obstructed by nearby modifications. This protocol describes the use of nano liquid chromatography (nLC) and mass spectrometry (MS) for accurate quantification of histone modifications. This method is designed to characterize a large variety of histone PTMs and the relative abundance of several histone variants within single analyses. In this protocol, histones are derivatized with propionic anhydride followed by digestion with trypsin to generate peptides of 5 - 20 aa in length. After digestion, the newly exposed N-termini of the histone peptides are derivatized to improve chromatographic retention during nLC-MS. This method allows for the relative quantification of histone PTMs spanning four orders of magnitude.
Introduction
Epigenetics is defined as the study of heritable changes in gene expression that arise by mechanisms other than altering the underlying DNA sequence1. Epigenetic regulation is critical during development as the organism undergoes dramatic phenotypic changes even though its DNA content does not change. There are several critical components required for proper epigenetic maintenance, including histone post-translational modifications (PTMs), histone variants, non-coding RNAs, DNA methylation and DNA binding factors, each of which affect gene expression through different mechanisms2. For example, while DNA methylation is a highly stable modification that represses gene translation3, histone variants and histone PTMs are much more dynamic and can influence chromatin in a variety of ways4.
Histone PTMs are mostly localized on the N-terminal tails, as they are the most exposed and flexible region of the protein. However, the nucleosome core is also heavily modified compared to average proteins5. Even though histone marks have been extensively characterized in the last decade, many links between known histone marks and their function are still unclear. This is largely due to the fact that most histone PTMs do not work alone, but rather function in tandem with other PTMs ("cross-talk") to alter a specific process such as transcription6,7. For instance, the combinatorial mark H3S10K14ac on the gene p21 activates its transcription, which would not occur with only one of the two PTMs8. The protein HP1 compacts chromatin by recognizing H3K9me2/me3 and spreading the modification to nearby nucleosomes. However, HP1 cannot bind H3K9me2/3 when the adjacent S10 is phosphorylated9. Acetylation of H3K4 inhibits binding of the protein spChp1 to H3K9me2/me3 in Schizosaccharomyces pombe10. Furthermore, the histone lysine demethylase PHF8 has the highest nucleosome binding efficiency when three PTMs H3K4me3, K9ac, and K14ac are present11. These examples highlight the importance of achieving a global overview of histone PTM changes rather than focusing on single modifications.
The presence of sequence variants also increases the complexity of histone analysis, as histone isotypes generally have highly similar sequences, but often have different roles in chromatin. For example, H2A.x has a C-terminal sequence which is more easily phosphorylated upon DNA damage compared to canonical H2A12, and it is required for inactivation of sex chromosomes in male mouse meiosis13; similarly, CENP-A substitutes canonical histone H3 in centromeres14. Despite their different functions, these variants share a large portion of their amino acid sequence with the respective canonical histone, making it difficult to identify and quantify them separately.
Antibody-based techniques such as western blotting have been extensively adopted to characterize histones. However, antibody-based approaches are limited for the following reasons: (i) they can only confirm the presence of a modification and cannot identify unknown PTMs; (ii) they are biased due to the presence of co-existing marks, which can influence binding affinity; (iii) they cannot identify combinatorial marks, as only very few antibodies are available for such purpose and (iv) they cross-react between highly similar histone variants or similar PTMs (e.g., di- and trimethylation of lysine residues). Egelhofer et al. described that more than 25% of commercial antibodies fail specificity tests by dot blot or western blot, and among specific antibodies more than 20% fail in chromatin immunoprecipitation experiments15. Mass spectrometry (MS) is currently the most suitable analytical tool to study novel and/or combinatorial PTMs, and it has been extensively implemented for histone proteins (reviewed in 16). This is mostly due to high sensitivity and mass accuracy of MS, and the possibility to perform large-scale analyses.
The bottom-up strategy is the most commonly used MS-based proteomics strategy for histone characterization and their PTMs, wherein the intact protein is enzymatically digested into short peptides (5 - 20 aa). This digestion facilitates both LC separation and MS detection. Masses in the range of 600 - 2,000 Da are commonly more easily ionized and identified with higher mass accuracy and resolution than larger masses. MS/MS fragmentation is also improved, as short peptides are generally well-suited for collision induced dissociation (CID). However, histones present a challenge for bottom-up MS as they are highly enriched in basic amino acid residues, namely lysine and arginine. Therefore, trypsin digestion leads to the generation of peptides that are too small for LC retention and unambiguous localization of the PTMs. To circumvent this issue, our protocol includes lysine and peptide N-terminal chemical derivatization17. The use of propionic anhydride is recommended for efficient chemical derivatization as compared to other reagents 18. Such derivatization blocks the ɛ-amino groups of unmodified and monomethyl lysine residues, allowing trypsin to perform proteolysis only at the C-terminal of arginine residues. Derivatized amines cannot exchange protons with the solution and thus the peptides are generally only doubly or triply charged, facilitating MS and MS/MS detection. Moreover, N-terminal derivatization increases peptide hydrophobicity and thus reversed-phase chromatographic retention. Here, we describe the workflow to purify histones and prepare them for PTM analysis via bottom-up proteomics (Figure 1). This strategy achieves quantification of single histone marks and combinatorial marks for histone PTMs that are relatively close in the amino acid sequence.
Protocol
1. Collection of Cells from Culture
- If cells were grown in suspension, collect cells by centrifugation at 300 rcf for 5 min. If adherent, aspirate and discard cell medium. Rinse the attached cells with PBS without Ca2+ and Mg2+ (referred to as PBS going forward). Incubate the cells in either trypsin or trypsin-EDTA (0.025% - 0.5% depending on the cell line) with enough volume to cover the surface of the plates at 37 °C until cells detach (time varies for different cell lines).
- Collect cells by centrifugation at 300 rcf for 5 min. Wash cells two more times in PBS and collect by centrifugation.
- Estimate the packed cell volume approximately from the graduations marked on the 1.8 ml tubes or 15 ml conical tubes.
Note: Cells from culture can be snap-frozen in liquid nitrogen and stored at -80 °C indefinitely at this stage.
2. Isolation of Nuclei from Intact Cells
- Thaw cells on ice.
- Thaw nuclear isolation buffer (NIB, Table 1).
- Prepare approximately 5 ml NIB buffer (Table 1) for every 100 µl packed cell volume. For every 1 ml NIB buffer, add protease inhibitors and stabilizing agents as follows: 1 µl of 1 M DTT, 2.5 µl of 200 mM AEBSF, 2 µl of 2.5 µM microcystin and 2 µl of 5 M sodium butyrate. NIB with inhibitors will be referred to as NIB from this point forward.
Note: If histone phosphorylation is studied, include EDTA free protease and phosphatase inhibitor cocktail. - Remove a fifth volume of NIB buffer thus prepared and add NP-40 Alternative (Table 1) to a final concentration of 0.2% . The remaining four-fifth volume will be used for washes.
- Wash cell pellet in 1:10 cell pellet to NIB without NP-40 Alternative ratio (v/v). Remove supernatant by centrifugation at 700 rcf for 5 min.
- Lyse the cell pellet by placing it on ice and adding 1:10 cell pellet to NIB with 0.2% NP-40 Alternative (v/v).
- If extracting from tissue samples, homogenize using mortar and pestle or dounce homogenizers. Cultured cells can be homogenized by gentle pipetting.
- Incubate homogenized cells on ice for 5 - 10 min. The cells will lyse and release the nuclei.
- Centrifuge at 1,000 rcf for 5 - 10 min at 4 °C. The pellet contains mostly cell nuclei, while the supernatant contains mostly cytoplasmic components. Save the cytoplasmic fraction if desired.
- Wash the nuclei pellet by gently resuspending it in 1:10 (v/v) NIB without NP-40 Alternative.
Note: This wash step is solely to remove traces of detergents prior to extracting histones from nuclei. - Centrifuge at 1,000 rcf for 5 min at 4 °C and remove supernatant.
- Repeat Step 2.10 - 2.11 at least two times to completely remove NP-40 Alternative. Removal of NP-40 Alternative is evident as gentle pipetting during the washing step no longer forms bubbles.
- For histone extraction from tissues:
- Rinse fresh or thawed frozen tissue in ice-cold NIB.
- Transfer tissue to a petri dish placed on ice with NIB, just enough to keep the tissue wet.
- Dice into smallest pieces (< 1 mm) with razor blade to increase the surface contact for nuclei isolation.
- Transfer minced tissue to a pre-chilled homogenizer and wash in NIB by pipetting up and down.
- Remove buffer by centrifuging at 300 rcf for 5 min.
- Add NIB containing NP-40 Alternative to the cells in a cells:buffer ratio of 1:10 (v/v) and homogenize by 5 - 10 strokes.
- Check for cell lysis and repeat homogenization as needed. A good indicator that cells have been lysed is the reduction of the pellet volume. The pellet should contain only nuclei.
- Centrifuge at 700 rcf for 5 min and save pellet. This pellet can be extracted 1 - 2 more times in 1:10 (v/v) of NIB containing NP-40 Alternative; at this stage, the histones are extracted out of chromatin and the pellet has shrunken considerably.
- Wash two times with 2 - 3 ml of NIB without NP40 Alternative to remove traces of the detergent.
Note: Interim stopping point: Sample can be resuspended in the minimum volume of NIB + 5% glycerol, and stored at -80 °C.
3. Extraction and Purification of Histones from Nuclei
Note: Histones are very rich in basic amino acid residues, allowing them to tightly interact with the phosphoric acid backbone of DNA. Histones are among the most basic proteins in the nucleus, thus allowing them to be extracted in ice-cold sulfuric acid (0.2 M H2SO4) with minimal contamination from non-histone proteins, which precipitate in strong acid. Highly concentrated TCA (to a final concentration of 33%) can then be used to precipitate histones from the sulfuric acid. TCA is stored as 100% in brown bottle at 4 °C.
- Resuspend cell nuclei in 1:5 (v/v) chilled 0.2 M H2SO4 (Table 1) by gentle pipetting.
- Incubate the sample with constant rotation or gentle shaking for 2 - 4 hr at 4 °C. Typically, for samples with more than 500 µl cell pellet, a 2 hr extraction is sufficient to extract histones; longer incubation may result in extraction of other basic proteins. For smaller nuclear pellets (< 200 µl), 4 hr extraction provides a better yield.
- Centrifuge at 3,400 rcf at 4 °C for 5 min.
- Transfer the supernatant to a new tube.
- Repeat Steps 3.3 - 3.4 to remove any insoluble material.
- To precipitate the histones, add chilled 100% TCA (Table 1) to the collected supernatant (now containing histones) in the ratio of 1:3 (v/v), in order to obtain a final TCA concentration of 33%. Mix by inverting the tube a few times.
Note: The samples will turn cloudy on addition of TCA, indicating the presence of histones. - Incubate the mixture on ice for at least 1 hr. For smaller starting pellet sizes, overnight precipitation is recommended.
- Centrifuge at 3,400 rcf for 5 min. The histones coat the sides of the tubes and also deposit at the bottom. A white insoluble pellet also forms at the very bottom of the tube, which mostly contains non-histone proteins and other biomolecules. Remove supernatant by aspiration, carefully without scraping the sides or the pellet.
- By using a glass Pasteur pipette, rinse the tube with ice-cold acetone + 0.1% HCl (Table 1) so as to cover the precipitated proteins coating the sides and bottom.
- Centrifuge at 3,400 rcf for 2 min and aspirate supernatant, carefully without scraping the sides or the pellet.
- Repeat Steps 3.9 - 3.10 using 100% ice-cold acetone.
- Dry pellet with air flow or with a vacuum centrifuge, or just by leaving the tube open. Acetone evaporates quickly.
- Dissolve the histones with ddH2O (double distilled water) in minimum volume possible to dissolve the white layer completely. Histones are readily soluble in water. For pellets in a 1.5 ml microcentrifuge tube, 100 µl ddH2O is usually enough to collect histones.
- Centrifuge at 3,400 rcf for 2 min and transfer the supernatant to a new tube.
4. Estimation of Protein Concentration and Purity
- For measuring protein concentration, use BCA, Bradford protein assay or amino acid analysis (AAA). Do not use techniques that adopt absorbance at 280 nm, as histones are poor in aromatic amino acid residues.
- Verify the purity of extracted histones by SDS-PAGE analysis with a 15% acrylamide gel and Coomassie staining (optional).
- If high purity single histone variants are desired, continue to HPLC-UV fractionation of histone variants (section 5). If not, skip directly to sample preparation for bottom-up histone PTM analysis (section 6).
5. Separation of Histone Variants by Reversed-phase HPLC (Optional)
Note: High purity histone variants can be obtained by fractionating the crude histone mixture using reversed-phase HPLC coupled to a UV detector. These purified histones are useful for studies that require higher sensitivity and purity. However, for standard histone PTM characterization, this step can be skipped because the analysis is sufficiently sensitive and exhaustive. Fractionation of intact histone variants ideally requires at least 100 - 300 µg of starting material.
- Connect an appropriate C18 5 µm column to an HPLC depending on the starting histone concentration: with about 100 µg of histones, use 2.1 mm x 250 mm column with a flow-rate of 0.2 ml/min; with about 300 µg histones, use 4.6 x 250 mm column with a flow-rate of 0.8 ml/min. Prepare Buffer A and B using dedicated glassware as follows:
- Prepare Buffer A: 5% HPLC grade acetonitrile, 0.1% TFA in HPLC grade water.
- Prepare Buffer B: 95% HPLC grade acetonitrile, 0.1% TFA in HPLC grade water.
- Connect the column to a UV detector, and set the absorbance to 210 - 220 nm.
- Acidify the histone sample dissolved in water with 100% TFA to achieve a final concentration of 0.1-1% TFA.
- Equilibrate the column with 100% buffer A for at least 15 min at the recommended flow-rate, which corresponds approximately to three column volumes. Use this signal to set the zero-absorbance level of the UV detector.
- Prepare appropriately sized tubes to collect fractions either manually or in an automatic sample collector.
- Inject sample at a concentration of approximately 1 µg/µl or higher. Samples dissolved in larger volumes might alter the equilibration of the column during loading and lead to lower retention.
- Run the gradient, programmed as follows: from 0 to 30% B in 1 min, 30 to 60% B in 90 min, and 60 to 90% B in 1 min.
- Collect fractions (example chromatogram shown in Figure 2) in 1 min intervals using an automatic fraction collector. Collect fractions in appropriate size tubes to contain the entire volume.
- Dry down fractionated samples in a vacuum concentrator.
Note: Interim stopping point: Dried histone fractions can be stored at room temperature for short periods (1 - 2 days) or in -80 °C freezer for long-term periods.
6. Chemical Derivatization of Histones Using Propionic Anhydride for Bottom-up Analysis
- Dissolve histone samples in 40 μl of 50 mM NH4HCO3, pH 8.0 (recommended amount: 50 - 100 μg). If samples were in pure ddH2O, add concentrated NH4HCO3 to make up 50 mM, pH 8.0.
- Wet a P10 pipette tip into the sample to check the pH using pH indicator strips without sample losses. NH4OH and formic acid can be used to adjust the pH to 8.0.
Note: The following part of the protocol (steps 6.3 - 6.7) should be done in batches of maximum three to four samples, in order to keep propionic anhydride reactive. - Use fume hood for the subsequent steps where propionic anhydride is used. Prepare fresh propionylation reagent by mixing propionic anhydride with acetonitrile in the ratio 1:3 (v/v). Add propionylation reagent to sample in 1:4 (v/v). For 40 µl histones, add 10 µl propionylation reagent.
Note: It is possible to observe white debris at this step. However, this mostly contains salts and propionic acid, and thus no specific action needs to be taken. - Quickly add NH4OH to re-establish pH 8.0 to the solution. Note: Propionic anhydride reacting with the free amines of the peptides produces propionic acid that decreases pH. Usually, adding NH4OH to the sample with a ratio of 1:5 (v/v) is appropriate to re-establish pH 8.0; e.g., 8 µl of NH4OH to 40 µl of sample.
- Mix immediately by vortexing.
- Check pH with the same procedure as Step 6.2.
Caution: When pH is greater than 10.0, labeling of other amino acid residues with higher pKa is possible. - Incubate samples at room temperature for 15 min.
- Repeat Steps 6.3 - 6.7, strictly performing the reaction for no more than 3 or 4 samples per batch of propionylation reagent.
- Dry samples down to 10 - 20 µl in a vacuum concentrator. This evaporates unreacted propionic anhydride, acetonitrile, acetic acid and ammonia gas released from NH4OH. If samples dry out completely, no significant sample losses occur.
Note: Isopropanol can be used instead of acetonitrile. However, acetonitrile has lower surface tension and thus more rapid evaporation. - Resuspend or dilute samples with ddH2O until 40 µl of final volume is achieved.
- Repeat Steps 6.2 - 6.9. A double round of histone propionylation ensures > 95% of reaction completion.
- Fill propionic anhydride bottle with argon gas so as to prevent formation of acetic acid in contact with moisture in the bottle.
Note: Interim stopping point: Sample can be stored at -80 °C reconstituted in ddH2O or dried.
7. Proteolytic Digestion with Trypsin
- Resuspend histones in 50 mM NH4HCO3 to achieve an optimal concentration of 1 μg/μl or higher. More diluted samples lead to lower trypsin efficiency.
Note: The histones at this step need to be at pH 8.0 If it still acidic, then add NH4HCO3 salt to sample by using pipet tip. - Add trypsin to histone samples at a 1:10 ratio (wt/wt).
- Incubate at 37 °C for 6 - 8 hr.
- Stop the digestion by freezing in -80 °C.
- Dry down the sample to 10 - 20 µl in a vacuum concentrator.
Note: Interim stopping point: Sample can be stored at -80 °C.
8. Propionylation of Histone Peptides at N-termini
Note: This section describes the derivatization of peptide N-termini generated from the trypsin digest. Such procedure improves HPLC retention of the shortest peptides (e.g., amino acid 3 - 8 of histone H3), as the propionyl group increases peptide hydrophobicity.
- Resuspend samples in 30 µl of 100 mM NH4HCO3.
- Repeat Steps 6.1 - 6.9.
Note: It is normal that drying of samples in vacuum takes a longer time at this step. - Resuspend or dilute samples with 50 - 100 µl ddH2O + either 0.1% TFA or 0.5% acetic acid. Note: Acetic acid is recommended for long storages, as TFA facilitates methionine oxidation in the long term. On the other hand, TFA is recommended if stage-tipping (section 9) is performed the same day, as TFA assists a better chromatographic retention.
Note: Interim stopping point: Sample can be stored at -80 °C.
9. Sample Desalting with Stage-tips
Note: At this stage, there is salt present in the sample. Salts impede HPLC-MS analysis because they ionize during electrospray, suppressing the signal from peptides. Salts can also form ionic adducts on peptides, reducing the signal intensity for the non-adducted peptide. As the adducted peptide will have a different mass, the peptide will not be properly identified or quantified.
- By using a P1000 pipette tip, punch a disk of C18 material from a solid phase extraction disk. Push the minidisk out of the P1000 tip by using a fused silica capillary and deposit the minidisk to the bottom of a P100 / 200 pipette tip. Ensure that the disk is securely wedged at the bottom of the tip (Figure 3).
Note: The P1000 tip has a rather small hole to punch the C18 disk. It is appropriate to cut the last centimeter of the tip in order to have a hole with larger diameter. - Use two C18 punches in the same P100/P200 tip if desalting over 25 µg of sample.
- Use a centrifuge adaptor to hold stage-tips in place in 1.5 ml or 2 ml microcentrifuge tubes. Use slow (300 - 400 rcf) rotation; the solvents normally pass through the resin in less than a minute.
- Flush the resin by spinning with 50 µl of 100% acetonitrile to activate the C18 material and remove potential contaminations.
- Equilibrate disk by flushing 80 µl of 0.1% TFA by slow centrifugation.
- Acidify the sample to pH 4.0 or lower with acetic acid. Check the pH with pH strips to minimize sample loss. Load sample onto the disk by slow centrifugation.
- Wash sample by flushing 70 - 80 µl of 0.1% TFA by slow centrifugation.
- Elute sample by flushing 70 µl 75% acetonitrile and 0.5% acetic acid by slow centrifugation. Collect the sample in a 1.5 ml tube.
- Dry sample in a vacuum concentrator.
Note: Interim stopping point: Sample can be stored at -80 °C.
10. Analysis of Histone Peptides
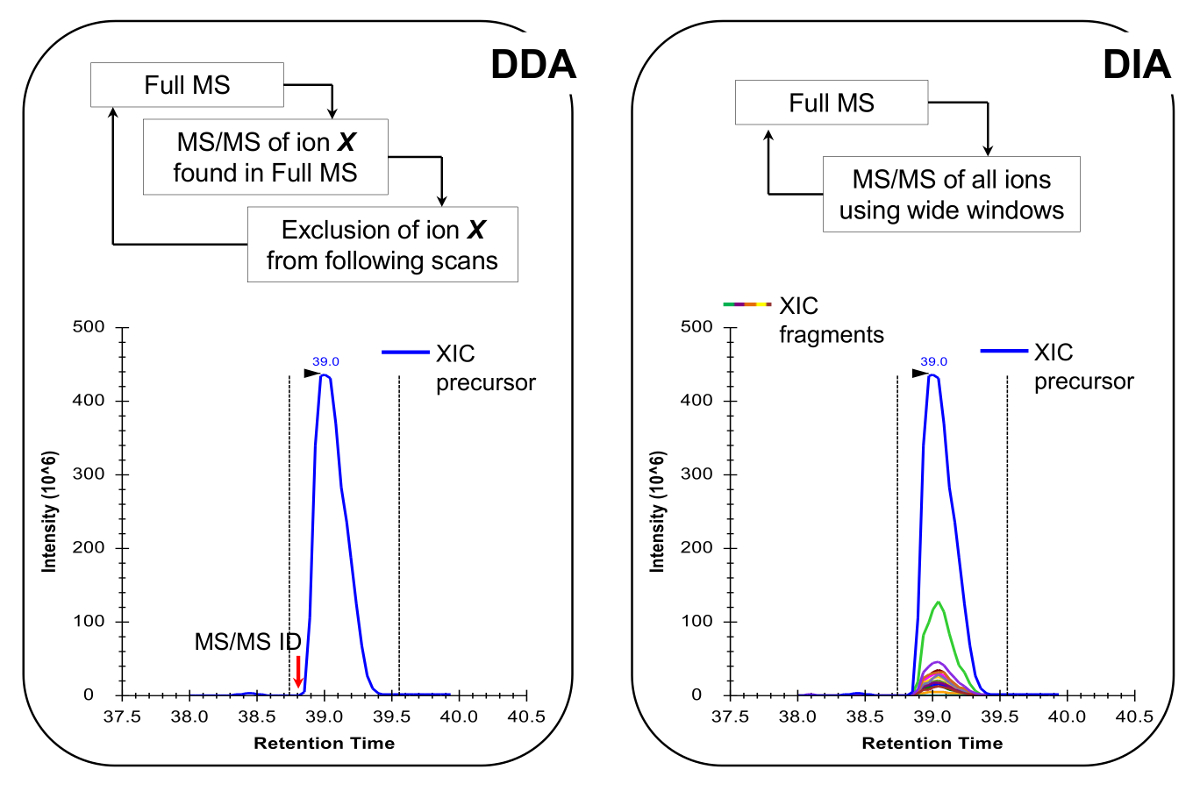
Note: The nLC-MS platform should be set up as done in traditional peptide analysis. The use of 200 - 300 nl flow column (75 µm ID analytical column, C18 particles) is recommended, as they are an excellent compromise between sensitivity and stability. The MS acquisition method can be either a combination of data-dependent acquisition (DDA) with targeted scans19 or a data-independent acquisition (DIA)20,21, both described in Representative Results and Figure 4.
- Prepare HPLC buffers — A: 0.1% formic acid in HPLC-grade water; B: 0.1% formic acid in HPLC-grade acetonitrile.
- Program the HPLC method as follows: from 0 to 30% buffer B in 30 min, from 30 to 100% B for the next 5 min and at isocratic 100% B for 8 min. If the HPLC is not programmed for automated column equilibration before sample loading, then include the following: gradient from 100 to 0% B in 1 min and isocratic flow at 0% B for 10 min. Set the flow rate of the analysis to 250 - 300 nl/min.
- Program the MS acquisition method to perform either DDA combined with targeted scans19 or DIA 20,21 (Figure 4). Make sure that the MS duty cycle allows one full MS scan every ~ 2 sec, in order to have enough data points to across the chromatographic peak, which enables more accurate quantification. Note: With C18 chromatography, the average baseline peak width is about 30 sec for the gradient described above.
- Load approximately 1 µg of sample onto the HPLC column.
- Run the HPLC-MS/MS method as programmed.
Note: In the protocol we do not recommend specifics of columns, MS instruments or MS parameter details, as any optimal setup that an individual proteomics lab developed will be suitable for the method. Proteomics laboratories should use their optimized setup, since histone peptides separate as traditional peptides.
11. Data Analysis
- Import the MS raw files into software to perform peak area integration. Note: EpiProfile22 is recommended, as it is optimized for histone peptides; by using the retention time knowledge of chromatographic elution it performs reliable peak area extraction of known histone peptides. Alternatively, Skyline23 is another ideal software for the purpose.
- Calculate the relative abundance of a given peptide by dividing its area by the total area of that peptide in all of its modified forms. Note: In case of discovery analysis Mascot is recommended to identify spectra of modified histone peptides. The performance of this tool has been described recently24. All other database search engines for proteomics are also usable, but upon our tests they provided lower performance.
Results
As an example, we analyzed histones extracted from human embryonic stem cells (hESCs) with and without retinoic acid (RA) stimulation, starting with 200 µl cell pellets. Presence of RA in cell culture leads to ESC differentiation. From the cell pellet, about 50 - 100 µg of histones were extracted, which is more than sufficient to perform multiple LC-MS injections of histone peptides. After derivatization, digestion, and desalting, the samples were loaded onto a 75 µm x 15 cm C18 column (particle diameter 3 µm, pore size 300 Å) in serial mode with a high-performance liquid nano chromatography system with microfluidic chips coupled to a hybrid linear trap quadrupole – orbitrap mass spectrometer. MS acquisition was performed using DIA. In parallel, samples were also analyzed with a DDA method using a nano-flow UHPLC coupled to a hybrid ion trap-orbitrap mass spectrometer (data not shown). In each cycle, one full MS orbitrap detection was performed with the scan range of 290 to 1,400 m/z, a resolution of 60,000 (at 200 m/z) and AGC of 106. Then, data dependent acquisition mode was applied with a dynamic exclusion of 30 sec. MS/MS scans were followed on parent ions from the most intense ones. Ions with a charge state of one were excluded from MS/MS. An isolation window of 2 m/z was used. Ions were fragmented using collision induced dissociation (CID) with collision energy of 35%. Ion trap detection was used with normal scan range mode and normal scan rate with AGC of 104.
Raw MS data were analyzed adopting software for the extraction of precursor and fragment ion chromatograms, namely Skyline23 and EpiProfile22. EpiProfile has been optimized for histone peptides, as it integrates intelligent peak area extraction due to previous knowledge of peptide retention time. On the other hand, Skyline is optimized for DIA analyses, and thus the DIA figures displayed (Figures 4 and 5A) are screenshots from this software. From the extracted ion chromatogram, the area under the curve is retrieved, and this is used to estimate the abundance of each peptide. The area of the chromatographic peak was calculated for the [M + H]+, [M + 2H]2+, and [M + 3H]3+ ions of the same peptide, even though in most cases the [M + 2H]2+ was the prevalent form. This provides the raw abundance of a given modified form of a peptide. In order to achieve the relative abundance of PTMs, the sum of all different modified forms of a histone peptide was considered as 100%, and the area of the particular peptide was divided by the total area for that histone peptide in all of its modified forms.
Histone peptides are present in a variety of isobaric forms (Figure 5). Isobaric peptides, e.g., K18ac and K23ac, can only be quantified at the MS/MS level, where their unique fragment ions are used to determine the ratio of the isobaric species (Figure 5A and 5B). This ratio is used to divide the area of the chromatographic peak between the two species. When using DDA, these isobaric forms were included in a list of targeted masses, because these peptides need to be selected for fragmentation through their entire elution, which would not occur in a standard DDA experiment. The discrimination of the relative abundance of the isobaric species is then performed by monitoring the elution profile of the fragment ions. On the other hand, DIA type of acquisition does not require any inclusion list. However, this type of acquisition method is not compatible with traditional database searching, and thus might prevent the discovery of unknown modified peptides.
Lysine acetylation (+ 42.011 Da) was discriminated from the nearly isobaric trimethylation (+ 42.047 Da) by using high resolution MS acquisition (> 30,000). Moreover, acetylation is more hydrophobic than trimethylation, leading to elution of acetylated peptides later than the respective trimethylated ones. The unmodified form of the same peptide elutes even later, due to the fact that the lysine is propionylated. In summary, the order of hydrophobicity for a peptide with one modifiable site is di- and trimethylated < acetylated < unmodified (propionylated) < monomethylated (propionylated).
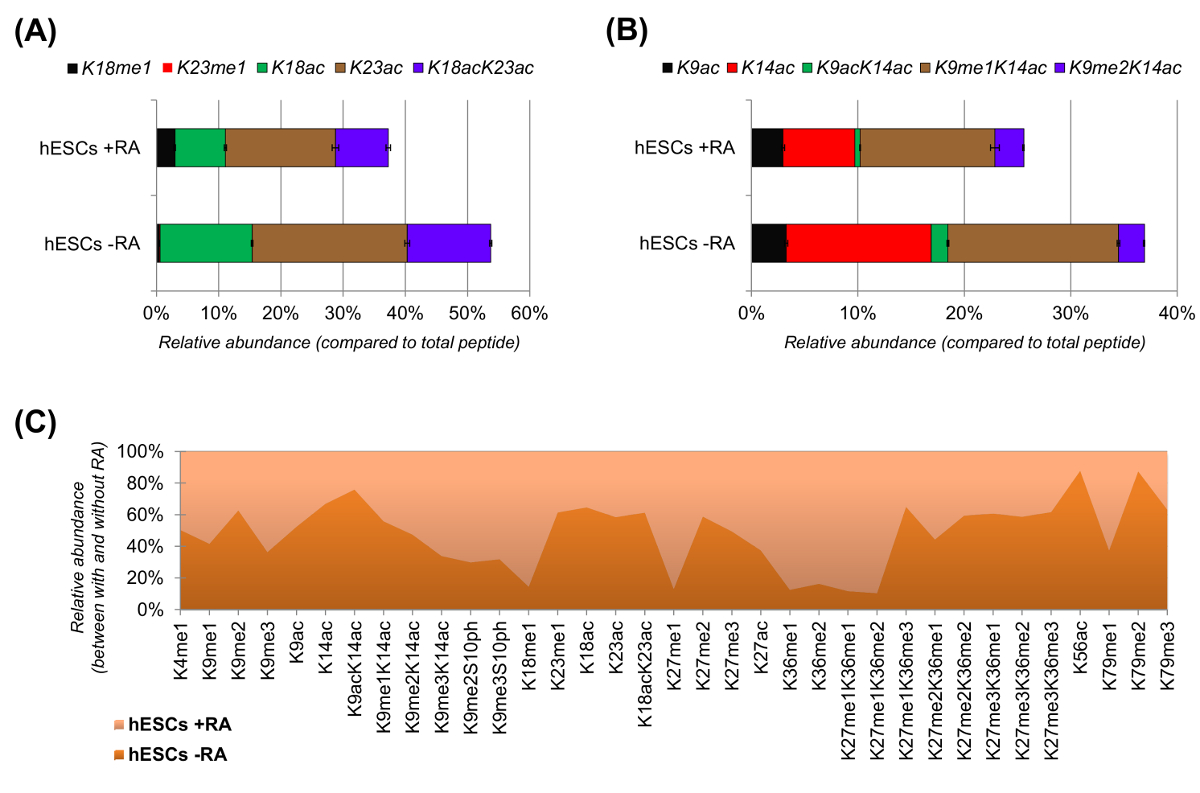
hESCs showed a clear reduction of acetylated peptides when stimulated for differentiation (Figure 6A and 6B). This was not surprising, as previous results reported higher acetylation in ESCs as compared to differentiating ones25, reflecting the generally permissive nature of the pluripotent chromatin. By focusing on histone H3, 35 different modified forms were quantified (Figure 6C). However, all histone proteoforms that can be investigated with this approach are more than 200, including all histone variants and low abundance modifications (data not shown). Moreover, our analysis showed that high reproducibility can be obtained between technical replicates, as evidenced by the small size of the error bars (representing ± standard deviation). Taken together, this section describes how to extract the relative abundance of histone modified peptides using nLC-MS data.

Figure 1: Workflow for Bottom-up MS/MS Histone Analysis. The ten steps for histone analysis are shown, including an estimation of the time required for each step. The section number is given in parenthesis as present in the manuscript. Section 5, describing sample fractionation to isolate the various histone variants, can be omitted unless there is a need for highly sensitive analysis of a given variant. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Reversed-Phase High Flow LC for Histone Variant Fractionation and Coomassie Gel. (A) LC-UV chromatogram representing intact histone separation. Histone H3 variants can be discriminated from one another according to their elution time. Fractions can be collected either manually or using an automated fraction collector. (B) Coomassie gel of three replicates of histone purification. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Making of Stage-tipping Plug. With a P1000 pipette tip, punch a disk made of C18 material from a solid phase extraction disk (second panel). The minidisk will stick in the tip (middle panel), so that it can be pushed out into a smaller P100/200 pipette tip using any kind of small capillary. In this example, we used a 700 µm external diameter fused silica tubing. The minidisk should be pushed to the bottom of the P100/200 pipette tip until it cannot go any further (last panel). The stage tip is ready for histone desalting, as it has sufficient capacity to retain enough sample material for numerous replicates. Specifically, one minidisk is enough for 15 - 20 µg of sample. If more sample is required, multiple disks can be packed on one another. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Schematic Representation of DDA and DIA Methods. When using DDA, the MS scan cycle is characterized by sequential selection of precursor ions for MS/MS fragmentation according to their intensity and charge state. Once a precursor ion has been fragmented it is placed into an exclusion list to avoid repetitive selection of the same peptide, so that the MS can "dig" into less abundant signals. This acquisition method is the technique of choice in proteomics for discovery mode. Quantification is achieved by integrating the full scan signal of a given ion next to the identified MS/MS spectrum. In DIA, the entire m/z range is fragmented at every scan cycle. This approach is less suitable for discovery mode, but it produces a chromatographic profile of all ions, precursors and products. This leads to more confident quantification and discrimination of isobaric forms. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Quantification of Isobaric Peptides. (A) Example of two isobaric peptides commonly abundant in histone analysis. The extracted ion chromatogram (XIC) of their precursor mass and relative isotopes (above) is identical. However, the XIC of the product ions (below) allows for discrimination of the two isobaric forms. Notably, only unique fragment ions should be used to estimate the relative abundance of the two species. (B) Representation of the unique fragment ions for the two described peptides (highlighted in red). (C) List of the commonly analyzed peptides in Homo sapiens having at least one isobaric equivalent. Sequence variants between the listed histone peptides are indicated. Please click here to view a larger version of this figure.
{kind=link}

Figure 6: Representative Results of Human Embryonic Stem Cells with and without Retinoic Acid Treatment. (A) Relative quantification of the histone H3 peptide KQLATKAAR (aa 18 - 26) in all of its modified proteoforms. The relative abundance was estimated using all proteoforms as 100% (the relative percentage of the unmodified peptide is not shown). (B) Relative quantification of the histone H3 peptide KSTGGKAPR (aa 9 - 17). (C) Relative abundance of detected peptides for canonical histone H3 with and without cell treatment with retinoic acid. The figure indicates in which of the two treatments the given modifications are more abundant (> 50%). Overall, we demonstrate that histone H3 acetylation decreases in most of the lysine residues upon induction of cell differentiation. Please click here to view a larger version of this figure.
{kind=link}
| Solution # | Composition | ||||||
| 1 | Nuclear Isolation Buffer (NIB) stock is made as follows and stored frozen as 100 ml aliquots at -20 °C; thawed NIB can be stored at 4 °C for few weeks: 15 mM Tris, 60 mM KCl, 15 mM NaCl, 5 mM MgCl2, 1 mM CaCl2, and 250 mM sucrose. The pH of the buffer is adjusted to 7.5 with HCl. | ||||||
| 2 | Protease inhibitors (add fresh to buffers prior to use): 1 M Dithiothreitol (DTT) in ddH2O (1,000x); 200 mM AEBSF in ddH2O (400x) | ||||||
| 3 | phosphatase inhibitor (add fresh to buffers prior to use): 2.5 µM Microcystin in 100% ethanol (500x) | ||||||
| 4 | HDAC inhibitor (add fresh to buffers prior to use): 5 M Sodium butyrate, made by titration of 5 M butyric acid using NaOH to pH 7.0 (500x) | ||||||
| 5 | NP-40 Alternative: 10% v/v in ddH2O | ||||||
| 6 | 0.2 M H2SO4 in ddH2O | ||||||
| 7 | Trichloroacetic acid (TCA): 100% w/v in ddH2O | ||||||
| 8 | Acetone+0.1% Hydrochloric acid (HCl): 0.1% v/v HCl in acetone | ||||||
Table 1. Solutions.
Discussion
The protocol described here is optimized considering costs, time, and performance. Other preparations are possible, but they have limitations, especially in the case of coupling with MS analysis. For instance, the high-salt extraction protocol can be used to purify histones26 instead of TCA precipitation (section 3). High-salt protocol is intrinsically milder, as it does not use strong acid. This preserves acid-labile PTMs and increases the yield of extracted histones, as TCA precipitation co-precipitates many other chromatin binding proteins. However, high-salt extraction leads to samples containing too concentrated salt for HPLC-MS/MS. In an alternative preparation, histone digestion can be performed without propionylation (section 6 - 8), for instance by reducing trypsin incubation time and the enzyme/substrate ratio27 or using ArgC as digestion enzyme28-30. However, derivatization with propionic anhydride is recommended, as it leads to the generation of more hydrophobic peptides, which are better retained during liquid chromatography.
For chemical derivatization, a variety of organic acid anhydrides have been evaluated and their merits comprehensively discussed18. Nonetheless, propionic anhydride proved to the best compromise between efficiency, minimized side products and improved peptide hydrophobicity. Potentially, propionic anhydride can be purchased in the isotopically labeled form; this allows for multiplexing analysis due to the possibility of mixing multiple samples and discriminate them at the MS level based on the different masses imparted from the heavy label. However, this analysis leads to an increased complexity of the LC-MS chromatogram and reduces the amount of sample that can be injected for each single condition.
In this regard, some critical aspects of the protocol should be highlighted. The following should be used as checklist to find errors in performing the procedure in case negative results are obtained. First, after nuclei precipitation the pellet should be carefully washed with NIB without NP-40 Alternative (section 2.10) until complete removal of the detergent (noticeable by the lack of bubbles during mixing). Failing to do so would compromise histone extraction with acids. Second, after histone precipitation with TCA (section 3.9) washes of the pellet with acetone is crucial. Presence of concentrated acid would harm the following step if propionylation and digestion (section 6.1) are directly performed. It would be not problematic in case histone fractionation is performed (section 5). Third, it is essential that the propionylation reaction is performed quickly (section 6.3 - 6.7). To do so, avoid using the same propionylation mix (propionic anhydride + acetonitrile) for more than 3 - 4 consecutive samples. Additionally, pH is the most important aspect of trypsin digestion (section 7). If not around 8.0 (7.5 - 8.5) the digestion will be ineffective. This can happen, as the sample will be rich in propionic acid at this step. NH4OH can be added until necessary. Also, for researchers familiar with proteomics workflows it will feel normal to acidify the sample to terminate trypsin digestion. This should not be done, as it will jeopardize the following reaction, i.e., propionylation of peptide N-termini (section 8.1). Finally, in the same issue, it is important to remember for data analysis that unmodified peptides are not actually unmodified; all free lysine residues and N-termini will be occupied by propionylation (56.026 Da). Thus, performing extracting ion chromatography of the mass corresponding uniquely to the peptide sequence would lead to no results.
The limitations of the method are mostly related to the inability of detecting combinatorial PTMs, due to the short peptide sequences, and the biases in achieving the true abundance of a modification, due to the fact that peptides in different modified forms might ionize with different efficiencies. The first issue can be solved by combining this technique with a middle-down or top-down approach (reviewed in 16). This type of analysis, even if technically more challenging, is ideal for studying co-existence frequencies of modifications. Moreover, it allows better discrimination of histone variants, which cannot always be achieved with bottom-up since some peptides have the same sequence in different histone variants. The second issue, related to the ionization efficiency, can be solved using a library of synthetic peptides31. This approach ensures a more accurate estimation of the relative abundance of histone PTMs. However, in most experiments, the desired outcome is the relative changes of given modifications between analyzed conditions. In this case, such correction is not necessary, due to the fact that all samples have the same bias.
In conclusion, this protocol allows for the analysis of histone PTMs that can be completed in 3 days using nLC coupled to tandem MS. Comparisons with techniques other than MS, i.e., using antibody based strategies as discussed in the Introduction, are not suitable, as they cannot achieve even nearly this level of throughput. In addition, antibody based techniques do not allow for the discovery of novel modifications, but they are exclusively based on confirming and quantifying predicted marks. We thus speculate that bottom-up proteomics on histone peptides will gain popularity in proteomics laboratories due to the intuitive advantages in knowing the regulation of histone marks, which are protagonists in tuning gene expression and thus affect the regulation of the proteome. Moreover, the protocol described includes recent improvements in the sample preparation and software for data analysis, which make histone analysis more trivial also for laboratories that never experienced characterization of this type of hypermodified peptides.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgements
This work was supported by funding from NIH grants (DP2OD007447, R01GM110174 and R01AI118891).
Materials
| Name | Company | Catalog Number | Comments |
| Trypsin 0.25% EDTA | Invitrogen | 25200056 | For harvesting cells |
| PBS | Invitrogen | 14200075 | |
| Tris | Roche | 77-86-1 | |
| Potassium Chloride | Fisher Scientific | BP366-500 | |
| Sodium Chloride | Sigma | S9888 | |
| Magnesium Chloride hexahydrate | Sigma | M9272 | |
| Calcium Chloride, anhydrous | Sigma | C1016 | |
| Sucrose | Fisher Scientific | BP220-1 | |
| DTT | Invitrogen | 15508-013 | |
| AEBSF | EMD Millipore Corp | 101500 | |
| Microcystin | Sigma | M4194 | |
| Sodium Butyrate | Sigma | B5887 | |
| Halt Protease and Phosphatase Inhibitor Cocktail, EDTA-free (100x) | Fisher Scientific | 78445 | |
| NP- 40 Alternative | CALBIOCHEM | 492016 | |
| Sulfuric Acid, ACS grade | Fisher Chemical | 7664-93-9 | |
| Trichloroacetic acid | Sigma | T6399 | |
| Acetone | Sigma | 179124 | |
| HCl | Fisher Chemical | A144-500 | |
| Bradford reagent | Biorad | 500-0006 | |
| 30% acrylamide/bis 29:1 — 500 ml | Biorad | 1610156 | |
| Coomassie | Fisher Scientific | 20278 | |
| C18 Column (5 µm) 2.1 mm x 250 mm | Grace | 218TP52 | |
| C18 Column (5 µm) 4.6 mm x 250 mm | Grace | 218TP54 | |
| HPLC grade acetonitrile | Fisher Chemical | A955-4 | |
| HPLC grade water | Fisher Scientific | W6 4 | |
| TFA | Fisher Scientific | A11650 | |
| Ammonium Bicarbonate | Sigma | A6141 | |
| ammonium hydroxide | Sigma | 338818 | |
| propionic anhydride | Sigma | 240311 | |
| Sequencing grade modified trypsin | Promega | PRV5113 | For digesting histones for MS |
| Acetic Acid | Sigma | 49199 | |
| C18 extraction disk | Empore | 2215 | |
| Formic Acid | Sigma | F0507 |
References
- Waddington, C. H. Canalization of development and the inheritance of acquired characters. Nature. 150, 563-565 (1942).
- Sharma, S., Kelly, T. K., Jones, P. A. Epigenetics in cancer. Carcinogenesis. 31 (1), 27-36 (2010).
- Reik, W., Dean, W., Walter, J. Epigenetic reprogramming in mammalian development. Science. 293 (5532), 1089-1093 (2001).
- Kouzarides, T. Chromatin modifications and their function. Cell. 128 (4), 693-705 (2007).
- Tessarz, P., Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Bio. 15 (11), 703-708 (2014).
- Fischle, W., Wang, Y. M., Allis, C. D. Histone and chromatin cross-talk. Curr Opin Cell Biol. 15 (2), 172-183 (2003).
- Lee, J. S., Smith, E., Shilatifard, A. The language of histone crosstalk. Cell. 142 (5), 682-685 (2010).
- Simboeck, E., et al. A Phosphorylation Switch Regulates the Transcriptional Activation of Cell Cycle Regulator p21 by Histone Deacetylase Inhibitors. J Biol Chem. 285 (52), 41062-41073 (2010).
- Hirota, T., Lipp, J. J., Toh, B. H., Peters, J. M. Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature. 438 (7071), 1176-1180 (2005).
- Xhemalce, B., Kouzarides, T. A chromodomain switch mediated by histone H3 Lys 4 acetylation regulates heterochromatin assembly. Genes Dev. 24 (7), 647-652 (2010).
- Vermeulen, M., et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. 142 (6), 967-980 (2010).
- van Attikum, H., Gasser, S. M. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol. 19 (5), 207-217 (2009).
- Fernandez-Capetillo, O., et al. H2AX is required for chromatin remodeling and inactivation of sex chromosomes in male mouse meiosis. Dev Cell. 4 (4), 497-508 (2003).
- Santaguida, S., Musacchio, A. The life and miracles of kinetochores. Embo J. 28 (17), 2511-2531 (2009).
- Egelhofer, T. A., et al. An assessment of histone-modification antibody quality. Nat Struct Mol Biol. 18 (1), 91-93 (2011).
- Sidoli, S., Cheng, L., Jensen, O. N. Proteomics in chromatin biology and epigenetics: Elucidation of post-translational modifications of histone proteins by mass spectrometry. J Proteomics. 75 (12), 3419-3433 (2012).
- Plazas-Mayorca, M. D., et al. One-Pot Shotgun Quantitative Mass Spectrometry Characterization of Histones. J Proteome Res. 8 (11), 5367-5374 (2009).
- Sidoli, S., et al. Drawbacks in the use of unconventional hydrophobic anhydrides for histone derivatization in bottom-up proteomics PTM analysis. Proteomics. 15 (9), 1459-1469 (2015).
- Lin, S., Garcia, B. A. Examining histone posttranslational modification patterns by high-resolution mass spectrometry. Methods Enzymol. 512, 3-28 (2012).
- Sidoli, S., et al. SWATH Analysis for Characterization and Quantification of Histone Post-translational Modifications. Mol Cell Proteomics. , (2015).
- Krautkramer, K. A., Reiter, L., Denu, J. M., Dowell, J. A. Quantification of SAHA-Dependent Changes in Histone Modifications Using Data-Independent Acquisition Mass Spectrometry. J Proteome Res. , (2015).
- Yuan, Z. F., et al. EpiProfile Quantifies Histone Peptides With Modifications by Extracting Retention Time and Intensity in High-resolution Mass Spectra. Mol Cell Proteomics. 14 (6), 1696-1707 (2015).
- MacLean, B., et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 26 (7), 966-968 (2010).
- Yuan, Z. F., Lin, S., Molden, R. C., Garcia, B. A. Evaluation of proteomic search engines for the analysis of histone modifications. J Proteome Res. 13 (10), 4470-4478 (2014).
- Tan, Y., Xue, Y., Song, C., Grunstein, M. Acetylated histone H3K56 interacts with Oct4 to promote mouse embryonic stem cell pluripotency. Proc Natl Acad Sci U S A. 110 (28), 11493-11498 (2013).
- Vonholt, C., et al. Isolation and Characterization of Histones. Methods Enzymol. 170, 431-523 (1989).
- Zhang, K. L., et al. Identification of acetylation and methylation sites of histone H3 from chicken erythrocytes by high-accuracy matrix-assisted laser desorption ionization-time-of-flight, matrix-assisted laser desorption ionization-postsource decay, and nanoelectrospray ionization tandem mass spectrometry. Anal. Biochem. 306 (2), 259-269 (2002).
- Jufvas, A., Stralfors, P., Vener, A. V. Histone Variants and Their Post-Translational Modifications in Primary Human Fat Cells. Plos One. 6 (1), e15960 (2011).
- Bonaldi, T., Imhof, A., Regula, J. T. A combination of different mass spectroscopic techniques for the analysis of dynamic changes of histone modifications. Proteomics. 4 (5), 1382-1396 (2004).
- Zhao, X. L., et al. Comparative Proteomic Analysis of Histone Post-translational Modifications upon Ischemia/Reperfusion-Induced Retinal Injury. J Proteome Res. 13 (4), 2175-2186 (2014).
- Lin, S., et al. Stable-isotope-labeled histone peptide library for histone post-translational modification and variant quantification by mass spectrometry. Mol Cell Proteomics. 13 (9), 2450-2466 (2014).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved