
Method Article
Single-molecule Super-resolution Imaging of Phosphatidylinositol 4,5-bisphosphate in the Plasma Membrane with Novel Fluorescent Probes
In This Article
Summary
PI(4,5)P2 regulates various cellular functions, but its nanoscale organization in the cell plasma membrane is poorly understood. By labeling PI(4,5)P2 with a dual-color fluorescent probe fused to the Pleckstrin Homology domain, we describe a novel approach to study the PI(4,5)P2 spatial distribution in the plasma membrane at the nanometer scale.
Abstract
Phosphoinositides in the cell membrane are signaling lipids with multiple cellular functions. Phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) is a determinant phosphoinositide of the plasma membrane (PM), and it is required to modulate ion channels, actin dynamics, exocytosis, endocytosis, intracellular signaling, and many other cellular processes. However, the spatial organization of PI(4,5)P2 in the PM is controversial, and its nanoscale distribution is poorly understood due to the technical limitations of research approaches. Here by utilizing single molecule localization microscopy and the Pleckstrin Homology (PH) domain based dual color fluorescent probes, we describe a novel method to visualize the nanoscale distribution of PI(4,5)P2 in the PM in fixed membrane sheets as well as live cells.
Introduction
Phosphoinositides contribute to a small portion of total membrane lipids, but play critical roles in a variety of cellular processes. They include seven members derived from reversible phosphorylation or dephosphorylation of the inositol rings on the 3rd, 4th and 5th positions1. Phosphatidylinositol 4-phosphate (PI4P) and phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) are two major phosphoinositides that function relatively independently as the determinant lipids of cell plasma membrane (PM) 2,3. Phosphatidylinositol (3,4,5)-trisphosphate (PIP3) is much less abundant than PI4P and PI(4,5)P2, but it has unique functions in different cellular processes including cancer4 and diabetes5. These lipids have complex molecular interactions with their effectors and many other proteins. Therefore, it is crucial to understand the spatial organization of these phosphoinositides in the PM at the nanometer scale.
Mounting evidence has shown that protein complexes or molecule clusters in the confined PM areas can serve as signaling hotspots6. For example, syntaxin 1a, a key protein that regulates membrane fusion7-9, displays cluster organization in the PM. Distinct from the consensus view of the cluster organization of syntaxin 1a, the spatial distribution of phosphoinositides in the PM is controversial. PI(4,5)P2 distribution patterns range from uniform10-12, large patches13,14, to dense clusters14-18, depending on cell types and experimental methods used. The spatial organization of PI(4,5)P2 at higher resolution is also inconsistent. A study using stimulated-emission-depletion (STED) microscopy 19 has revealed a large number of dense PI(4,5)P2 nano-clusters (~73 nm in diameter) in the PM sheets of PC-12 cells 20. This result is different from studies using quick freezing electron microscopy (EM)21,22, an approach that preserves the intact PM structure of live cells much better than chemical fixation. The latter showed distinct pools of PI(4,5)P2; relatively concentrated PI(4,5)P2 in caveolae and coated pits as well as uniform distribution on the flat PM region. Moreover, nanoscale PI(4,5)P2 organization in membrane sheets may differ in live and fixed cells. Our recent work investigated this issue in both fixed and live INS-1 cells using single-molecule localization microscopy (SMLM)23.
SMLM is based on stochastically switching on only a small subset of fluorophores at any given time so that individual fluorophores can be localized with high precision. Many super-resolution imaging approaches have been developed using similar principles to surpass the diffraction limit of conventional light microscopy, such as photoactivation localization microscopy (PALM)24, fluorescence photoactivation localization microscopy (FPALM)25, stochastic optical reconstruction microscopy (STORM)26,27 and direct STORM (dSTORM)28. With photo-switchable or photoactivatable fluorophores (dyes or fluorescent proteins), SMLM techniques allow scientists to image biological structures at nanometer resolution24,29,30 with video-rate in living cells31,32.
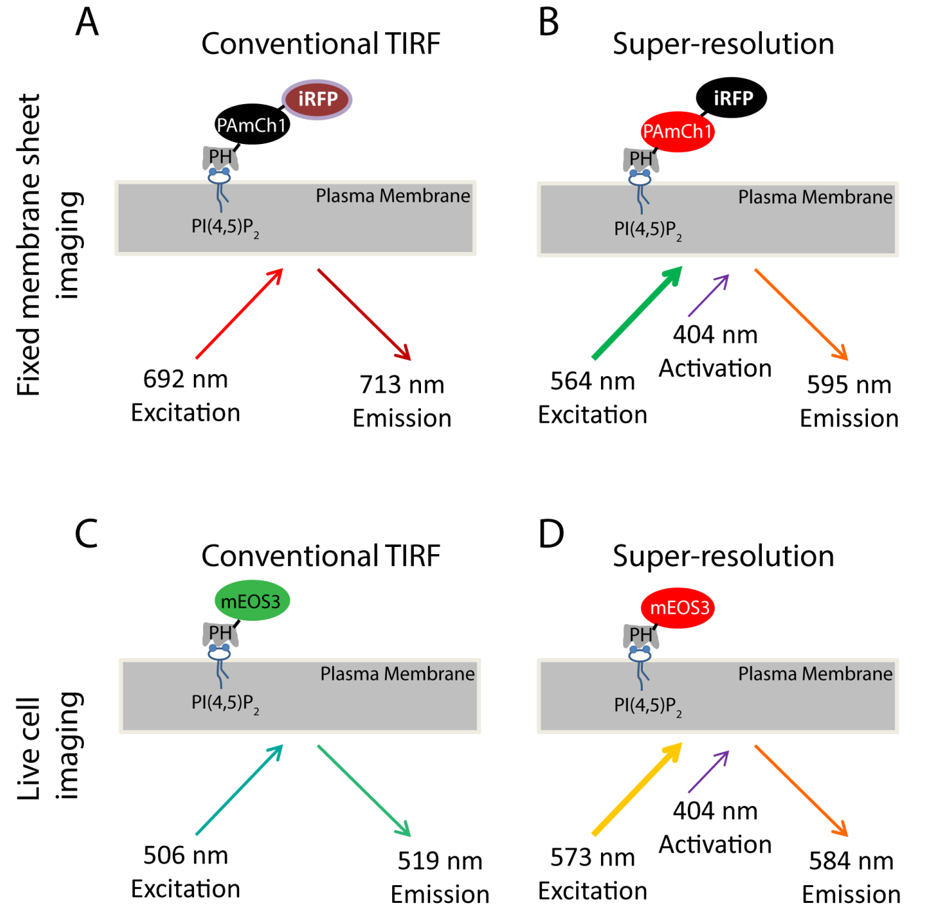
Using PI(4,5)P2 as an example, we introduced the SMLM approach to study the nanoscale distribution of phosphoinositides on the PM. The PH domain of PLCδ1 (Phospholipase C δ1) that specifically binds to PI(4,5)P2 is a well-established probe for imaging PI(4,5)P2 sub-cellular distribution and dynamics33,34 in the PM. We have genetically tagged this domain with two fluorescent proteins, PAmCherry135 and iRFP36 to produce a dual-color fusion protein (iRFP-PAmCherry1-PHPLCδ1) (Figure 1A-B). PAmCherry1 serves as the photoactivatable probe of PH domain for PALM and iRFP serves as a general indicator to identify transfected cells before PALM acquisition. We apply this dual-color fluorescent probe for SMLM imaging in the fixed membrane sheets. For live PALM imaging, we tagged mEOS337 instead of PAmCherry1 to the PHPLCδ1 domain to generate the mEOS3.1-PHPLCδ1 probe for its better photon efficiency and brightness (Figure 1C-D).
SMLM imaging with these novel probes in the PM of insulin-secreting INS-1 cells38 has uncovered homogeneous labeling of PI(4,5)P2 in the majority of the PM regions as well as concentrated PI(4,5)P2 microdomains that are sparsely intermixed in the flat PM and some filopodia-like structures23. The nanoscale distribution of PI(4,5)P2 provides a structural base for rethinking how it functions in living cells.
Protocol
1. Membrane Sheet Preparation and Fixation
- Prepare membrane sheets labeled with iRFP-PAmCherry1-PHPLCδ1
- Prepare the DNA plasmid encoding iRFP-PAmCherry1-PHPLCδ123 using standard molecular biology techniques.
- Culture INS-1 cells on #1.5 18 mm round coverslips pre-coated with 30 µg/ml fibronectin to 50-70% confluency following standard INS-1 cell culture protocols38,39. Transfect the cells a day after 50 to 70% confluency is reached.
- Transfect cells with iRFP-PAmCherry1-PHPLCδ1 using liposome transfection reagent following manufacturer's protocols. After transfection, allow the cells to grow for 48 hr.
- On the day of the experiment, coat coverslips (on one-side) with ~0.5 - 1 ml of 500 µg/ml Poly-D-Lysine (PDL; diluted in dH2O) for 1-2 hr. Then drain PDL by placing a tissue paper on the edge of the coverslip. Place the coverslips on a pre-chilled (4 °C) metal plate for later use.
- Wash pre-transfected INS-1 cells growing on coverslips with ~0.5 - 1 ml ice-cold phosphate-buffered saline (PBS) containing 1 mM EGTA. Repeat the wash three times and drain PBS.
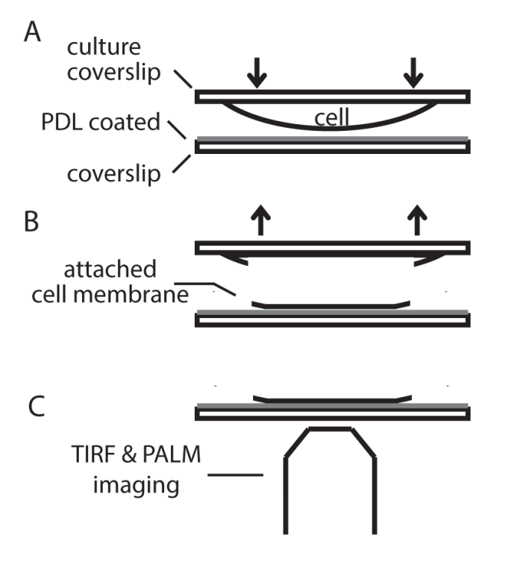
- Place the coverslips with cultured cells (cell-side facing down) on the PDL-coated coverslips on a pre-chilled metal plate with a pair of tweezers. Leave the plate in a refrigerator (4 °C) for 7 - 10 min to allow cells to attach to the PDL-coated coverslips (Figure 2).
NOTE: The incubation time should be in the range of 7 - 10 min for better attachment of the cells to the PDL-coated surface. The refrigerator environment is dry and incubation should not be too long. - Remove the metal plate from the refrigerator. Gently peel off the coverslip containing the pre-transfected cells using tweezers. This produces a thin layer of cell membrane sheet on the PDL-coated coverslip. Gently wash the membrane sheets with ~0.5 - 1 ml ice-cold PBS and then fix with ~0.5 - 1 ml ice-cold 4% paraformaldehyde (PFA) + 0.2% glutaraldehyde (GA) in PBS for 15 min at 4 °C.
Caution: Paraformaldehyde and glutaraldehyde are toxic. Handle them in a fume hood with skin and eye protection. - After fixation, image the membrane sheets immediately (see section 3) or store in ~0.5 - 1 ml PBS at 4 °C.
NOTE: After fixing the membrane sheet, there will be no equilibrium binding of PH probes from cytosol to PI(4,5)P2. The bound PH probes will gradually dissociate from the membrane sheet and diffuse into the solution (although it might take days to weeks). Therefore, it is recommended to image the fixed samples right after sample preparation.
- Prepare membrane sheets for PI(4,5)P2 specific antibody labeling
- Culture INS-1 cells on #1.5, 18 mm round coverslips pre-coated with 30 µg/ml fibronectin to 50%-70% confluence. Transfect cells with DNA plasmid the next day as described in step 1.1.3.
- Repeat steps from 1.1.4 to 1.1.7 as described above.
NOTE: Perform the following procedures at 4 °C. - Wash the coverslips containing fixed PM sheets three times with ~0.5 - 1 ml ice-cold PBS containing 50 mM NH4Cl. Quench the sheets with ~0.5 - 1 ml 0.1% sodium borohydride in PBS for 7 min, and wash with ~0.5 - 1 ml PBS (without 50 mM NH4Cl).
- Block the samples with ~0.5 - 1 ml blocking solution (PBS solution containing 5% (v/v) normal goat serum, 5% (v/v) bovine serum albumin and 50 mM NH4Cl) for 45 min. Then, incubate with ~0.5 - 1 ml primary PI(4,5)P2 antibody (1:300 dilution) in blocking solution for 1 hr. Wash three times (10 min each time) with ~0.5 - 1 ml PBS containing 50 mM NH4Cl.
- Incubate samples with ~0.5 - 1 ml secondary F(ab')2-goat-anti-mouse antibody conjugated with fluorophores (1:300) in blocking buffer for 1 hr. Wash three times with ~0.5 - 1 ml PBS.
- Post-fix samples with 4% PFA + 0.2% GA for 15 min, then wash samples three times with PBS (7 min each time). Proceed to Section 3 for imaging or store in ~0.5 ~ 1 ml PBS at 4 °C for later use.
NOTE: Prepare samples at 4 °C. Use 4% PFA + 0.2% GA fixative to preserve the physiological distribution of phosphoinositides in the PM. RT preparation or fixation with 4% PFA alone may cause significant artifacts (Figure 4).
2. Cell Culture Preparation for Live-cell Imaging with mEOS3.1-PHPLCδ1
- Prepare the DNA plasmid encoding the mEOS3.1-PHPLCδ1 as described previously 23.
- Culture INS-1 cells on #1.5 18 mm round coverslips pre-coated with 30 µg/ml fibronectin until they reach 50%-70% confluency following standard INS-1 cell culture protocols38,39.
- The following day, transfect INS-1 cells with the mEOS3.1-PHPLCδ1 DNA plasmid using liposome transfection reagent following manufacture's protocol. Incubate for 48 hr before imaging.
3. PALM Image Acquisition of Membrane Sheets and Live Cells
- PALM image acquisition of membrane sheets
NOTE: Here, a total internal reflection fluorescence (TIRF) microscope based SMLM system (100X oil, APO, NA = 1.49, WD 0.12 mm) 23 is used for all image acquisition.- Before imaging, dilute 1 µl of fluorescent bead solution in 10 ml PBS + 50 mM MgCl2. Add 200 µl diluted solution into the imaging chamber containing cell samples (from steps 1.1.8/1.2.6) for 10 min. Then, wash with PBS three times.
- Start the PALM imaging system. In the associated imaging software, select the "iRFP channel" button (642 nm laser excitation, 700/75 nm emission). Find the cell membrane expressing PH probes in the iRFP channel.
NOTE: The membrane sheet should have a fluorescent bead nearby. This is required for drift correction later. - Use the "Capture" button to collect a conventional TIRF image of the cell membrane in iRFP channel as a reference for future image reconstruction. Set a normal TIRF angle (i.e., after reaching the critical angle, turn another 1.5 degrees) by typing in 2140 in the TIRF angle setting for imaging. Use this angle setting for all steps unless otherwise noted.
NOTE: In the TIRF system described here, 3,500 is the vertical up angle, 2,200 is the TIRF critical angle, and 2140 is the normal TIRF angle, which is 1.5 degree more after TIRF critical angle. The narrow TIRF angle mentioned below in 3.2.3 is 2120, which is another 2 degree after reaching TIRF critical angle. - Switch to PAmCherry1 channel by clicking the RFP channel button (561 nm excitation, 600/50 nm emission filter). Adjust the scroll bar in the "AOTF" pad all the way to the right to have full power illumination of 561 nm laser for 10-20 sec to bleach the background membrane fluorescence.
- Set the optimal camera setting (2x2 binning) in the "Format" tab and the fast acquisition protocol in "ND sequence acquisition" tab (typically 10,000-40,000 images at 20 Hz).
- Start acquisition of PAmCherry1 images (561 nm laser at full power, 50 msec/frame) with simultaneous 405 nm laser activation at a low level (0.1% - 1%) by clicking the "Run now" button. Adjust the intensity of 405 nm laser so that spatially isolated individual points in each frame are easily identifiable.
NOTE: The number of activatable PAmCherry1 molecules decreases gradually during acquisition. 405 nm laser intensity should be gradually increased to keep the optimal density of individual molecule signals in each frame. - Continue acquiring images until no PAmCherry1 single molecule signal is activated.
- PALM imaging in live cells
- Before imaging, dilute fluorescent beads into the imaging chamber for 10 min as described in step 3.1.1. Then start PALM imaging by opening the associated imaging software.
NOTE: Use a magnetic quick-release imaging chamber for live-cell imaging. Maintain the center of the imaging field at 35 °C with a temperature controller under constant perfusion with extracellular buffer (Extracellular Solution: 135 mM NaCl, 5.6 mM KCl, 2.6 mM CaCl2, 1.2 mM MgCl2, 3 mM glucose, and 20 mM HEPES; pH =7.3). - Choose the GFP channel button (525/50 nm emission). Identify the cell membrane expressing fluorescent probes based on green fluorescent from mEOS3 under 488 nm laser excitation. Make sure that fluorescent beads nearby are also included during imaging as this is required later for offline drift correction.
- Click the "Capture" button to collect a TIRF image of the cell membrane in mEOS3 (green) channel as a reference image. Use a shallow evanescent wave excitation illumination (after reaching the critical angle, turn another 2 degrees) by typing in 2120 in the TIRF angle setting for live-cell imaging.
NOTE: This minimizes the fluorescence from the unbound mEos3-PH in the cytosol adjacent to the plasma membrane. - Switch to the "RFP channel" button (561 nm excitation, 600/50 nm emission). Use the full power 561 nm laser to bleach background fluorescence of the membrane sheets for 10 ~ 20 sec with the scrollbar in the "AOTF pad" (see 3.1.4).
- Start image acquisition by clicking the "Run Now" button in the red channel (561 nm full power, set at 10 msec/frame in the "camera" setting tab) with simultaneous 405 nm laser activation. Adjust the 405 nm laser intensity to activate spatially separated points in each frame.
NOTE: As acquisition proceeds, un-converted mEOS3.1-PHPLCδ1 molecule number slowly decreases. Gradually increase the 405 nm laser power to optimize the single molecule signal. The imaging acquisition length depends on the experimental purpose. Acquire images continuously for 5 min under normal conditions. If a longer acquisition is required, collect multiple image stacks instead of a single, large image stack. The file size of each image stack should not exceed 4 GB or it will affect software analysis later.
- Before imaging, dilute fluorescent beads into the imaging chamber for 10 min as described in step 3.1.1. Then start PALM imaging by opening the associated imaging software.
4. SMLM Image Processing and Reconstruction
- Transfer the image files (.tif image stacks) to an image analysis station. Launch the image reconstruction program (custom-written) in Matlab as described previously37 and load the image stack by clicking the "File" tab in the main menu, then "open", then "new file".
- Identify and localize individual molecular events from each frame. Set filtering intensity threshold with a threshold number (1-10) in the "wavelet". Verify the optimal parameter settings for point detection visually before the reconstruction. Then go to the "PALM" tab and click "one step process" to start image reconstruction.
NOTE: Intensity threshold for point detection is arbitrary and depends on personal experience. Several trials may be required to generate an optimal detection threshold that neither picks up too many unqualified (dim) points nor filters out too many qualified (bright) points. Adjust other parameters to optimize data analysis. Here, neighborhood distance (fluorescence points in the neighboring frames within a distance are combined) is set as 65 nm (1/2 pixel width) and fitted as a single molecule. Gap frames (individual molecule events that occurred within these frames and neighborhood distance were combined and fitted as a single molecule event) is set as 26 frames (1.3 sec) for PAmCherry1 imaging40. Adjust these parameter according to the properties of single molecule probes and acquisition rate to avoid over-counting40 in SMLM. Apply drift correction with fluorescent beads during the reconstructions.- For imaging the fixed membrane sheets labeled by iRFP-PAmCherry1-PHPLCδ1, reconstruct the whole image stacks from the same membrane sheet into a single super-resolution image37.
- For live cell samples labeled by mEOS3.1-PHPLCδ1, separate the whole image stack into several smaller stacks of 1,000 frames so that 1,000-consecutive-frame stack is reconstructed as a single super-resolution image, which has a temporal resolution of 10 sec. In the end, combine all the time series images into the final time series stack23.
NOTE: In live-cell PALM imaging, a small number of activated probes may move or dissociate from PI(4,5)P2 in the PM before bleaching. To account for the oversampling of the probes, combine individual molecule signals in the neighboring frames within 130 nm (instead of 65 nm) into a single emission event during image analysis.
- After obtaining the reconstructed images, carry out further image analysis with the custom-written program in Matlab to quantify molecule density, micro-domain density/size, and cluster analysis with pair-correlation41.
Results
The localization uncertainty (σ) of our super-resolution system is 14.73 nm23. Direct comparisons between TIRF and PALM images demonstrated a significant improvement of spatial resolution. Figure 3A-B shows the representative PI(4,5)P2 TIRF images labeled with PI(4,5)P2 antibody and iRFP-PAmCherry1-PHPLCδ1 in typical membrane sheets and live cells. The images with conventional TIRF microscopy in membrane sheets are remarkably similar to those in intact live cells labeled with the enhanced green fluorescence protein (EGFP) tagged PH domains (EGFP-PHPLCδ1) (Figure 3C). All samples showed an even distribution of probes. In contrast, non-optimal fixation of the samples resulted in sharp dense PI(4,5)P2 clusters and a decrease in signal intensity (Figure 4). Under optimal fixation conditions, the super-resolution images of PI(4,5)P2 in fixed cells (Figure 5) revealed a homogenous distribution of probes in a significant portion of the PM with only limited concentration gradients. Some membrane patches enriched with PI(4,5)P2 probes were sparsely distributed and had various sizes. Live cell PALM images display a similar spatial distribution as fixed cells (Figure 6). Detailed analysis of PI(4,5)P2 signals over time results in fast dynamics in local areas, without significant changes of their abundance in broad areas.

Figure 1. Scheme for fluorescent probes used in this study. (A-B) iRFP-PAmCherry1-PHPLCδ1 probe used in the fixed membrane sheet experiments. During conventional TIRF imaging (A), a 640 nm laser is used to excite the iRFP (Ex: 692 nm; Em: 713 nm). TIRF image taken in this condition serves as a TIRF reference image for the super-resolution imaging obtained by PALM imaging (B). A 405 nm laser is used to photo-activate the PAmCherry1 fluorophore and a 561 nm laser is used to excite PAmCherry1 (Ex: 564 nm; Em: 595 nm) for PALM imaging. (C-D) mEos3.1-PHPLCδ1 probe used in the live cell experiments. (C) During conventional TIRF imaging, a 488 nm laser is used to excite mEOS3.1 (Ex: 506 nm; Em: 519 nm). (D) After photoconversion by a 405 nm laser, mEos3.1 turns into the red form (Ex: 573 nm; Em: 584 nm) and a 561 nm laser is used for PALM acquisitions. Please click here to view a larger version of this figure.
{kind=link}

Figure 2. Scheme for membrane sheet preparation from INS-1 cells. (A) Place the coverslip with cultured cells facing down onto a PDL-coated coverslip and wait for 7 ~ 10 min at 4 °C to allow cell attachment to the PDL-coated coverslip. (B) Peel off the top coverslip with tweezers and fix the membrane sheet attached to the PDL pre-coated coverslip. (C) Image the samples with TIRFM and PALM. Please click here to view a larger version of this figure.
{kind=link}

Figure 3. PI(4,5)P2 spatial organization is similar between membrane sheets and intact live cells under conventional TIRF microscope. (A) A typical TIRF image of membrane sheets fixed at 4 °C with 4% PFA and 0.2% GA. PI(4,5)P2 was labeled with PI(4,5)P2 specific antibody. (B) A membrane sheet from the INS-1 cell expressing iRFP-PAmCherry1-PHPLCδ1. (C) TIRF image of two intact live cells expressing EGFP-PHPLCδ1 at different levels. Scale bars: (A): 3 µm; (B) and (C): 5 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 4. PI(4,5)P2 spatial organization in membrane sheets is sensitive to common fixation conditions. (A) Membrane sheets fixed at 37 °C with 4% PFA alone and labeled with PI(4,5)P2 specific antibody (as in Figure 3A). (B) Membrane sheet fixed at RT with PFA alone and labeled with PI(4,5)P2 specific antibody. Note that the dense clusters of PI(4,5)P2 probes are clearly visible under TIRF microscope, in contrast to the much more even fluorescence images shown in Figure 3A. Scale bars: A-B: 3 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 5. PALM imaging of PI(4,5)P2 probes reveals their nanometer scale distribution in an INS-1 cell PM. (A) iRFP TIRF image of a membrane sheet from an INS-1 cell expressing iRFP-PAmCherry1-PHPLCδ1. (B) Corresponding PALM image of PM in the same region based on PAmCherry1 signal reconstruction. Note the homogeneous PI(4,5)P2 spatial distribution in major PM regions and several PI(4,5)P2 microdomains. (C) An enlarged view of the boxed region in (B). Arrows indicate sparsely distributed PM microdomains enriched with PI(4,5)P2 probes. Scale bars: A and B: 3 µm; C: 500 nm. Please click here to view a larger version of this figure.
{kind=link}

Figure 6. PALM imaging in live INS-1 cells. (A) TIRF image of PI(4,5)P2 in a live INS-1 cell expressing mEos3.1-PHPLCδ1. The image was rapidly acquired in the green channel before PALM acquisition (35 °C). (B) Sequential live-cell PALM images at 10 sec interval. Insets show the intensity profiles of the local PI(4,5)P2 density along the same straight line 1 position at different times in (B) and (C). Note their large local intensity changes within 10 sec. (D) Time course of the average intensity changes of PI(4,5)P2 in the large area (box2, 3x3 µm) and small circles (3, 4, and 5, 500 nm diameter) in (B) during 5 min of PALM imaging (frame/10 sec). Note the rapid intensity fluctuations of local PI(4,5)P2 probes (circle 3, 4 and 5) compared to very small changes in the broad area (box 2). (E) Enlarged PALM images of the box2 region in (B) at indicated times. Arrows indicate PI(4,5)P2 enriched membrane patches under physiological conditions. Scale bars: C: 3 µm; E: 500 nm. Please click here to view a larger version of this figure.
{kind=link}
Discussion
For troubleshooting, two processes need extra attention: membrane sheet production and sample fixation. As described in the protocol, the incubation time of the coverslips in step 1.1.6 is important for membrane sheet production. Optimal incubation time under our experimental condition is 7-10 min (Figure 2). Longer than 10 min incubation will produce intact cells instead of the membrane sheets on PDL coverslips and shorter incubation will lead to less or no membrane sheets on the PDL-coated coverslips. As described in the protocol, the fixatives and temperature during fixation are critical for maintaining the PI(4,5)P2 distribution in the PM. Fixation at RT or use of 4%-PFA alone could distort the normal lipid distribution in the PM.
By applying PALM microscopy to membrane lipid research, we are able to observe the nanometer scale distribution of PI(4,5)P2, a key phosphoinositide that mediates many fundamental cellular activities. This spatial distribution of PI(4,5)P2 with limited concentration gradients in INS-1 cells provides a framework for rethinking lipid-protein interactions and local signaling events of PI(4,5)P2 in these cells. Moreover, the methods developed in this work can also be applied to other membrane phospholipid research with proper probes, thereby, offering novel tools to study phosphoinositides in biological processes.
The use of membrane sheets in this work bypasses two major concerns in phospholipid morphological studies: detergent treatment and potential signal contamination from the cytosol. Detergents often cause clustering and significant loss of PM phospholipid signal. The contamination of cytosolic signal is particularly problematic in the case of low abundance phosphoinositides on the PM23, such as PI(3,4,5)P3 and PI(3,4)P2. Membrane sheet samples are able to circumvent these problems without significantly disrupting structures associated with the plasma membrane, such as cortical actin meshwork and clathrin-coated pits42,43. The relative homogenous distribution of PI(4,5)P2 in the PM is in good agreement with other quick freezing EM studies using GST-PHPLCδ1 probes in the fibroblast membrane44.
It is important to note that improper sample processing conditions can generate misleading results. First, it is critical to perform the fixation steps at a lower temperature (4 °C) and use the fixative GA for membrane sheet production. As shown in Figure 3, warm temperature and PFA fixation without GA is not sufficient to fix phosphoinositides in cell PM. This could distort the intact PI(4,5)P2 distribution and generate sharp clusters that are not observed in live cells under physiological conditions. Second, the use of PAmCherry1 as the SMLM probe, rather than other probes, is pivotal for quantitative PALM imaging. The benefit of PAmCherry1 application comes from its well-characterized single molecular photo-physical properties35,40,45, such as brightness, high photo-activation efficiency and most of all, very limited photo-blinking. These properties enable us to eliminate potential cluster artifacts from photo-blinking and quantitatively analyze the molecular density of membrane PI(4,5)P2.
This approach also has its limitations. First, the membrane sheet method used in this study may not fully mimic the physiological distribution of PI(4,5)P2 because the cell is disrupted before imaging. However, our live PALM imaging shows similar relatively homogeneous distribution of PI(4,5)P2, supporting the results observed with membrane sheet samples. Second, as we discussed in our previous work23, SMLM requires imaging expertise and extra attention to avoid imaging artifacts that might arise from different processes, including the probes used, sample preparation and fixation, image sampling and reconstruction. Lastly, though the PH domain based probes and antibodies have been widely used in phosphoinositide studies46,47 it remains possible that not all PI(4,5)P2 in the membrane can be detected by this approach. For example, PI(4,5)P2 bound by other endogenous proteins may not be accessible to PH probes or antibodies, and this may cause an underestimation of PI(4,5)P2 due to the space hindrance of probe themselves. An alternative way of labeling PI(4,5)P2 would be using Top-Fluor PI(4,5)P248, a pre-labeled PI(4,5)P2 analog with a modification on the tail of original PI(4,5)P2. However, it can be converted rapidly into other phosphoinositide subtypes by fast live cell metabolism since its inositol ring is the same as endogenous PI(4,5)P2. This raises the concern whether this pre-labeled PI(4,5)P2 analog in live cells can faithfully represent PI(4,5)P2 rather than its metabolic products. Therefore, despite some limitations, PH domain based probes are still among the best probes that have been widely used to monitor PI(4,5)P2 distribution and dynamics on the PM of cells.
The future application of this methodology can be extended to other phosphoinositide studies, such as PI(3,4,5)P3 and PI(3,4)P2. In summary, the novel SMLM approach used here opens new ways to study phosphoinositide in cells. Using PI(4,5)P2 as an example, we demonstrate the unique properties of PALM imaging in the morphological and quantitative study of cell membrane molecules, as well as its drawbacks. This approach can be adapted to other molecules of interests and will have wide applications in cell biology.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work is supported by the National Institutes of Health (NIH) grants R01DK093953 (X.L.), P30NS069271, BRFSG-2014-07 (X.L.). C.J. is partially supported by American Heart Associate pre-doctoral fellowship (14PRE20380168, to C.J.).
Materials
| Name | Company | Catalog Number | Comments |
| Microscope | Nikon | Ti-U | |
| sCMOS camera | Andor | Neo | |
| Spinning disk | Yokogawa | CSU X-1, 10,000 rpm | |
| High Power Monolithic Laser Combiner SP with 405, 488, 561 and 642 nm lasers. | Agilent | MLC400 | laser original powers & powers at the end of optical fiber) 405 nm: 15 mW & 13 mW; 488 nm: 45 mW & 42 mW; 561 nm: 45 mW & 40 mW; 640 nm: 35 mW & 16 mW. |
| 100X Objective | Nikon | APO 100X Oil, NA 1.49, WD 0.12 mm | |
| Nikon acquisition and analysis software (NIS Element) | Nikon | ||
| Matlab | MathWorks | ||
| Coverslip | Warner | 64-0714 | Round 18 mm coverslip, #1.5 |
| Fibronectin | Millipore | FC010-5MG | |
| Lipofectamin 3000 (transfection reagent) | Invitrogen | L3000-008 | |
| Glutaraldehyde | Electron Microscopy Science | #16120 | |
| PI(4,5)P2 1st antibody | Santa Cruz | sc-53412 | |
| secondary antibody F(ab’)2-goat-anti-mouse Alexa Fluor 647 | Invitrogen | A-21237 | |
| Tetraspeck beads | Invitrogen | T7279 | |
| magnetic quick release imaging chamber | Warner Instruments | Cat#641994 | |
| temperature controller | Warner Instruments | TC-344C | |
| Poly-D-Lysine | Sigma | P6407 | |
| Phosphate buffer saline | Sigma | P4417-100TAB | |
| EGTA | Sigma | 3780 | |
| NH4Cl | Sigma | A0171 | |
| Goat serum | Sigma | G9023 | |
| bovine serum albumin | Sigma | A7906 | |
| NaCl | Sigma | S5886 | |
| KCl | Sigma | P5405 | |
| CaCl2•2H2O | Sigma | 223506 | |
| MgCl2•6H2O | Sigma | M9272 | |
| Glucose | Sigma | G7021 | |
| HEPES | Sigma | H4034 | |
| Paraformaldyhyde | Sigma | P6148 |
References
- Di Paolo, G., De Camilli, P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 443, 651-657 (2006).
- Hammond, G. R. V., et al. PI4P And PI(4,5)P(2) Are Essential But Independent Lipid Determinants Of Membrane Identity. Science. 337, 727-730 (2012).
- Nakatsu, F., et al. PtdIns4P synthesis by PI4KIIIalpha at the plasma membrane and its impact on plasma membrane identity. J Cell Biol. 199, 1003-1016 (2012).
- Cantley, L. C. The phosphoinositide 3-kinase pathway. Science. 296, 1655-1657 (2002).
- Czech, M. P. Dynamics of phosphoinositides in membrane retrieval and insertion. Annu Rev Physiol. 65, 791-815 (2003).
- Spira, F., et al. Patchwork organization of the yeast plasma membrane into numerous coexisting domains. Nat Cell Biol. 14, 640-648 (2012).
- Barg, S., Knowles, M. K., Chen, X., Midorikawa, M., Almers, W. Syntaxin clusters assemble reversibly at sites of secretory granules in live cells. Proc Natl Acad Sci U S A. 107, 20804-20809 (2010).
- Sieber, J. J., et al. Anatomy and dynamics of a supramolecular membrane protein cluster. Science. 317, 1072-1076 (2007).
- Knowles, M. K., et al. Single secretory granules of live cells recruit syntaxin-1 and synaptosomal associated protein 25 (SNAP-25) in large copy numbers. Proc Natl Acad Sci U S A. 107, 20810-20815 (2010).
- Milosevic, I., et al. Plasmalemmal phosphatidylinositol-4,5-bisphosphate level regulates the releasable vesicle pool size in chromaffin cells. J Neurosci. 25, 2557-2565 (2005).
- van Rheenen, J., Jalink, K. Agonist-induced PIP(2) hydrolysis inhibits cortical actin dynamics: regulation at a global but not at a micrometer scale. Mol Biol Cell. 13 (2), 3257-3267 (2002).
- Hammond, G., Schiavo, G., Irvine, R. Immunocytochemical techniques reveal multiple, distinct cellular pools of PtdIns4P and PtdIns (4, 5) P2. Biochem J. 422, 23-35 (2009).
- Huang, S., et al. Phosphatidylinositol-4,5-bisphosphate-rich plasma membrane patches organize active zones of endocytosis and ruffling in cultured adipocytes. Mol Cell Biol. 24, 9102-9123 (2004).
- James, D. J., Khodthong, C., Kowalchyk, J. A., Martin, T. F. Phosphatidylinositol 4,5-bisphosphate regulates SNARE-dependent membrane fusion. J Cell Biol. 182, 355-366 (2008).
- Laux, T., et al. GAP43, MARCKS, CAP23 modulate PI(4,5)P(2) at plasmalemmal rafts, and regulate cell cortex actin dynamics through a common mechanism. J Cell Biol. 149, 1455-1472 (2000).
- Aoyagi, K., et al. The activation of exocytotic sites by the formation of phosphatidylinositol 4,5-bisphosphate microdomains at syntaxin clusters. J Biol Chem. 280, 17346-17352 (2005).
- Kabachinski, G., Yamaga, M., Kielar-Grevstad, D. M., Bruinsma, S., Martin, T. F. CAPS and Munc13 utilize distinct PIP2-linked mechanisms to promote vesicle exocytosis. Mol Biol Cell. 25, 508-521 (2014).
- Wang, J., Richards, D. A. Segregation of PIP2 and PIP3 into distinct nanoscale regions within the plasma membrane. Biol Open. 1, 857-862 (2012).
- Hell, S. W., Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt Lett. 19, 780-782 (1994).
- van den Bogaart, G., et al. Membrane protein sequestering by ionic protein-lipid interactions. Nature. 479, 552-555 (2011).
- van Rheenen, J., Achame, E. M., Janssen, H., Calafat, J., Jalink, K. PIP2 signaling in lipid domains: a critical re-evaluation. Embo J. 24, 1664-1673 (2005).
- Sato, K., et al. Differential requirements for clathrin in receptor-mediated endocytosis and maintenance of synaptic vesicle pools. Proc Natl Acad Sci U S A. 106, 1139-1144 (2009).
- Ji, C., Zhang, Y., Xu, P., Xu, T., Lou, X. Nanoscale Landscape of Phosphoinositides Revealed by Specific Pleckstrin Homology (PH) Domains Using Single-molecule Superresolution Imaging in the Plasma Membrane. J of Biol Chem. 290, 26978-26993 (2015).
- Betzig, E., et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 313, 1642-1645 (2006).
- Hess, S. T., Girirajan, T. P., Mason, M. D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys J. 91, 4258-4272 (2006).
- Rust, M. J., Bates, M., Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods. 3, 793-795 (2006).
- Xu, K., Zhong, G., Zhuang, X. Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science. 339, 452-456 (2013).
- Wombacher, R., et al. Live-cell super-resolution imaging with trimethoprim conjugates. Nat Methods. 7, 717-719 (2010).
- Szymborska, A., et al. Nuclear pore scaffold structure analyzed by super-resolution microscopy and particle averaging. Science. 341, 655-658 (2013).
- Pertsinidis, A., et al. Ultrahigh-resolution imaging reveals formation of neuronal SNARE/Munc18 complexes in situ. Proc Natl Acad Sci U S A. 110, 2812-2820 (2013).
- Shim, S. -. H., et al. Super-resolution fluorescence imaging of organelles in live cells with photoswitchable membrane probes. Proc Natl Acad Sci. 109, 13978-13983 (2012).
- Huang, F., et al. Video-rate nanoscopy using sCMOS camera-specific single-molecule localization algorithms. Nat Methods. 10, 653-658 (2013).
- Balla, T., Várnai, P. Visualization of Cellular Phosphoinositide Pools with GFP-Fused Protein-Domains. Curr Protoc Cell Biol. 24, 24 (2009).
- Xu, C., Watras, J., Loew, L. M. Kinetic analysis of receptor-activated phosphoinositide turnover. J Cell Biol. 161, 779-791 (2003).
- Subach, F. V., et al. Photoactivatable mCherry for high-resolution two-color fluorescence microscopy. Nat Methods. 6, 153-159 (2009).
- Filonov, G. S., et al. Bright and stable near infra-red fluorescent protein for in vivo imaging. Nature Biotechnol. 29, 757-761 (2011).
- Zhang, M., et al. Rational design of true monomeric and bright photoactivatable fluorescent proteins. Nat Methods. 9, 727-729 (2012).
- Hohmeier, H. E., et al. Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes. 49, 424-430 (2000).
- Dyachok, O., Isakov, Y., Sågetorp, J., Tengholm, A. Oscillations of cyclic AMP in hormone-stimulated insulin-secreting β-cells. Nature. 439, 349-352 (2006).
- Nan, X., et al. Single-molecule superresolution imaging allows quantitative analysis of RAF multimer formation and signaling. Proc Natl Acad Sci U S A. 110, 18519-18524 (2013).
- Sengupta, P., et al. Probing protein heterogeneity in the plasma membrane using PALM and pair correlation analysis. Nat Methods. 8, 969-975 (2011).
- Morone, N., et al. Three-dimensional reconstruction of the membrane skeleton at the plasma membrane interface by electron tomography. J Cell Biol. 174, 851-862 (2006).
- Peters, K. R., Carley, W. W., Palade, G. E. Endothelial plasmalemmal vesicles have a characteristic striped bipolar surface structure. J Cell Biol. 101, 2233-2238 (1985).
- Fujita, A., Cheng, J., Tauchi-Sato, K., Takenawa, T., Fujimoto, T. A distinct pool of phosphatidylinositol 4,5-bisphosphate in caveolae revealed by a nanoscale labeling technique. Proc Natl Acad Sci U S A. 106, 9256-9261 (2009).
- Durisic, N., Laparra-Cuervo, L., Sandoval-Álvarez, &. #. 1. 9. 3. ;., Borbely, J. S., Lakadamyali, M. Single-molecule evaluation of fluorescent protein photoactivation efficiency using an in vivo nanotemplate. Nat Methods. 11, 156-162 (2014).
- Thomas, C. L., Steel, J., Prestwich, G. D., Schiavo, G. Generation of phosphatidylinositol-specific antibodies and their characterization. Biochem Soc Trans. 27, 648-652 (1999).
- Várnai, P., Balla, T. Live cell imaging of phosphoinositides with expressed inositide binding protein domains. Methods. 46, 167-176 (2008).
- Honigmann, A., et al. Phosphatidylinositol 4,5-bisphosphate clusters act as molecular beacons for vesicle recruitment. Nature Struct Mol Biol. 20, 679-686 (2013).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved