Bu içeriği görüntülemek için JoVE aboneliği gereklidir. Oturum açın veya ücretsiz deneme sürümünü başlatın.
Method Article
DNA İçerik Ölçüm tarafından Memeli Çoğaltma Zamanlama genom Tayini
Bu Makalede
Özet
We describe here a relatively fast and simple approach for mapping genome-wide mammalian replication timing, from cell isolation to the basic analysis of the sequencing results. A genomic map of a representative replication program will be provided following the protocol.
Özet
genomun replikasyonu, DNA kopyalama doğruluğunu sağlayan bir yüksek ölçüde düzenlenmiş bir işlemde, hücre döngüsünün S fazında oluşur. Her genomik bölge çoğaltma birden kökenleri eşzamanlı aktivasyonu yoluyla S fazında belirgin bir anda çoğaltılır. çoğaltma (İş Tanımı) Zaman birçok genomik ve epigenetik özellikleri ile ilişkilidir ve mutasyon oranları ve kanser ile bağlantılıdır. Sağlıkta ve hastalıkta, çoğaltma programın tam genomik görünümü kavramak önemli bir gelecek hedefi ve mücadeledir.
, Memeli hücrelerinin genomu geniş Tanımı eşleştirmek için basit bir yaklaşım: Bu makalede ayrıntılı olarak (CNR-İş Tanımı burada denir) yöntemini "Çoğaltma genomik Zaman haritalama için S / G1 Kopya sayısı Oranı" açıklar. yöntem S fazı hücreleri ve G1 faz hücreleri arasındaki kopya sayısı farklılıklara dayanmaktadır. CNR-tor yöntemi 6 adımda gerçekleştirilir: propidyum iyodür (PI) hücre ve boyama hazırlanması 1.; 2. Sorting G1 ve sıralama floresan aktive hücre kullanılarak S fazı hücreleri (FACS); 3. DNA saflaştırılması; 4. Sonication; 5. Kütüphane hazırlık ve sıralama; ve 6. Biyoinformatik analizi. CNR-Şartname yöntemi ayrıntılı çoğaltma haritalarda sonuçlanan hızlı ve kolay bir yaklaşımdır.
Giriş
Memeli DNA replikasyonu, hücre döngüsü sırasında tam olarak bir kez her kromozomun tam çoğaltma sağlamak için sıkıca düzenlenir. Çoğaltma oldukça düzenlenmiş sıraya göre gerçekleşir - Birden fazla büyük genomik bölgeler (~ Mb) diğer genomik bölgelerde orta ya da geç S fazı (orta ve geç kopyalayan etki) daha sonra çoğaltmak ise 1 (erken etki kopyalayan) S fazına başında çoğaltırlar. Genomun 50%, kanser dönüşüm 5 içinde farklılaşması 3, 4 sırasında ve daha az bir ölçüde, dokular 2 arasındaki ToR'u bilgileri - genomunun en% 30 ise, tüm dokulardan (yapıcı Tanımı etki) aynı anda çoğaltır . Ayrıca, bazı genomik bölge senkronize olmayan bir 6, 7, 8, yani bir fark vardır çoğaltmaiki allel arasındaki Tanımında.
Tanımı transkripsiyon seviyeleri, GC içeriği, kromatin durum, genin yoğunluk, vb 1, 9 da dahil olmak üzere pek çok genomik ve Epigenomik özellikleri ile ilişkilidir. Tanımı mutasyon oranları ve tip 10, 11 ve bu nedenle şaşırtıcı olmayan ilişkili, çoğaltma programı pertürbasyonlar kanseri 12, 13 ile bağlantılıdır. Şartname ve kromatin yapısı arasındaki nedensel ilişki henüz anlaşılamamıştır. Açık kromatin erken çoğaltma kolaylaştırır mümkündür. Bununla birlikte, alternatif bir model, 14 kromatin çoğaltma sırasında bir araya getirilen ve farklı bir kromatin başlangıçta mevcut düzenleyiciler ve S fazında kurşun ucu erken ve geç replike bölgeleri 1 ambalajını diferansiyel düşündürmektedir . Son zamanlarda Tanımı genomik bölgelerden 11 oluşur mutasyon tipini etkileyerek GC içeriğine şekiller göstermiştir.
In situ hibridizasyon (FISH) floresan bağımsız lokuslar Tor ölçmek için ana yöntem. Tek FISH sinyallerini genel belirli bir alel 15, 16 çifti arasında bir yüzdesini sergiler S fazı hücrelerin yüzdesi sayarak basit gerçekleştirilir. Alternatif bir yöntem, S boyunca çoklu zaman noktaları DNA içeriğine göre olan hücreler, sıralama BrdU DNA etiketleme BrdU içeren DNA immüno-çökeltilmesinin ve qPCR 17, çökelen DNA bolluk kontrol darbe oluşur.
Genomik Tanımı eşleme iki yöntemle elde edilebilir. İlk yöntem, yukarıda tarif edilen BrdU-IP tabanlı yöntemin genomik versiyonu olduğu bir miktarının ölçümüHer fraksiyonda Çökelen DNA mikrodizilerinin melezleştirme yoluyla tüm genom ya da derin dizilemesi ile eş zamanlı olarak yapılır. İkinci yöntem, CNR-tor G1 hücrelerdeki DNA içeriği ile S fazı hücreleri ve normalize her genomik bölgenin kopya sayısını ölçülmesi esasına dayanır. Bu yöntemde, hücreler, replike olmayan (G1 fazı) ve replike (S fazı) grupları (Şekil 1) içine FACS ile sıralanır. G1 Hücreler tüm genomik bölgelerde aynı kopya sayısını ve böylece onların DNA içeriği aynı olmalıdır. Öte yandan, S DNA kopya sayısı Geç replike bölgeleri dolayısıyla DNA içeriği olacak hücrelerin çoğunda yapılmamış tekrarlanıp değil ise, erken replike bölgeler çok hücre replikasyonu yapılmış ve bu nedenle DNA içeriği iki olduğundan, Şartname bağlıdır G1 hücre edilene benzer. Bu nedenle DNA içeriği G1 oranı S Şartname göstergesidir. Her bir genomik bölge için DNA miktarı ile hibridizasyon yoluyla ölçülürmikroarrayler veya derin sıralama 2, 8 ile. CNR-Şartname yöntemin avantajları daha ayrıntılı olarak ele alınacaktır.
Şekil 2'de tarif edildiği gibi, kağıt genomik Tanımı eşleme CNR-tor yöntemi tarif etmektedir. Kağıt sonuçlarının temel analiz ve genomik Şartname haritalarının oluşturulması kadar hücrelerin toplanması kadar tüm sürecin ince ayrıntıları anlatılır. Bu yazıda anlatılan protokol başarıyla kültüründe yetişen çeşitli hücre tipleri üzerinde yapılmıştır. Bu protokolün gelecek iyileştirmeler in vivo ToR'un haritalama ve nadir hücre tiplerinde yol açabilir.
Protokol
Not: Şartname sadece büyüyor, eşitlenmemiş hücreler üzerinde ölçülebilir. Genellikle S fazında ~ 1 x 10 5 hücre neden olur 2 x 10 6 hızlı büyüyen hücreler, (oran sınırlayıcı adım) - prosedür en az 1 ile başlamalıdır. Iki ya da üç kez tekrarlanmış kullanılarak her bir deney için tavsiye edilir. CNR-ToR'un tüm süreci bir hafta içinde tamamlanabilir - iki gün bir veya iki gün sıralama için gerekli olan ve ek bir gün ilk veri analizi için gerekli olan, kütüphane hazırlanmasına kadar tüm adımlar adanmış olmalıdır.
Kültür hücreleri 1. Koleksiyonu
NOT: protokolü (yaklaşık 2 içeren - 5 x 10 6 hücre) 10 cm'lik plakalar kültür büyüyen hücreler için yazılmıştır, ancak kolayca diğer platformlara ayarlanabilir.
- süspansiyon içinde büyütüldü hücreler için, fiksasyon (bölüm 2) geçin.
- Yapışık hücreler, aspirat ve pl yıkama içinCa 2+ ve Mg 2+ olmadan 3 ml PBS ile yedi.
- PBS atın ve hücreler ayrılana kadar 1 ml ticari bir tripsin-EDTA ile 37 ° C 'de 5 dakika boyunca hücreler inkübe edin.
Not: tripsin tedavi süresi, her hücre türü için ayarlanmalıdır. - Tripsin nötralize ve 15 ml konik tüp veya 5 ml polistiren tüp hücreleri toplamak için 3 ml kültür ortamı ekleyin. buz üzerinde tutun.
2. Sabitleme
Not: Bu parça için, tüm adımlar 4 ° C'de yapılmalıdır.
- 4 ° C'de 5 dakika boyunca 300 x g'de santrifüjleyin hücreleri.
- Aspire edin ve 1 ml soğuk PBS ile iki kez yıkama hücreleri.
- 250 uL (toplam) soğuk PBS içinde süspanse edin hücreleri.
- tüp hafifçe vorteks birlikte, yavaş yavaş Cı% 100 etanol -20 ° damla damla 800 mL ekleyin. % 80 - Bu 70 bir son etanol konsantrasyonu yol açar.
NOT: Yüksek saflıkta etanol bu aşamada tavsiye edilir. - fo buz üzerinde inkübe hücreleriR 30 dk.
NOT: Bu aşama, hücreler 4 ° C'de ya da -20 ° C'de bir kaç ay için bir kaç gün için muhafaza edilir ve bu da.
3. PI Boyama
- 4 ° C'de 10 dakika boyunca 500 xg'de hücreleri santrifüjleyin.
- dikkatle süpernatant aspirat ve 1 ml soğuk PBS ile iki kez hücreleri yıkayın.
- Aspire, aşağıdaki karışım ile her bir örnek tekrar süspansiyon ve 1 ml PBS, 5 uL 10 mg / mL RnazA, 50 uL 1 mg / ml propidium iodide (PI, kullanımdan önce şişe karıştırıldı). ~ 2 x 10 6 hücre / ml) kadar konsantrasyonunu ayarlayın.
NOT: Işık uzak tutun - PI duyarlı ışıktır. - 5 ml polistiren tüp 35 mikron örgü süzülmeye ve Parafilm ile yakın.
- 30 dakika - 15, karanlıkta, oda sıcaklığında inkübe edin.
NOT: Hücreler artık FACS analizi için hazırız. Gerekirse, lekeli hücreler, karanlıkta 4 ° C'de en az 24 saat süre ile depolanabilir.
4. Sıralama
- FACS makinesi kullanılarak Sıralama hücreleri. Bizekendi PI yoğunluğuna dayalı hücreleri ayırt etmek için 561 nm lazer e. 488 nm veya 532 gibi 535 nm uyarma maksimum yakın diğer lazerler FACS makine konfigürasyonuna bağlı olarak kullanılabilir.
- En iyi sonuçlar için, (çoğu hücreleri 85 mikron için) belirli bir hücre boyutu için önerilen en küçük meme kullanın. verim üzerinde saflık modları öncelik. 45 psi kılıf basıncı ile, genellikle yukarı 300-500 olaylar / s, sürekli yavaş akış kullanın.
- Yolluk kullanarak, SSC vs FCS çizimi ile ölü hücreleri ve hücre içi enkaz (düşük FCS ve yüksek SSC) ayırt. PI-H (çiftli aynı H-değere sahip olacaktır vs canlı hücreler bir FSC-W FSC-H arsa vs ve PI W tarafından takip SSC-Genişlik (W) vs SSC-Boy (H) çizerek çiftleri ayırt ama daha büyük W-değer). canlı tek hücrelerin hücrelerin DNA içeriğini temsil eden PI-Alan (A) yoğunluk histogramı çizin.
- Şekil 1 'de gösterildiği gibi, G1 ve S evreye kriteri hücreleri,. S Yolluk genişliğinde ve G1 yolluk dar ve bildiğim kadarıyla S mümkün olduğunca gerekirken, G1 ve G2 fazları davetsiz gerekir.

Şekil 1. Hücre döngüsü faz belirleme PI yoğunluğuna göre. fare embriyonik fibroblast (MEF) nüfusun (PI-Alan ile ölçülen) hücresel DNA içeriğinin dağılımını gösteren histogram. Işaretlenmiş bölgeleri kullanılarak, 4N DNA muhtevasına) - DNA içeriği, iki alt-popülasyonları i) G1 hücreleri (2N DNA içeriği) ve ii) S fazı hücreleri (2N içine nüfus sıralamak için kullanılmaktadır. Bu rakamın büyük halini görmek için lütfen buraya tıklayınız.
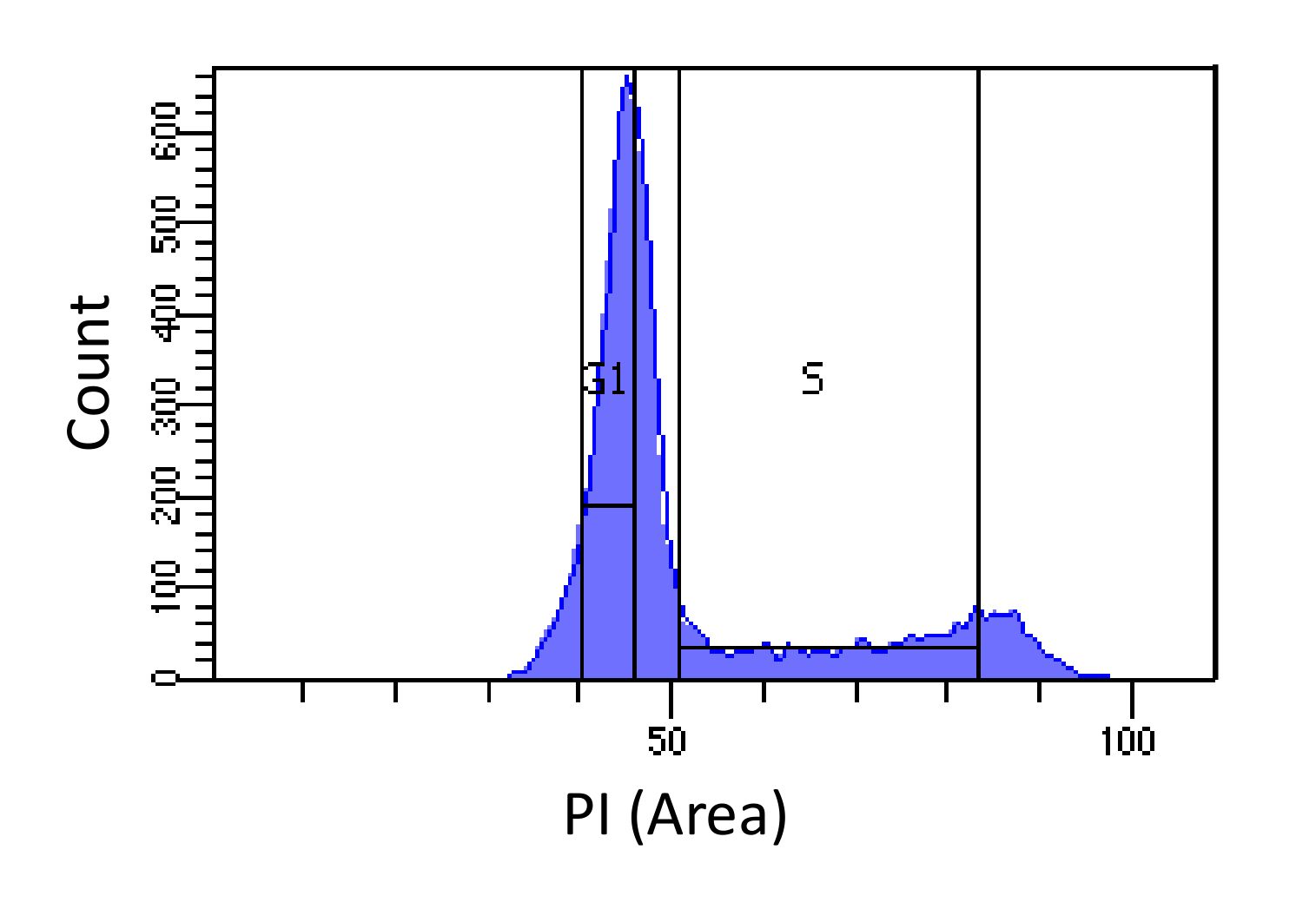{kind=link}
NOT: G1 hücreleri toplanması amacı farklı genomik bölgeler arasında sıralama verimliliği önyargıları hesaba etmektir. Alternatif uygulamaRoach, aynı hücre tipinden G1 tutuklandı hücreleri kullanmaktır. Bu yaklaşım temizleyici sonuçlar verir (o S fazı kontaminasyonu minimize beri) ama tutuklandı hücreleri ve ölçülen hücreler arasındaki genetik farklılıklardan kaynaklanan önyargıları tanıtmak olabilir.
- soğuk koşullarda sıralama ve 1.5 / 2 ml tüpler içine sıralanan hücreleri toplamak. sıralama aşağıdaki buz üzerinde tüpler tutun.
Not: 4 ° C'de 18 ° C'de 3 saat - 1 -% 5 BSA, DNA kurtarma geliştirmek için, düşük bağlanma tüpleri kullanmak için ya da 4 ile kaplanmış tüpler kullanımı daha iyidir.
5. DNA Saflaştırma
- Her bir örnek (G1 ve G) için bir DNA saflaştırma kiti kullanılarak DNA arındırmak.
Not: ticari kit kullanılarak üreticinin tavsiye ettiği gibi, yüksek verim için 2 ml tüp içine 400 uL yıkama tamponu ile yıkayın. - fluorometreyle DNA konsantrasyonu kontrol edin.
NOT: FACS tarafından toplanan 100.000 memeli hücrelerden, bir DNA ~ 1 mikrogram almalısınız. birt Bu aşamada DNA 4 ° C'de veya uzun depolama için -20 ° C'de birkaç gün muhafaza edilebilir.
6. Sonication
- kullanılan manyetik standı ile uyumlu olan bir 1.7 mL tüp içine DNA transferi.
- manyetik bir stand kullanılarak üreticinin talimatlarına göre 2x SPRI boncuklar kullanılarak DNA Konsantre ve 50 uL yıkama tamponu içinde elüte.
- 250 bp bir ortalama hedef pik boyutuna odaklanmış ultrasonikatör yamultma DNA. 50 uL DNA örneği için aşağıdaki ayarları kullanın: 50 W,% 20 Duty faktörü, patlama başına 200 Cycles, 20 ° C, 120 s.
- elektroforez ile kesilmiş DNA boyutunu kontrol edin. ~ 250 bp bir zirve ile 700 bp - önerilen boyut dağılımı 200.
NOT: Bu aşama, DNA, 4 ° C'de veya uzun depolama için -20 ° C'da bir kaç gün için muhafaza edilir ve bu da.
7. Kütüphane Hazırlık ve Sıralama
NOT: farklı Birçok kütüphane hazırlık setleri veent sıralama platformları malzemeler bölümünde bizim tarafımızdan kullanılan ve söz konusu olanları benzer şekilde çalışması gerekir. Aslında, geçmişte, Tanımı harita mikrodizi platformları 2 ile çok benzer bir metot kullanılarak elde edildi.
- herhangi bir ticari kütüphane hazırlık kiti kullanılarak kütüphaneleri hazırlayın.
- 800 bp - kütüphane hazırlık sonunda, 300 manyetik boncuklar kullanılarak boyutunu seçin.
- kütüphaneler hazırladıktan sonra, fluorometreyle DNA konsantrasyonu ölçmek.
- elektroforezi kullanılarak DNA boyutunu ölçmek.
- Herhangi bir platformda sıralama yapın.
Not: Ardışık en az 10 m önerilir numune başına okur. Bu derinlik okuma yaklaşık her 300 bp (3 ~ için Gb boyutu genomu) eşdeğerdir ve 50 çözünürlükte Tanımı ölçümleri için yeterlidir - 100 kb. pencere boyutunda bir azalmaya neden olur derinliği artırılması ve böylece daha yüksek bir kesinlikle çözünürlüğünü artırma sağlayacaktır. Eşli uç sıralama gerekli değildirSadece kapsama bilgi beri bu protokol toplanır. Ancak, tekrar eden diziler içeren okur yerini çözümünde yardımcı olabilir.
8. Analiz
NOT: Veri analizi A. Koren ve arkadaşları tarafından kullanılan yönteme dayanmaktadır. 19.
- bowtie2 ya da herhangi bir güvenilir kısa okumak hizalama kullanılarak ilgili genom Harita dizi verileri. değişen boyutu, 200 kapsamına giren kesimlerine eşit kapsama kromozomal pencereler G1 fraksiyonunda okur tanımlayın ve saymak S fazı aynı pencerelerde okur.
- her pencere için S / G1 oranını hesaplayın. Bu genom (Şekil 3) boyunca S / G1 oranında büyük dalgalanmalar bir harita oluştursun. Tanımı ölçümlerin güvenilirliği için iyi bir denetim çok düz olmalıdır (G1 iki ayrı ölçümlerinden) G1 / G1 oranına Bu haritayı karşılaştırmaktır.
- 0 veriler ortalama normalleştirmek ve her v çıkarılarak 1 SD(X kromozomu hariç) ve tüm pencereleri S / G1 standart sapma ile sonuçları bölünmesi tüm pencerelerin ortalama değerini Alue. Bu z skorları dönüştürmek ve farklı deneyler arasında karşılaştırma sağlamak amacıyla yapılır.
- UCSC genom tarayıcısı yanı sıra 15'den az veri pencereler içeren kalan her arası boşluk fragmanı listelenen tüm boşluk bölgeleri çıkarın.
- 10 bir parametre ile Matlab fonksiyonu csaps üzerinden kübik yumuşatma eğri kalan parçaları Düzgün - 16 ve ayar noktaları her 100 kb enterpolasyon.
NOT: yumuşatma ve interpolasyon parametreleri veri derinliğine göre ayarlanmalıdır. Diğer uygun düzeltme yöntemleri ve işlevleri vardır ve kullanılabilir. - görsel olarak her çoğaltmak güvenilirliğini onayladıktan sonra, tüm okur birleştirme ve bu verilere yukarıda açıklanan aynı işlemi gerçekleştirerek daha derin bir çözünürlük profili hesaplar.
Sonuçlar
Tipik Tanımı harita fare embriyonik fibroblastlar için Şekil 3 (MEF'ler) 'de gösterilmiştir. gösterir çünkü bu rakam analiz sürecini gösteren bireysel pencereler (adım 8.3) için normalize S / G1 oranı olan noktalar, yanı sıra kübik yumuşatma ve enterpolasyon (adım 8.5) sonucunda oluşan çizgidir.
i) erken aynı anda (TO = sabit Tanımı bölgeler) çoğaltmak bir megabase sırasın...
Tartışmalar
CNR-Tanımı (Rhind N. ve Gilbert DM 20 tarafından incelenmiştir) S FACS ve G1 aşamaları ile ayrılabilir bir ökaryotik proliferatif hücre popülasyonu ilke olarak gerçekleştirilebilir. Burada tarif edilen yöntem, insan ve fare gibi ~ 3 Gb bir genom boyutuna sahip memeli hücrelerine ayarlanmıştır. (Hücre hazırlama ve dizileme derinliğinde) CNR-tor protokol küçük değişiklikler ökaryotlarda için ayarlamak için, ihtiyaç vardır. bu hız sınırlayıcı bir adımdır çünkü...
Açıklamalar
No conflicts of interest declared.
Teşekkürler
Biz rakamları oluşturmada yardım için Oriya Vardi teşekkür ederiz. BS grubunda çalışma, İsrail Bilim Vakfı (hibe No 567/10) ve Grant (# 281306) Başlangıç Avrupa Araştırma Konseyi tarafından desteklenmiştir.
Malzemeler
| Name | Company | Catalog Number | Comments |
| PBS | BI (Biological Industries) | 02-023-1A | |
| Trypsin-EDTA | BI (Biological Industries) | 03-052-1B | |
| 15 mL conical tube | Corning | 430790 | |
| 5 mL Polystyrene round Bottom tube with cell strainer cap | BD-Falcon | 352235 | |
| Ethanol | Gadot | 64-17-5 | |
| RNAse-A 10 mg/mL | Sigma | R4875 | |
| Propidiom iodide 1 mg/mL | Sigma | P4170 | |
| Parafilm | Parafilm | PM-996 | |
| 1.5 mL DNA LoBind Eppendorf tubes | Eppendorf | 22431021 | |
| BSA | Sigma | A7906 | |
| 1.7 mL MaxyClear tube | Axygen | MCT-175-C | |
| magnetic beads - Agencourt AMPure XP | Beckman Coulter | A63881 | |
| Ultrasonicator | Covaris | M-series -530092 | |
| 50 µL microTUBE AFA Fiber Screw-Cap 6 x 16 mm | Covaris | 520096 | |
| Qubit fluorometer | Invitrogen | ||
| Qubit dsDNA High Sensitivity (HS) Assay Kit | Invitrogen | Q32854 | |
| Electrophoresis 2200 Tape station system | Agilent | D1000 ScreenTape | |
| Seqeuncing - Illumina NextSeq system | Illumina | SY-415-1001 | |
| Dneasy kit for DNA purification | Qiagen | 69504 | |
| PureProteom Magnetic Stand | Millipore | LSKMAGS08 | |
| Anti-BrdU/FITC | DAKO | F7210 | |
| FACS sorter | BD | FACSARIA III | |
| FACS software | BD | FACSDiva v 8.0.1 |
Referanslar
- Farkash-Amar, S., Simon, I. Genome-wide analysis of the replication program in mammals. Chromosome Res. 18 (1), 115-125 (2010).
- Yaffe, E., et al. Comparative analysis of DNA replication timing reveals conserved large-scale chromosomal architecture. PLoS Genet. 6 (7), e1001011 (2010).
- Hiratani, I., et al. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 6 (10), (2008).
- Rivera-Mulia, J. C., et al. Dynamic changes in replication timing and gene expression during lineage specification of human pluripotent stem cells. Genome Res. 25 (8), 1091-1103 (2015).
- Ryba, T., et al. Abnormal developmental control of replication-timing domains in pediatric acute lymphoblastic leukemia. Genome Res. 22 (10), 1833-1844 (2012).
- Farkash-Amar, S., et al. Global organization of replication time zones of the mouse genome. Genome Res. 18 (10), 1562-1570 (2008).
- Koren, A., McCarroll, S. A. Random replication of the inactive X chromosome. Genome Res. 24 (1), 64-69 (2014).
- Mukhopadhyay, R., et al. Allele-specific genome-wide profiling in human primary erythroblasts reveal replication program organization. PLoS Genet. 10 (5), e1004319 (2014).
- McNairn, A. J., Gilbert, D. M. Epigenomic replication: linking epigenetics to DNA replication. Bioessays. 25 (7), 647-656 (2003).
- Sima, J., Gilbert, D. M. Complex correlations: replication timing and mutational landscapes during cancer and genome evolution. Curr Opin Genet Dev. 25, 93-100 (2014).
- Kenigsberg, E., et al. The mutation spectrum in genomic late replication domains shapes mammalian GC content. Nucleic Acids Res. 44 (9), 4222-4232 (2016).
- Woo, Y. H., Li, W. H. DNA replication timing and selection shape the landscape of nucleotide variation in cancer genomes. Nat Commun. 3, 1004 (2012).
- Liu, L., De, S., Michor, F. DNA replication timing and higher-order nuclear organization determine single-nucleotide substitution patterns in cancer genomes. Nat Commun. 4, 1502 (2013).
- Goren, A., Cedar, H. Replicating by the clock. Nat Rev Mol Cell Biol. 4 (1), 25-32 (2003).
- Selig, S., Okumura, K., Ward, D. C., Cedar, H. Delineation of DNA replication time zones by fluorescence in situ hybridization. EMBO J. 11 (3), 1217-1225 (1992).
- Smith, L., Thayer, M. Chromosome replicating timing combined with fluorescent in situ hybridization. J Vis Exp. (70), e4400 (2012).
- Simon, I., et al. Asynchronous replication of imprinted genes is established in the gametes and maintained during development. Nature. 401 (6756), 929-932 (1999).
- Phi-Wilson, J. T., Recktenwald, D. J. Coating agents for cell recovery. Google Patents. , (1993).
- Koren, A., et al. Differential relationship of DNA replication timing to different forms of human mutation and variation. Am J Hum Genet. 91 (6), 1033-1040 (2012).
- Rhind, N., Gilbert, D. M. DNA replication timing. Cold Spring Harb Perspect Biol. 5 (8), a010132 (2013).
- Koren, A., et al. Genetic variation in human DNA replication timing. Cell. 159 (5), 1015-1026 (2014).
Yeniden Basımlar ve İzinler
Bu JoVE makalesinin metnini veya resimlerini yeniden kullanma izni talebi
Izin talebiDaha Fazla Makale Keşfet
This article has been published
Video Coming Soon
JoVE Hakkında
Telif Hakkı © 2020 MyJove Corporation. Tüm hakları saklıdır