Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Microscopie multicolore localisation des protéines de la Membrane unique dans les organites des cellules mammifères vivants
Dans cet article
Résumé
Nous présentons ici un protocole pour la localisation multi-couleur des protéines membranaires unique dans les organites des cellules vivantes. Pour fixer les fluorophores, étiquetage des protéines sont utilisées. Protéines, situés dans des compartiments de différentes membranes de l’organite même, peuvent être localisées avec une précision d’environ 18 nm.
Résumé
Connaissance de la localisation des protéines cellulaires sous-compartiments est crucial de comprendre leur fonction spécifique. Nous présentons ici une technique de Super-résolution qui permet pour la détermination de la microcompartments qui sont accessibles pour les protéines en générant la localisation et le suivi des cartes de ces protéines. En outre, par microscopie de localisation multi-color, la localisation et le suivi des profils des protéines dans différents sous-compartiments sont obtenus simultanément. La technique est spécifique pour les cellules vivantes et est basée sur l’imagerie répétitive des protéines membranaires mobile unique. Protéines d’intérêt sont génétiquement fusionnées avec des tags auto étiquetage spécifiques, ce que l'on appelle. Ces balises sont des enzymes qui réagissent avec un substrat de manière covalente. Conjugué à ces substrats sont des colorants fluorescents. Réaction des protéines enzymatiques-tag avec la fluorescence étiqueté résultats de substrats en protéines marquées. Ici, tétraméthylrhodamine (TMR) et silicium Rhodamine (SiR) sont utilisés comme colorants fluorescents attachés aux substrats des enzymes. En utilisant des concentrations de substrat dans la MP à éventail nM, Sub stoechiométrique d’étiquetage est réalisé qui se traduit par des signaux distincts. Ces signaux est localisés avec ~ 15 – 27 précision nm. La technique permet d’imagerie multicolore de molécules simples, auquel cas le nombre de couleurs est limité par les colorants disponibles membrane perméable et le répertoire de l’étiquetage automatique des enzymes. Nous montrer la faisabilité de la technique de déterminer la localisation de l’enzyme de contrôle de la qualité (Pten)-induite par la kinase 1 (PINK1) dans différents compartiments mitochondriales au cours de son traitement par rapport à d’autres protéines de la membrane. Le critère de véritables interactions physiques entre les protéines simples différemment étiquetées par seule molécule frette ou suivi Co est limité, cependant, parce que les degrés d’étiquetage faibles réduisent la probabilité d’avoir deux protéines adjacentes marquées en même temps. Alors que la technique est forte pour l’imagerie des protéines dans les compartiments de la membrane, dans la plupart des cas il n’est pas appropriée pour déterminer la localisation des protéines solubles très mobiles.
Introduction
Le but du présent protocole est de fournir une méthode d’imagerie pour localiser et suivre les protéines de la membrane unique à l’intérieur des cellules vivantes. Nous appelons cette méthode de suivi et de localisation microscopie (TALM)1,2. Comme la microscopie de Reconstruction optique stochastique (tempête)3 et la microscopie de Fluorescence Photoactivation localisation ((F) PALM)4,5, TALM est une technique de localisation unique axée sur la molécule de fluorescence. Toutefois, il est distinct de la manière que la mobilité des protéines de la membrane en combinaison avec l’imagerie répétitive du même étiqueté molécule à différentes positions révèle le microcompartiment qui est accessible pour la protéine mobile. En d’autres termes, les localisations possibles de la protéine sont définies par l’architecture de l’organite et par la mobilité de la protéine1. La méthode est complémentaire à divers autres super-résolution techniques6,7,8 , parce qu’il révèle la localisation et la trajectoire des cartes par des protéines d’imagerie mobiles. L’étiquetage est basé sur l’utilisation des protéines de fusion génétiquement modifiées qui sont en soi non fluorescent. Ces protéines de fusion sont étiquetage automatique d’enzymes qui réagissent de façon covalente avec un substrat conjugué à un colorant. Cette procédure a l’avantage que le degré d’étiquetage peut être contrôlées par la quantité de substrat ajouté. En outre, il permet de varier la couleur de la fluorescence, selon le colorant choisi conjugué. Plusieurs étiquettes enzyme étiquetage automatique sont disponibles9. Un autre avantage d’utiliser l’étiquetage automatique des balises enzyme est, que les colorants conjugués sont généralement plus stable et plus brillante que protéines fluorescentes1 et protéines individuelles par conséquent peuvent être enregistrés plus et plus précisément jusqu'à ce qu’ils sont blanchis. Cela permet l’enregistrement des trajectoires des protéines mobiles et l’extraction des coefficients de diffusion10,11.
Ici, nous avons démontré la faisabilité de TALM avec des protéines de la membrane mitochondriale, mais il peut aussi être appliqué aux autres protéines de la membrane intra - et extra-cellulaires, y compris les cellules différents types12,13. Nous montrons que multi-couleur TALM plus permet la distinction simultanée des protéines dans différents sous-compartiments en complémentation existants Super-résolution fluorescence microscopy techniques14,15, 16. TALM est compatible avec17d’imagerie de cellules vivantes. La photo-physique des rhodamines choisis tétraméthylrhodamine (TMR) et silicium-Rhodamien (SiR), en particulier leur luminosité et leur stabilité, permet aux protéines de la membrane unique record sur plusieurs périodes de fourniture de cartes de localisation (et trajectoire). Cependant, TALM est limitée pour la localisation des protéines solubles avec les coefficients de diffusion élevée car le flou est trop élevé et les photons recueillis par image sont trop faibles pour la localisation correcte. En outre, TALM nécessite moins d’énergie que par exemple tempête ou émission de stimulé l’appauvrissement (STED) microscopie6,7, excitation réduisant effets phototoxiques. C’est important, car le stress phototoxique affecte souvent morphologie organellar18 et, partant, mobilité analyse19. En somme, nous présentons TALM multicolore dans les cellules vivantes comme une technique qui comble une lacune entre les méthodes de microscopie de localisation STORM/STED/PALM (F) et les techniques qui analysent la mobilité des protéines telles que la récupération de fluorescence après photoblanchiment (FRAP)20 ,21, corrélation de fluorescence spectroscopy (FCS)22et fluorescence croisent corrélation spectroscopie (SCCC)11,23.
Protocole
Le protocole suivant suit les directives de la Commission d’éthique de recherche établissement local.
1. méthodes
-
Culture cellulaire
- Cultiver des cellules, par exemple les cellules HeLa (carcinome du col de l’utérus humain), dans un flacon de culture cellulaire T25 contenant 5 mL de milieu de culture à 37 ° C et 5 % de CO2.
Remarque : Pour l’imagerie, fractionner les cellules sur des lamelles préparés (voir étapes 1.3 et 1.4) et conserver au moyen de l’imagerie.
- Cultiver des cellules, par exemple les cellules HeLa (carcinome du col de l’utérus humain), dans un flacon de culture cellulaire T25 contenant 5 mL de milieu de culture à 37 ° C et 5 % de CO2.
-
Transfection de cellules
Remarque : Utiliser des lignées de cellules qui expriment stablement les protéines étiquetées si possible24 afin d’éviter une forte surexpression. Pour la transfection transitoire, adapter la quantité d’ADN utilisée pour la transfection plasmidique. Par exemple, lorsqu’on utilise Ca2 + phosphate transfection25 , transfecter cellules (confluence de 80 à 90 %) dans une boîte de Petri de cellule de 3,5 cm avec 2,5 – 5 µg d’ADN plasmidique. Lorsque vous effectuez une transfection double, utilisez 2,5 µg par chaque construction de plasmide.- Pour des expériences de couleur double, utilisez une lignée cellulaire avec expression stable d’une protéine de l’étiquetage et transitoirement transfecter avec le plasmide codant les autres protéines étiquetage automatique17.
NOTE : Ici, pour des expériences de couleur double, les cellules HeLa ont été utilisés qui stablement exprime les protéines auto étiquetage PINK1-Halo-Tag et Tom20-fSNAP-Tag.
- Pour des expériences de couleur double, utilisez une lignée cellulaire avec expression stable d’une protéine de l’étiquetage et transitoirement transfecter avec le plasmide codant les autres protéines étiquetage automatique17.
-
Nettoyage des lamelles couvre-objet
- Placer les lamelles dans un bécher. Ajouter 30 mL d’H2O dans le bécher contenant les lamelles et secouer doucement pour enlever la poussière de leur surface.
- Rassembler les lamelles couvre-objet avec des pincettes et séchez-les avec un courant d’azote.
- Enlever toute contamination organique à la surface des lamelles, par exemple, par le nettoyage de plasma.
Remarque : Pour éviter la contamination du matériau verre, porter des gants lors de la manipulation des lamelles.
ATTENTION : Quand les lamelles sont nettoyés par plasma nettoyage, seulement la partie supérieure des lamelles est nettoyée ; utiliser ce côté de revêtement avec la poly-L-Lysine-polyéthylène glycol-glycine-aspartate d’arginine (PLL-PEG-RGD) (section 1.4) et la cellule ensemencement (section 1.5).
-
Revêtement de la lamelle couvre-objet avec PLL-PEG-RGD
NOTE : PLL-PEG-RGD est un dérivé de (PLL) de poly-L-Lysine attaché avec un polyéthylène glycol (3 000 Da) et un peptide (CGRGDS) cysteine-glycine-arginine-glycine-aspartate-serine. PLL se lie à la surface de verre chargée négativement et forme une brosse de PEG. Cela réduit considérablement la liaison non-spécifique de colorants fluorescents chargées. En outre, le motif RGD imite le peptide signal du récepteur intégrine et favorise ainsi l’intégrine adhérence de cellules qui autrement ne pas facilement adhèrent.- Préparer les PLL-PEG-RGD comme décrit précédemment26. En bref, dissoudre 0,8 mg de PLL-PEG-RGD dans 1 mL de PBS. Ajouter 10 µL de la solution de PLL-PEG-RGD sur la face supérieure d’une lamelle couvre-objet propre.
- Prendre une seconde lamelle et placez-le avec sa surface propre à l’envers sur la première lamelle (qui a la goutte de PLL-PEG-RGD sur le dessus) ; Cela se traduit en intercalant la solution PLL-PEG-RGD entre deux lamelles couvre-objet.
- Placez le couvre-objet en sandwich dans un bécher délicatement et incuber pendant 1 heure à température ambiante dans un endroit sec et sans poussière.
- Après 1 h, ajouter 30 mL d’H2O dans le bécher de recouvrir entièrement les lamelles couvre-objet avec de l’eau.
- Agiter doucement le récipient jusqu'à ce que les lamelles se détachent les uns des autres.
- Pince à épiler permet de recueillir les lamelles hors de l’eau et les faire sécher sous un courant d’azote gazeux.
Remarque : Les lamelles enduits peuvent être stockés dans un sec en verre stérile Pétri avec couvercle pour quelques jours.
-
Préparation de l’éprouvette pour l’imagerie
- Transférer les lamelles couché seul dans une boîte de Petri de cellule de 35 mm, avec la surface recouverte de PLL-PEG-RGD vers le haut et ajouter 2 mL d’imagerie médium sur le dessus.
- Ajouter ~ 500 000 cellules trypsinisés (200 – 500 µL) qui expriment les balises auto étiquetage sur les protéines de la membrane respectifs à 2 mL moyen dans le plat de culture cellulaire avec la lamelle revêtu d’imagerie. Secouer doucement à la main pour assurer une répartition homogène des cellules afin d’obtenir une couche de cellules uniformément.
- Incuber les cellules à 37 ° C et 5 % de CO2 jusqu'à confluence de 80 % est atteint.
Remarque : Les échantillons cellulaires devraient être semés à 3 jours avant l’imagerie et 1 jour avant la transfection. Les cellules, qui stablement expriment la protéine d’intérêt, peuvent être semés 2jours avant l’imagerie. Plus tard, seulement les cellules cultivées sur la lamelle couvre-objet sont imagés.
-
Marquage des protéines étiquetées
NOTE : Substrats fluorescents plus doivent être dissous dans le DMSO sans eau. Nous conseillons d’utiliser des solutions mères du substrat fluorescent de 1 µM, lorsque la concentration finale d’étiquetage est de 0,2 à 30 nM17. Pour l’imagerie des protéines de la membrane à l’intérieur des cellules, utilisez substrats fluorescents perméable membrane.- Moyen d’imagerie ou à 37 ° C dans un bain d’eau d’échauffement.
- Pipette 1 mL de milieu d’imagerie pré chauffé dans un tube de 2 mL avec couvercle. Ajouter 0,2 – 30 µL de substrats fluorescents à partir de solutions mères 1 µM pour préparer la solution d’étiquetage finale (concentration finale : 0,2 à 30 nM).
- Vortex la solution d’étiquetage pour 10 s.
- Remplacer le support dans la boîte de Petri 35 mm avec les cellules sur un lamelle couvre-objet (Voir l’étape 1.5) par 1 mL de prêt solution d’étiquetage.
- Incuber les cellules dans la solution d’étiquetage à 37 ° C et 5 % de CO2 pendant 20 à 30 min.
- Laver les cellules avec 2 mL de PBS une fois, puis 2 ml d’imagerie moyen deux fois. Enfin, pipette de 1 mL de milieu d’imagerie frais dans le plat de la cellule et remettre l’échantillon dans l’incubateur à 37 ° C et 5 % de CO2 pendant au moins 1 h. Avant l’imagerie, échanger une fois de plus le moyen d’imagerie.
Remarque : Lorsque vous exécutez l’expérience pour la première fois, confirmer le ciblage correct des protéines Self marquées aux membranes organellar par coloration les organites avec organelle disponibles dans le commerce des colorants spécifiques27,28. Dans ce cas, également utiliser 100 – 200 nM de substrat pour les enzymes d’étiquetage automatique pour produire des signaux forts.
-
Préparation d’un échantillon de billes fluorescentes
Remarque : Afin de déterminer la dérive optique et d’aligner les images des différents canaux, multicolores perles fluorescentes (0,1 µm) sont utilisés. Les images enregistrées, une matrice de transformation affine pour les canaux de deux émission sera générée.- Diluer la solution de perles à 1 % avec pure H2O.
- Déposer 5 gouttes de la solution préparée avec les perles de fluorescence à cinq positions différentes sur une lamelle couvre-objet nettoyé (Voir l’étape 1.3).
- Sécher l’échantillon fluorescent perle sur un banc propre.
Remarque : L’échantillon peut être réutilisé ; par conséquent, couvrir l’échantillon avec le papier d’aluminium pour éviter la contamination et le conserver à une température 4 ° C.
2. microscopie
-
Montage expérimental
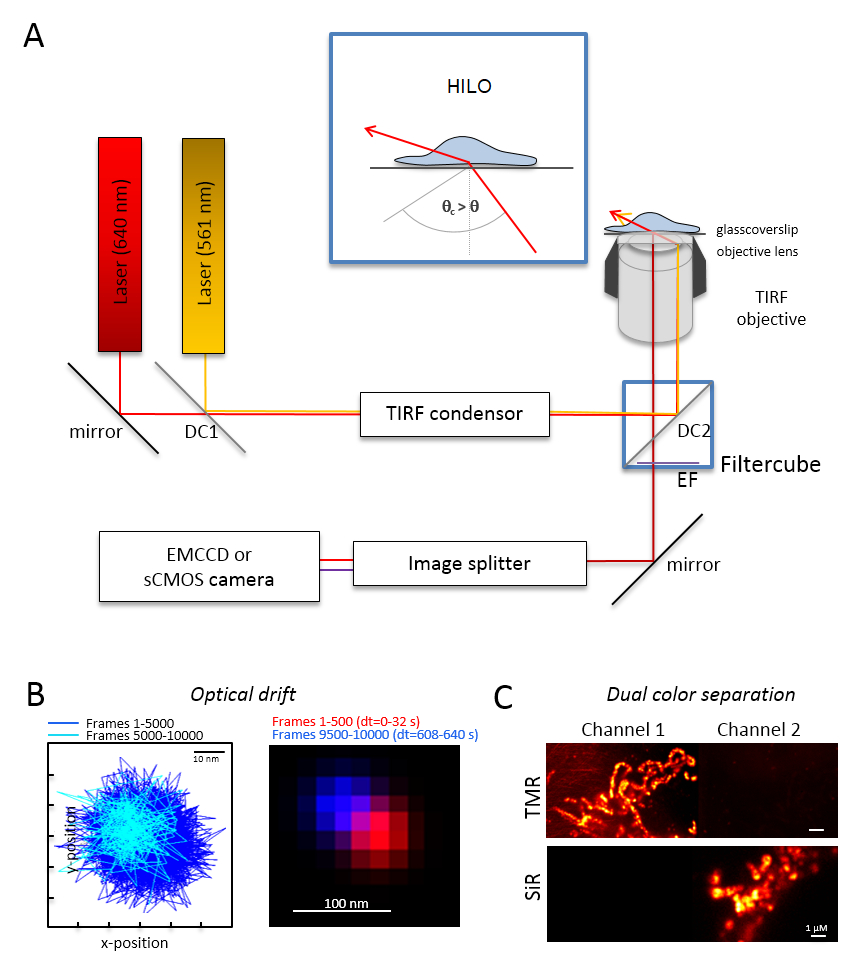
Remarque : Un système de base de la microscopie pour l’imagerie bicolores seule molécule repose sur un microscope inversé : il est équipé de deux lasers couplés via une polarisation multi-mode-optique maintien fibre monomode dans une seule réflexion totale interne (TIR) condenseur, un objectif à immersion d’huile conçu pour FRBR, un filtre de d’émission polyband, un séparateur d’image et une caméra très sensibles (Figure 1). Un condensateur TIR est nécessaire qui permet un réglage continu de l’angle d’incidence pour basculer entre l’épi-, fortement incliné et optique laminé (éclairage mince très inclinée (HILO)29) et le mode d’excitation de la FRBR avec optimisé profondeur de pénétration. Images sont obtenues avec un système de détecteur refroidi très sensibles, par exemple, un rétro-éclairé multipliant les électrons chargés appareil couplé (EMCCD) camera (efficacité quantique QE > 90 %) ou un appareil photo sCMOS (QE > 80 à 90 %).- Déterminer la dérive optical Imaging perles fluorescentes (Voir l’étape 2.2) dans les mêmes conditions que celles qui sera utilisé plus tard pour l’expérience, par exemple, si 10 000 cadres sont enregistrées dans l’expérience, enregistrent également 10 000 cadres avec l’échantillon de perle. Pour la détermination de la dérive optique, comparer la position des billes dans le cadre de la première et la dernière image acquise (Figure 1 b). Si nécessaire, par la suite corriger les séries d’images pour dérive optique30 et/ou utiliser dérive un environnement stable.
- Équiper le cube de filtre avec le séparateur de faisceau dichroïque approprié, par exemple, pour fluorescence orange et rouge, plus les filtres adéquats d’émission de fluorescence orange et la fluorescence rouge. Équiper le séparateur d’image avec les filtres adéquats. Recherchez la fuite possible de signaux d’un canal à l’autre canal en enregistrant des échantillons de couleur unique dans les deux canaux (Figure 1).

Figure 1 : Disposition optique pour multicolore suivi et localisation la microscopie (TALM) avec les émetteurs orange et rouges. (A) installation microscope inversé avec au moins deux lasers à excitation, un condenseur FRBR, un objectif approprié FRBR, un séparateur d’image et une caméra sensible. En médaillon : pour exciter des organites à l’intérieur des cellules, l’angle du faisceau incident doit être définie plus petit que l’angle critique pour FRBR atteindre fortement incliné et feuilleté illumination optique feuille (HILO). DC1 : Miroir dichroïque 1 ; DC2 : Miroir dichroïque 2. EF : le filtre d’émission. (B) essai sur la dérive optical Imaging postes d’une perle fluorescente pour 10 000 images avec la même cadence depuis les expériences suivantes (ici : 15 Hz). Postes connectés les 500 premiers trames et les 500 derniers montrent la dérive. En outre, une image fusionnée avec la position de la première et la dernière image en rouge et bleu montrent une dérive minimale. La dérive est que la distance entre le centre des signaux divisé par la durée totale d’enregistrement, ici 125 h/s. (C) vérification de la séparation claire des signaux, ici TMR et SiR. Pour les deux canaux, images de la somme cumulée de 3 000 cadres (TMR en canal 1) et SiR dans canal 2 ont été générés. SiRHTL était rattaché à Tom20-HaloTag et TMRHTL à OxPhos complexe V-HaloTag. Les couleurs sont fausses couleurs. Barreaux de l’échelle = 100 nm (B) et 1 µm (C). S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
-
Images générées par un alignement physique du répartiteur de l’image
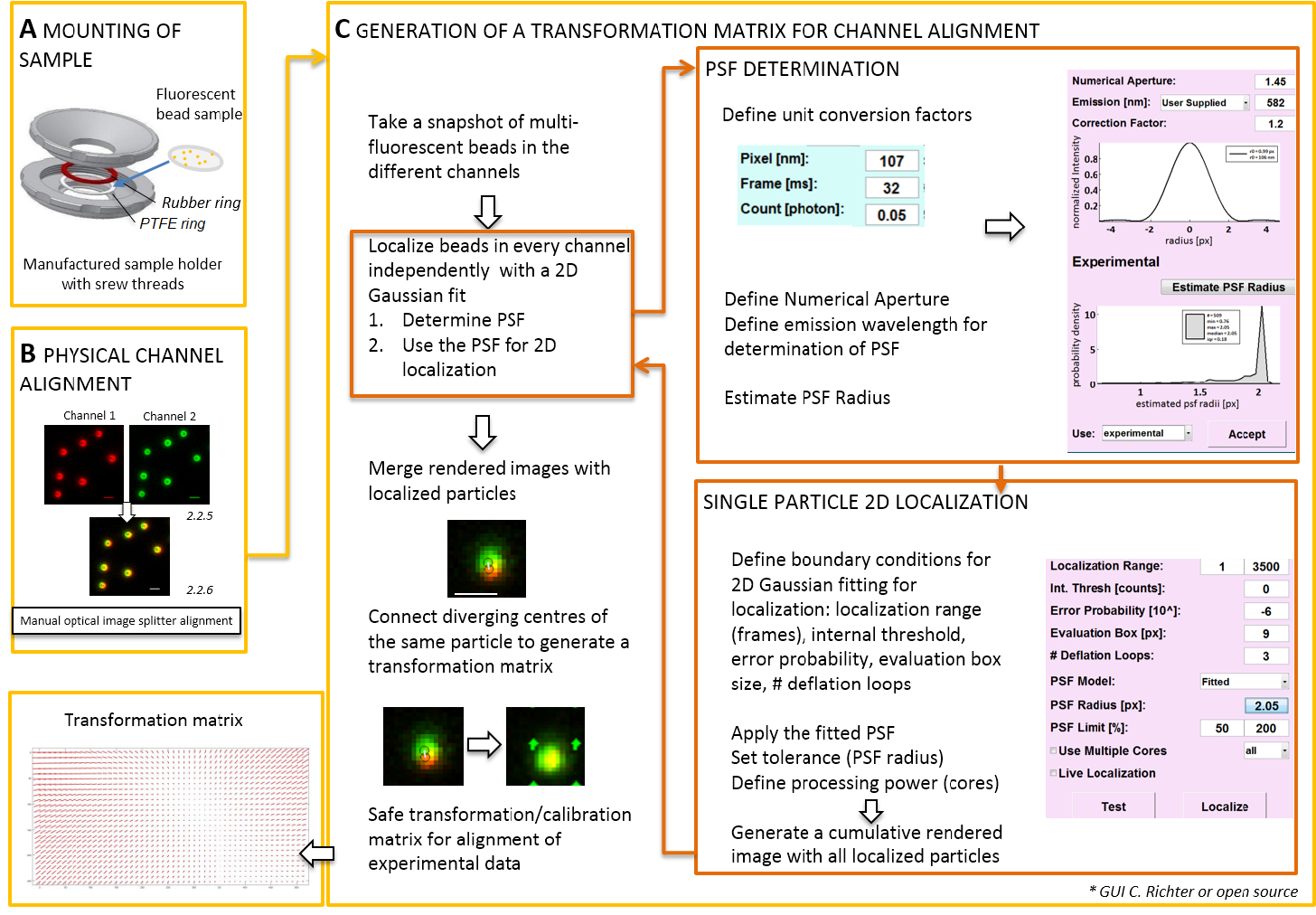
Remarque : Pour le montage de l’échantillon préparé sur un lamelle couvre-objet, un self-made porte-échantillon peut être utilisé (Figure 2 a). Pour éviter la poussière, etc. , tomber dans l’exemple, placez le couvercle de la boîte de Petri souplement sur le dessus de la chambre, lorsqu’il est monté. Le porte-échantillon même peut être utilisé pour monter la lamelle de perles fluorescentes ou de cellules ; Lorsque les cellules sont imagés, ajouter 0,5 à 0,8 mL de milieu d’imagerie. Le séparateur d’image divise l’image en deux ou plusieurs spectralement séparés des canaux et projette côte-à-côte sur le même appareil. Ce processus potentiellement introduit des distorsions systématiques entre les canaux en raison des chemins optiques distincts traversés et entrave l’analyse directe de colocalisation. Par conséquent, tout d’abord effectuer l’alignement physique et autre part, après correction de l’alignement avec une matrice de transformation. Pour les deux processus d’alignement, perles fluorescentes devraient être distribués homogène dans tout le champ de vision.- Monter l’échantillon préparé avec les perles fluorescentes dans le porte-échantillon entre le polytétrafluoroéthylène (PTFE)-anneau et l’anneau de caoutchouc rouge (Figure 2 a).
- Démarrer le microscope, tous les composants matériels et tous les logiciels nécessaires pour la microscopie.
- Nettoyez l’objectif et le fond de la lamelle avec une lingette de tissu non pelucheux imbibée d’alcool isopropylique. Puis séchez les deux éléments avec un tissu non pelucheux frais. Déposer une goutte d’huile à immersion sur la pupille de l’objectif.
- Placez le porte-échantillon avec l’échantillon de perle sur la platine du microscope afin que le fond de la lamelle entre en contact avec l’huile. Mettre l’accent sur des billes à l’aide de lumière de transmission ou une ligne laser.
- Régler la puissance des deux lasers excitation pour atteindre l’intensité du signal similaire dans les deux canaux de fluorescence. Recherche d’un lieu avec plusieurs signaux fluorescents distinctes.
- Générer un affichage fusionné des canaux fluorescence en utilisant le logiciel de contrôle de la caméra. Puis utilisez les vis dans le séparateur d’image pour basculer manuellement les miroirs internes entre le séparateur et image pour atteindre la meilleure superposition des signaux des deux canaux fluorescents (Figure 2 b).
Remarque : Attention ! Ne pas dépasser la plage dynamique de la caméra.
-
Alignement des canaux spectralement séparés par le logiciel en exécutant une transformation spatiale
Remarque : La partie suivante montre la correction après l’alignement et la procédure de localisation avec notre logiciel plugin (disponible sur demande).- Démarrez le microscope TIRF contrôlant logiciel et choisir d’afficher les canaux individuels dans le mode de flux en direct. Prendre une fluorescence excitant image de capture instantanée dans tous les canaux (Figure 2).
- Cette image de capture instantanée permet de produire la matrice de transformation (voir la Figure 2).
Remarque : La matrice de transformation est utilisée pour une transformation spatiale, typiquement un affine, qui corrige la traduction (divergence des signaux d’une source ponctuelle unique entre deux canaux). - Démarrez le logiciel analyse plugin (peuvent être obtenus sur demande auprès de notre laboratoire, voir Figure 2).
- Charger les images préalablement enregistrées bicolore (Voir l’étape 2.2) de perles fluorescentes dans le logiciel. Choisissez l’orientation utilisée des canaux fluorescents. Ensuite cliquez sur « Oui » quand on vous demande « calibrer images » et sélectionnez l’instantané pris précédemment.
- Ouvrir le « gestionnaire d’unité » pour définir les facteurs de conversion d’unité (taille du pixel, cadence, facteur de conversion photon).
- Ouvrir le « gestionnaire de localisation ». Déterminer que le point spread function (PSF) tout d’abord. Appuyez sur le bouton : « Rayon de PSF ». Dans la fenêtre « PSF estimateur » qui s’ouvre, définissez l’ouverture numérique et l’émission maximale. Démarrer « estimation PSF rayon » en cliquant sur. Accepter l’obtenu expérimental polyesters. définir la zone d’évaluation, nombre de boucles de déflation, et le nombre de noyaux de l’ordinateur est utilisé pour le calcul. Appuyez sur « localiser » pour commencer le montage de la distribution de l’intensité des particules unique par une fonction 2D de Gauss symétrique (Figure 2).
- « Accepter » l’obtenu expérimental polyesters. définir la zone d’évaluation, nombre de boucles de déflation, et le nombre de noyaux de l’ordinateur est utilisé pour le calcul. Appuyez sur « localiser » pour commencer le montage de la distribution de l’intensité des particules unique par une fonction 2D de Gauss symétrique (Figure 2).
- Ouvrir le « gestionnaire de calibrage ». Rendu de l’image fusionné des deux canaux, les signaux originaux et les centres localisés sont divulgués. Choisissez le mode « affine ». Connecter manuellement les paires correspondantes de centres localisés dans les deux chenaux qui proviennent de la même bille fluorescente en traçant une ligne de connexion.
- Connecter les signaux correspondants répartis partout dans le champ de vision. Après cela, appuyez sur « accepter ». Enregistrez l’étalonnage.
Remarque : La transformation spatiale est échantillonnée à chaque perle fluorescente et interpolée entre les deux. La fonction de transformation extrait représente une Δr(x,y) de champ de déplacement qui sert à corriger par la suite les localisations expérimental bicolores seule molécule afin qu’ils se superposer la précision de leur localisation. La matrice de transformation spatiale est typiquement un affine qui corrige la traduction, mise à l’échelle et la rotation entre les canaux avec une précision nanométrique, et il peut être déduit de ce mappage un à un manuel (Figure 2).

Figure 2 : "Workflow" pour alignement bicolore. (A), la lamelle avec les perles fluorescentes est monté dans un porte-échantillon entre un PTFE et un anneau en caoutchouc. Puis la partie supérieure et inférieure de la chambre sont boulonnées ensemble. (B) l’alignement physique des vues canal qui sont générés par le séparateur d’image. Enregistré les signaux fluorescents de perles (0,1 µm) à deux canaux (vert et rouge, fausses couleurs) sont fusionnées. Les vis correspondantes sur le répartiteur optique de l’image sont activés manuellement jusqu'à atteindre la meilleure superposition des signaux différents (couleur jaune, panneau inférieur). (C) de génération d’une matrice de transformation pour l’alignement de canaux post-processive. Pour la localisation précise d’une particule, il est nécessaire de déterminer que le point spread function (PSF) en fonction de la longueur d’onde d’émission et de l’ouverture numérique de l’objectif. Le centre d’un PSF peut être déterminé par son profil d’intensité analysé par une gaussienne bidimensionnelle symétrique s’adapter. La localisation résultante de la crête du signal est ensuite projetée sur les signaux originaux, floues. Dans une image fusionnée, les centres localisés des signaux des deux canaux sont connectés pour générer une matrice de transformation qui sera utilisée ultérieurement pour l’alignement post-processive des données expérimentales. Barreaux de l’échelle = 1 µm (B, C). S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
-
Imagerie de la seule molécule des protéines de la membrane mitochondriale
Remarque : Toutes les expériences sont réalisées à température ambiante. T-cellules ou les cellules non adhérentes doivent être immobilisés dans l’agarose avant imaging31.- Monter le spécimen avec cellules adhérentes sur la lamelle entre le caoutchouc et les anneaux PTFE (Figure 3 a). Remplir la chambre de 0,5 à 0,8 mL moyen d’imagerie.
- Répétez les étapes 2.2.2–2.2.5.
- Régler le microscope de la FRBR angle éclairage contrôle logiciel pour créer un angle d’incidence inférieur à l’angle critique pour le mode TIR exciter la région spécifique d’intérêt via une feuille de HILO32 (mode de HILO, Figure 1 a).
ATTENTION : Évitez le contact visuel direct avec le faisceau laser ! - Réglez le gain de l’EM et choisir un temps de pose approprié pour l’expérience qui recueille suffisamment photons par image.
- Régler la puissance du laser pour atteindre un haut rapport signal sur bruit (S/N) (Figure 3 b), étant donné que la précision de localisation correspond directement au S/N33 (Figure 3).
- Trouver un terrain à la périphérie de la cellule avec les mitochondries allongées, sans chevauchement et des signaux de molécule unique (Figure 3D; Vidéo complémentaire 1). Si aucun signal de la seule molécule n’est visibles, attendre jusqu'à ce que le blanchiment fait apparaître des signaux de molécules simples (Figure 3E).
- Enregistrer jusqu'à ce que le nombre de signaux est trop faible pour poursuite raisonnable (généralement 1 000 – 10 000 trames selon le comportement de blanchiment du colorant fluorescent, Figure 3F).
- Commencer l’imagerie traitement logiciel et vérifier les structures mitochondriales en générant une image somme rendu cumulée d’au moins 1 000 images enregistrées (Figure 3).
NOTE : La plus rapide possible de la cadence est dictée par la zone de lecture. Le champ de vision pour un seul canal est réduit par un splitter image bicolores (512 x 512 pixels) à 256 x 512 pixels et d’une couleur quadri à 256 x 256 pixels. Ainsi, pour utiliser un séparateur d’image pour deux couleurs, c’est 30 Hz. Réglez le mode de transfert de trame pour atteindre le temps d’initialisation possible le plus bas. - Démarrez le logiciel analyse plugin et charge des données brutes. Sélectionnez les images de l’orientation et la charge du canal. Utilisez la matrice de transformation de l’étape 2.3.9 interrogé pour « Calibrer des images ». Canaux s’afficheront séparément.
- Ouvrir le « gestionnaire de l’unité » comme avant pour définir les facteurs de conversion d’unité pour chaque canal. Ouvrez le gestionnaire de « localisation » pour chaque canal. Définir la zone d’évaluation, nombre de boucles de déflation, ajoutez ensuite la FSP théorique pour les conditions utilisées et le nombre de noyaux de l’ordinateur est utilisé pour le calcul de la valeur. Enfin, appuyez sur « localiser » pour obtenir des particules unique localisées (Figure 3 H; Vidéo supplémentaire 2).
- Note que le programme va générer enfin une image superrésolution cumulatif montrant tous localisés des particules (Figure 3I).
- Effectuer une analyse, par exemple, de logiciels open source ou notre logiciel disponible sur demande.
- Suivre les molécules simples dans les deux voies localisées, par exemple, avec le traceur multicible10
NOTE : Étape 2.4.13 doit préliminaire (expérimental) connaître la diffusibilité des protéines d’intérêt est correctement réglé les conditions aux limites. Trouver les bonnes conditions de limite est généralement un processus itératif.

Figure 3 : Étapes au cours de la microscopie de localisation unique molécule. (A) A lamelle avec le spécimen est monté entre la partie supérieure et inférieure (gris) du porte-échantillon fait maison (conçu par J. Bereiter-Hahn). Un anneau de caoutchouc (rouge) et un anneau PTFE (blanc) sceller le système au-dessus et en dessous de la lamelle couvre-objet, lorsque les porte-échantillon des pièces sont boulon ensemble. (B) Rapport Signal sur bruit du signal TMR. (C) calculé histogramme de précision de localisation pour les particules toutes localisées. (D) choix d’une région raisonnable d’imagerie, ici, de la périphérie de la cellule avec les mitochondries clairement séparés. (E) enregistrement et traitement d’image : une seule image avec signaux distincts seule molécule est indiquée (ici, les molécules simples de CV-HaloTag/TMRHTL ont été enregistrés). Intensité de TMR (F) sur la durée d’enregistrement. Image de somme cumulée (G) de 3 000 cadres, non transformés. (H) des particules de CV-HaloTag/TMRHTL localisée avec une fonction gaussienne 2D d’une seule image. (j’ai) Cumulative, rendu l’image somme de toutes les particules de CV-HaloTag/TMRHTL localisées de 3 000 cadres. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
Résultats
Analyse de l’imagerie et la colocalisation multicolore peut aider à déterminer la localisation sup-organites des protéines. Nous l’a démontré plus haut avec la phosphatase cytosolique et tensine homologue, PINK1, qui a différents endroits sub-mitochondriale en raison de son traitement par les protéases mitochondrial17. PINK1 est un facteur important de garantir la fonctionnalité mitochondriale34,35
Discussion
Ici, une technique pour la localisation seule molécule bicolore des protéines membranaires mobile a été présentée. Suivant le protocole, les protéines membranaires sont fondues à étiquetage automatique des protéines qui réagissent avec les colorants rhodamine TMR et SiR conjugué à leurs substrats respectifs. Rhodamine dyes sont lumineuse et photostable et permettent ainsi répétitifs d’imagerie1. Pour une performance réussie, plusieurs conditions et des sujets critiques doivent ga...
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Les auteurs aimeraient remercier le groupe de biophysique et Jacob Piehler à l’Université d’Osnabrück pour un soutien continu, Wladislaw Kohl pour assistance technique et la préparation du matériel et le Conseil de CellNanOs pour fournir des microscopes pour utilisation. Le projet a été financé par la 944 SFB.
matériels
| Name | Company | Catalog Number | Comments |
| (2-(4-(2-hydroxyethyl)-1-piperazinyl)-ethanesulfonic acid, 1 M) (HEPES) | Biochrom | #1104E | |
| DC1: Dichroid beam splitter | Chroma | 640 dcxr | NC506031 |
| DC2: Polychroic Mirror, beamsplitter | Chroma | zt405/488/561/640rpc | discontinued |
| Dulbecco´s Phosphate-Buffered Saline (PBS) 1x (w/o Ca & Mg) | Sigma-Aldrich & Co. | #RNBF8311 | |
| Earle´s MEM without phenol red, without L-Glutamine and without NaHCO3 containing 1% FBS, 0.1% HEPES, 0.1% NEAA, 0.1% Alanyl-L-Glutamine and 34.78% sodium hydrogen carbonate (NaHCO3 0.75g/l) | Imaging medium | ||
| Earle´s minimum essential medium (MEM) with phenol red, containing 1% Fetal Bovine Serum Superior (FBS), 0.1% HEPES (2-(4-(2-hydroxyethyl)-1-piperazinyl)-ethanesulfonic acid, 1 M), and 0.1% non-essential amino acids (NEAA) | Growth medium | ||
| EF: Emission filter quadbandpass | AHF analysentechnik | F72-866 | Brightline HC 446 nm/523 nm/600 nm/677 nm |
| EMCCD camera | Andor | Andor iXON 897 | EMCCD camera |
| Emission filter QuadView filter cubes, orange | AHF analysentechnik | F39-637 | bandpass 582 - 619 nm |
| Emission filter QuadView filter cubes, red | Chroma | bandpass 655 - 725 nm (HQ 690/70) | |
| FBS (Fetal bovine serum) superior | Biochrom | S0615 | |
| Fluorescent beads: TetraSpeck™ Microspheres, 0.1 µm, fluorescent blue/green/orange/dark red | Thermo Fisher Scientific | T7279 | fluorescent microspheres |
| Glutamine | Biochrom | #0951C | |
| HeLa cells | DSMZ | ACC-57 | Cervical carcinoma cells from patient Henrietta Lacks |
| Hela cells CI::paGFP, stable | Muster et al., PLOSOne 2010 | ||
| Hela cells CV g::Halo7-Tag, stable | Appelhans et al., NanoLett 2012 | ||
| Hela cells Tom20::Halo7-Tag, stable | Appelhans et al., NanoLett 2012 | ||
| Image splitter | Photometrics | Dual-View QV2 | image splitter emission |
| Imaging processing software | ImageJ2 / Fiji | freeware | |
| Immersion Oil - ImmersolTM 518 F (ne = 1.518, ve = 45) | Carl Zeiss Jena GmbH | 444960-0000-000 | |
| Inverted epifluorescence microscope | Olympus IX-71/73/83 | ||
| Laser 561 nm, 200 mW | CrystaLaser | CL-561-200 | 561 nm emission |
| Laser 642 nm, 140 mW | Omicron | Luxx-642-140 | 642 nm emission |
| MATLAB | MathWorks | version R2013a | |
| MEM with Earle's Balanced Salt Solution 2.2 g/L NaHCO3, stable glutamine w/o PR | Biochrom | FG-0385 | |
| MEM with Earle's Balanced Salt Solution with 2.2 g/L NaHCO3, stable glutamine, Phenolred | Biochrom | FG-0325 | |
| MitoTracker® Deep Red FM | Thermo Fisher Scientific | M22426 | dye |
| MitoTracker® Green FM | Thermo Fisher Scientific | M7514 | dye |
| Multi-mode-optical polarization maintaining monomode fiber | Pointsource/Qioptiq | KineFLEX | |
| NHS-PEG-MAL, Rapp Polymer | Rapp Polymere GmbH Tübingen | coverslip coating | |
| non-essential amino acids (NEAA) | Biochrom | #0802E | |
| PEG 800 (Polyethylene glycol) 10 % | Carl Roth GmbH | Art No. 0263.1 | coverslip coating |
| Penicillin/Streptomycin | Biochrom | #0122E | |
| Plasmid for PINK1-Halo7-Tag expression | Beinlich et al., ACS Chemical Biology 2015 | ||
| Poly-L-lysine (1.2 mg/ml) | Sigma-Aldrich & Co. | Cat. No.P9155 | coverslip coating |
| RGD Peptide (Ac-CGRGDS-COOH) | Coring System Diagnostix GmbH, Gernsheim | coverslip coating / Intergrin receptor motif | |
| Silicon Rhodamine linked to HaloTag®-Ligand (SiRHTL) | personal gift from Kai Johnson | dye | |
| Software analysis plugin | self-written C. P. Richter, Biophysik Osnabrück | SLIMFAST 16g | |
| Tetramethylrhodamine / SNAP-Cell® TMR-Star linked to SNAP-Ligand (TMRstar) | New England Biolab® | S9105S | dye |
| Tetramethylrhodamine linked to HaloTag®-Ligand (TMRHTL) | Promega | G8251 | dye |
| TIRF condensor | Olympus | Cell^TIRF MITICO System | TIRF condensor |
| TIRF microscope controlling software | Olympus cellSens 1.12 | ||
| TIRF objective | Olympus | 150x oil objective (N.A. 1.45; Olympus UAPO) | |
| Trypsin/EDTA 10x | Biochrom | #0266 | |
| Water H2O 99,5 % Rotipuran® Low organic | Carl Roth GmbH | Art. No. HN57.1 |
Références
- Appelhans, T., Richter, C., Wilkens, V., Hess, S., Piehler, J., Busch, K. Nanoscale organization of mitochondrial microcompartments revealed by combining tracking and localization microscopy. Nano Letters. 12 (2), 610-616 (2012).
- Appelhans, T., Busch, K. Single Molecule Tracking and Localization of Mitochondrial Protein Complexes in Live Cells. Methods Mol Biol. 1567, 273-291 (2017).
- Rust, M. J., Bates, M., Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods. 3 (10), 793-795 (2006).
- Gould, T. J., Verkhusha, V. V., Hess, S. T. Imaging biological structures with fluorescence photoactivation localization microscopy. Nat Protoc. 4 (3), 291-308 (2009).
- Pennacchietti, F., Gould, T. J., Hess, S. T. The Role of Probe Photophysics in Localization-Based Superresolution Microscopy. Biophys J. 113 (9), 2037-2054 (2017).
- Wegel, E., Göhler, A., Lagerholm, B. C., Wainman, A. Imaging cellular structures in super-resolution with SIM, STED and Localisation Microscopy: A practical comparison. Scientific reports. , (2016).
- Pellett, P., et al. Two-color STED microscopy in living cells. Biomedical Optics Express. 2 (8), (2011).
- Ishigaki, M., et al. STED super-resolution imaging of mitochondria labeled with TMRM in living cells. Mitochondrion. 28, 79 (2016).
- Liss, V., Barlag, B., Nietschke, M., Hensel, M. Self-labelling enzymes as universal tags for fluorescence microscopy, super-resolution microscopy and electron microscopy. Scientific Reports. 5, 17740 (2015).
- Sergé, A., Bertaux, N., Rigneault, H., Marguet, D. Dynamic multiple-target tracing to probe spatiotemporal cartography of cell membranes. Nat Methods. 5 (8), (2008).
- Appelhans, T., Busch, K. B. Dynamic imaging of mitochondrial membrane proteins in specific sub-organelle membrane locations. Biophysical reviews. 9 (4), 345-352 (2017).
- Wilmes, S., et al. Triple-color super-resolution imaging of live cells: resolving submicroscopic receptor organization in the plasma membrane. Angewandte Chemie. 51 (20), 4868-4871 (2012).
- Niewidok, B., et al. Single-molecule imaging reveals dynamic biphasic partition of RNA-binding proteins in stress granules. J Cell Biol. , (2018).
- Wurm, C. A., Jakobs, S. Differential protein distributions define two sub-compartments of the mitochondrial inner membrane in yeast. FEBS Lett. 580 (24), 5628-5634 (2006).
- Schmidt, R., Wurm, C. A., Punge, A., Egner, A., Jakobs, S., Hell, S. W. Mitochondrial cristae revealed with focused light. Nano Lett. 9 (6), 2508-2510 (2009).
- Kukat, C., Wurm, C. A., Spahr, H., Falkenberg, M., Larsson, N. G., Jakobs, S. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proceedings of the National Academy of Sciences of the United States of America. 108 (33), 13534-13539 (2011).
- Beinlich, F., Drees, C., Piehler, J., Busch, K. Shuttling of PINK1 between Mitochondrial Microcompartments Resolved by Triple-Color Superresolution Microscopy. ACS chemical biology. 10 (9), 1970-1976 (2015).
- Shim, S., et al. Super-resolution fluorescence imaging of organelles in live cells with photoswitchable membrane probes. Proceedings of the National Academy of Sciences. 109 (35), 13978-13983 (2012).
- Sbalzarini, I., Mezzacasa, A., Helenius, A., Koumoutsakos, P. Effects of Organelle Shape on Fluorescence Recovery after Photobleaching. Biophysical Journal. 89 (3), 1482-1492 (2005).
- Reits, E., Neefjes, J. From fixed to FRAP: measuring protein mobility and activity in living cells. Nature Cell Biology. 3 (6), E145-E147 (2001).
- Goehring, N., Chowdhury, D., Hyman, A., Grill, S. FRAP Analysis of Membrane-Associated Proteins: Lateral Diffusion and Membrane-Cytoplasmic Exchange. Biophysical Journal. 99 (8), 2443-2452 (2010).
- Bacia, K., Haustein, E., Schwille, P. Fluorescence correlation spectroscopy: principles and applications. Cold Spring Harbor protocols. 2014 (7), 709-725 (2014).
- Sukhorukov, V., Dikov, D., Busch, K., Strecker, V., Wittig, I., Bereiter-Hahn, J. Determination of protein mobility in mitochondrial membranes of living cells. Biochimica et biophysica acta. 1798 (11), 2022-2032 (2010).
- Kim, T. K., Eberwine, J. H. Mammalian cell transfection: the present and the future. Anal Bioanal Chem. 397 (8), 3173-3178 (2010).
- Graham, F. L., van der Eb, A. J. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology. 52 (2), 456-467 (1973).
- Wedeking, T., et al. Spatiotemporally Controlled Reorganization of Signaling Complexes in the Plasma Membrane of Living Cells. Small. 11 (44), 5912-5918 (2015).
- Mironov, S. L., Ivannikov, M. V., Johansson, M. [Ca2+]i signaling between mitochondria and endoplasmic reticulum in neurons is regulated by microtubules. From mitochondrial permeability transition pore to Ca2+-induced Ca2+ release. J Biol Chem. 280 (1), 715-721 (2005).
- Poot, M., et al. Analysis of mitochondrial morphology and function with novel fixable fluorescent stains. J Histochem Cytochem. 44 (12), 1363-1372 (1996).
- Tokunaga, M., Imamoto, N., Sakata-Sogawa, K. Highly inclined thin illumination enables clear single-molecule imaging in cells (vol 5, pg 159). Nature Methods. 5 (5), 455 (2008).
- Elmokadem, A., Yu, J. Optimal Drift Correction for Superresolution Localization Microscopy with Bayesian Inference. Biophys J. 109 (9), 1772-1780 (2015).
- Barlag, B., et al. Single molecule super-resolution imaging of proteins in living Salmonella enterica using self-labelling enzymes. Sci Rep. 6, 31601 (2016).
- Tokunaga, M., Imamoto, N., Sakata-Sogawa, K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nature Methods. 5 (2), 159-161 (2008).
- Mortensen, K. I., Churchman, L. S., Spudich, J. A., Flyvbjerg, H. Optimized localization analysis for single-molecule tracking and super-resolution microscopy. Nat Methods. 7 (5), 377-381 (2010).
- Jin, S., Youle, R. J. PINK1- and Parkin-Mediated Mitophagy at a Glance. Journal of Cell Science. 125, 795-799 (2013).
- Yamano, K., Youle, R. J. PINK1 is degraded through the N-end rule pathway. Autophagy. 9 (11), 1758-1758 (2013).
- Wiedemann, N., et al. Machinery for protein sorting and assembly in the mitochondrial outer membrane. Nature. 424 (6948), 565-571 (2003).
- Thompson, R., Larson, D., Webb, W. Precise Nanometer Localization Analysis for Individual Fluorescent Probes. Biophysical Journal. 82 (5), 2775-2783 (2002).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.