
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Microscopia di multi-colore localizzazione delle proteine di membrana singola in organelli delle cellule di mammiferi dal vivo
In questo articolo
Riepilogo
Qui, presentiamo un protocollo per la localizzazione di multi-colore delle proteine di membrana singola in organelli delle cellule vive. Per allegare fluorofori, proteine self-etichettatura sono usate. Proteine, trova in scomparti di diverse membrane dell'organello stesso, possono essere localizzati con una precisione di ~ 18 nm.
Abstract
Conoscenza circa la localizzazione delle proteine in subcomparti cellulare è fondamentale per capire la loro funzione specifica. Qui, presentiamo una tecnica di Super-risoluzione che permette per la determinazione del microcompartments che sono accessibili per le proteine generando localizzazione e tracciamento di mappe di queste proteine. Inoltre, da microscopia localizzazione multi-colore, la localizzazione e tracciamento profili delle proteine in diversi subcomparti sono ottenuti simultaneamente. La tecnica è specifica per le cellule vive e si basa sull'imaging ripetitivo delle proteine di membrana mobile singolo. Le proteine di interesse sono geneticamente fuso con specifici, i cosiddetti self-etichettatura tag. Questi tag sono enzimi che reagiscono con un substrato in modo covalente. Coniugato di questi substrati sono coloranti fluorescenti. Reazione delle proteine enzima-etichetta con la fluorescenza etichettato risultati di substrati in proteine con etichettate. Qui, tetrametilrodamina (TMR) e silicio rodamina (SiR) vengono utilizzati come coloranti fluorescenti attaccati ai substrati degli enzimi. Utilizzando concentrazioni di substrato nel pM a Nm, Sub-stechiometrico etichettatura è raggiunto che si traduce in segnali distinti. Questi segnali sono localizzati con ~ 15 – 27 nm precisione. La tecnica permette per l'imaging multi-colore di singole molecole, per cui il numero di colori è limitato dalla disponibili coloranti membrana permeabile e il repertorio di enzimi con etichetta automatica. Per determinare la localizzazione dell'enzima di controllo di qualità (Pten) mostreremo la fattibilità della tecnica-indotta della chinasi 1 (PINK1) in diversi compartimenti mitocondriali durante l'elaborazione in relazione alle altre proteine di membrana. Il test per vere interazioni fisiche tra proteine diversamente con etichetta singole di singola molecola FRET o co-rilevamento è limitato, però, perché i gradi d'etichettatura Bassi diminuiscono la probabilità di avere due proteine adiacenti con l'etichetta allo stesso tempo. Mentre la tecnica è forte per l'imaging di proteine nei compartimenti di membrana, nella maggior parte dei casi non è opportuno determinare la localizzazione delle proteine solubili altamente mobile.
Introduzione
L'obiettivo del presente protocollo è quello di fornire un metodo di imaging per localizzare e tenere traccia di proteine di membrana singola all'interno di cellule vive. Noi chiamiamo questo metodo di rilevamento e localizzazione microscopia (TALM)1,2. Come microscopia di ricostruzione ottica stocastica (tempesta)3 e microscopia a fluorescenza fotoattivazione localizzazione ((F) PALM)4,5, TALM è una tecnica di localizzazione singola molecola-basato fluorescenza. Tuttavia, è distinto in modo che la mobilità delle proteine di membrana in combinazione con formazione immagine ripetitiva dello stesso etichettato molecola alle diverse posizioni rivela la microcompartment che è accessibile per la proteina mobile. In altre parole, le possibili localizzazioni della proteina sono impostate dall'architettura dell'organello e dalla mobilità della proteina1. Il metodo è complementare a vari altri super-risoluzione tecniche6,7,8 , perché rivela la localizzazione e la traiettoria mappe dalle proteine mobile imaging. L'etichettatura è basato sull'utilizzo di proteine di fusione geneticamente ingegnerizzati che sono di per sé non fluorescenti. Queste proteine di fusione sono enzimi che reagiscono in modo covalente con un substrato coniugato a un colorante con etichetta automatica. Questa procedura ha il vantaggio che il grado d'etichettatura può essere controllata dalla quantità di substrato aggiunto. Inoltre, permette di variare il colore della fluorescenza, a seconda il colorante coniugato selezionato. Diversi auto-etichettatura degli enzimi-tag sono disponibili9. Un altro vantaggio dell'utilizzo di self-etichettatura degli enzimi-tag è, che le tinture coniugate sono solitamente più stabile e più proteine fluorescenti1 e singole proteine pertanto possono essere registrati più brillante e più precisamente fino a quando essi vengono sbiancati. Questo consente la registrazione delle traiettorie delle proteine cellulari e l'estrazione di diffusione coefficienti10,11.
Qui, noi dimostrare la fattibilità di TALM con proteine di membrana mitocondriale, ma può essere applicato anche per altre proteine di membrana intra - ed extracellulari, compresi tipi differenti delle cellule12,13. Mostriamo che multi-colore TALM ulteriormente consente la distinzione simultanea delle proteine in diversi subcomparti a complementazione a esistente super-risoluzione fluorescenza microscopia tecniche14,15, 16. TALM è compatibile con live cell imaging17. La foto-fisica del rhodamines selezionate tetrametilrodamina (TMR) e silicio-Rhodamien (SiR), in particolare loro luminosità e stabilità, permette di proteine di membrana singolo record in più fotogrammi fornendo mappe di localizzazione (e traiettoria). Tuttavia, TALM è limitata per la localizzazione delle proteine solubili con i coefficenti di diffusione alta poiché il motion blur è troppo alto e i fotoni raccolti per ogni frame sono troppo bassi per la corretta localizzazione. Inoltre, TALM richiede meno energia di eccitazione di ad esempio tempesta o svuotamento di emissione stimolata (STED) microscopia6,7, la riduzione di effetti fototossici. Questo è importante, poiché lo stress fototossico colpisce spesso organellari morfologia18 e così mobilità analisi19. In somma, presentiamo TALM multicolore in cellule viventi come una tecnica che colma una lacuna tra i metodi di microscopia di localizzazione STORM/STED/PALM (F) e le tecniche che analizzano la mobilità di proteine come il recupero di fluorescenza dopo photobleaching (FRAP)20 ,21, fluorescenza correlazione spettroscopia (FCS)22e fluorescenza cross correlazione spettroscopia (FCC)11,23.
Protocollo
Il seguente protocollo segue le linee guida del comitato etico di ricerca istituzione locale.
1. metodi
-
Coltura cellulare
- Coltivare le cellule, per esempio le cellule HeLa (carcinoma della cervice umana), in un matraccio di cultura cellulare T25 contenente 5 mL di medium di crescita a 37 ° C e 5% CO2.
Nota: Per l'imaging, dividere le celle su preparati vetrini coprioggetti (Vedi punti 1.3 e 1.4) e tenere in mezzo di imaging.
- Coltivare le cellule, per esempio le cellule HeLa (carcinoma della cervice umana), in un matraccio di cultura cellulare T25 contenente 5 mL di medium di crescita a 37 ° C e 5% CO2.
-
Transfezione delle cellule
Nota: Utilizzare linee cellulari che esprimono stabilmente le proteine etichettate quando possibile24 per evitare forte sovraespressione. Per trasfezione transiente, adattare l'importo utilizzato per la transfezione del DNA del plasmide. Ad esempio, quando viene utilizzato il Ca2 + fosfato transfezione25 , transfect cellule (confluency di 80 – 90%) in una piastra di coltura cellulare di 3,5 cm con 2,5-5 µ g di DNA plasmidico. Quando si esegue la transfezione doppia, utilizzare 2,5 µ g per ogni costrutto di plasmide.- Per gli esperimenti di doppio colore, utilizzare una linea cellulare con espressione stabile di una proteina self-etichettatura e transitoriamente transfect con il plasmide che codifica l'altre proteine self-etichettatura17.
Nota: Qui, per esperimenti di doppio colore, cellule HeLa sono state usate che esprime stabilmente le proteine self-etichettatura PINK1-Halo-Tag e Tom20-fSNAP-Tag.
- Per gli esperimenti di doppio colore, utilizzare una linea cellulare con espressione stabile di una proteina self-etichettatura e transitoriamente transfect con il plasmide che codifica l'altre proteine self-etichettatura17.
-
Pulizia dei vetrini coprioggetti
- Posizionare i vetrini coprioggetti in un becher. Aggiungere 30 mL di H2O nel becher contenente i coprioggetti e agitare delicatamente per rimuovere la polvere dalla loro superficie.
- Raccogliere i coprioggetti con pinzette e asciugarle con un flusso di azoto.
- Rimuovere qualsiasi contaminazione organica sulla superficie delle lamelle, per esempio, dalla pulizia al plasma.
Nota: Per evitare ulteriori contaminazioni del materiale vetro, indossare i guanti durante la manipolazione di lamelle.
Attenzione: Quando le lamelle sono pulite di pulizia al plasma, solo il lato superiore delle lamelle è pulito; utilizzare questo lato per rivestimento con poli-L-lisina-polietilene glicole-arginina-glicina-aspartato (PLL-PEG-RGD) (sezione 1.4) e semina (punto 1.5) cellulare.
-
Vetrino coprioggetti rivestimento con PLL-PEG-RGD
Nota: PLL-PEG-RGD è un derivato di poli-L-lisina (PLL) collegato con un polietilene glicole (Da 3.000) ed un peptide (CGRGDS) cysteine-glycine-arginine-glycine-aspartate-serine. PLL si lega alla superficie del vetro caricato negativamente e forma un pennello di PEG. Questo riduce drasticamente il legame aspecifici di caricate coloranti fluorescenti. Inoltre, il motivo RGD imita il peptide di segnale del recettore integrina e promuove l'adesione integrina-mediata delle cellule che altrimenti non facilmente aderire in.- Preparare PLL-PEG-RGD come descritto in precedenza26. In breve, sciogliere 0,8 mg di PLL-PEG-RGD in 1 mL di PBS. Aggiungere 10 µ l della soluzione PLL-PEG-RGD sul lato superiore di un vetrino coprioggetto pulito.
- Prendere un secondo vetrino coprioggetto e posizionarla con la sua superficie pulita upside-down sul primo vetrino coprioggetti (che ha la goccia di PLL-PEG-RGD sulla parte superiore); in questo modo che racchiude la soluzione PLL-PEG-RGD tra due vetrini coprioggetti.
- Posizionare i vetrini coprioggetti sandwich in un becher e incubare per 1 h a temperatura ambiente in un ambiente privo di polvere secco.
- Dopo 1 h, aggiungere 30 mL di H2O Becher per coprire integralmente i coprioggetti con acqua.
- Agitare delicatamente il bicchiere fino a lamelle staccano da altro.
- Utilizzare pinzette per raccogliere i coprioggetti fuori dall'acqua e asciugarli sotto un getto di gas azoto.
Nota: Lamelle rivestite possono essere memorizzate in un bicchiere asciutto, sterile capsula di Petri con coperchio per un paio di giorni.
-
Preparazione del campione per l'imaging
- Trasferire singole lamelle rivestite in una piastra di coltura cellulare di 35 mm, con la superficie ricoperta di PLL-PEG-RGD rivolta verso l'alto e aggiungere 2 mL di imaging medio sulla parte superiore.
- Aggiungi ~ 500.000 cellule tripsinizzate (200 – 500 µ l) che esprimono i tag auto-etichettatura presso le proteine di membrana rispettivi a 2 mL medio nella piastra di coltura delle cellule con il coprioggetto rivestito di imaging. Agitare delicatamente a mano per garantire una distribuzione omogenea delle cellule per ottenere un uniforme strato di cellule.
- Incubare le cellule a 37 ° C e 5% CO2 sino al confluency di 80%.
Nota: I campioni delle cellule dovrebbero essere seminati 3 giorni prima di formazione immagine e 1 giorno prima della trasfezione. Cellule, che esprimono stabilmente la proteina di interesse, possono essere seminate 2 giorni prima di formazione immagine. Più tardi, solo le cellule coltivate sul coprioggetto sono Imaging.
-
Etichettatura di proteine etichettate
Nota: Substrati più fluorescente devono essere sciolto in DMSO privo di acqua. Si consiglia di utilizzare soluzioni di riserva di substrato fluorescente di 1 µM, quando la concentrazione di etichettatura finale è di 0,2 – 30 nM17. Per l'imaging di proteine di membrana all'interno delle cellule, utilizzare substrati fluorescenti permeabile di membrana.- Riscaldare a 37 ° C in un bagno d'acqua medio imaging.
- Pipettare 1 mL di terreno imaging pre-riscaldato in una provetta da 2 mL con coperchio. Aggiungere 0,2 – 30 µ l di substrati fluorescenti dalle soluzioni di riserva di 1 µM per preparare la soluzione di etichettatura finale (concentrazione finale: 0,2 – 30 nM).
- Vortex la soluzione di etichettatura per 10 s.
- Sostituire il mezzo nella piastra di coltura di 35mm con le cellule su un vetrino coprioggetti (Vedi punto 1.5) da 1 mL di preparato soluzione di etichettatura.
- Incubare le cellule nella soluzione di etichettatura a 37 ° C e 5% di CO2 per 20 – 30 min.
- Lavare le cellule con 2 mL di PBS una volta, poi con 2 mL di mezzo di imaging due volte. Infine, Pipettare 1 mL di terreno nuovo imaging per il piatto di cella e rimettere il campione in incubatore a 37 ° C e 5% CO2 per almeno 1 h. Prima di formazione immagine, scambiare ancora una volta il mezzo di imaging.
Nota: Quando si esegue l'esperimento per la prima volta, confermare la corretta destinazione di self-etichettate proteine alle membrane organellari macchiando gli organelli con coloranti specifici disponibili in commercio organello27,28. In questo caso, anche uso 100 – 200 nM del substrato per gli enzimi auto-etichettatura per produrre segnali forti.
-
Preparazione di un campione di perlina fluorescente
Nota: Al fine di determinare la deriva ottica e per allineare le immagini dei diversi canali, multi-colore perline fluorescenti (0,1 µm) sono usati. Con le immagini registrate, verrà generata una matrice di trasformazione affine per i canali di due emissione.- Diluire la soluzione di perline all'1% con pura H2O.
- Posto 5 gocce della soluzione preparata con le perline di fluorescenza in cinque posizioni diverse su un vetrino coprioggetti pulito (Vedi punto 1.3).
- Lasciare il campione di perlina fluorescente asciugare su un banco pulito.
Nota: Il campione può essere ri-utilizzato; di conseguenza, coprire il campione con foglio di alluminio per evitare la contaminazione e tenerlo a 4 ° C.
2. microscopia
-
Messa a punto sperimentale
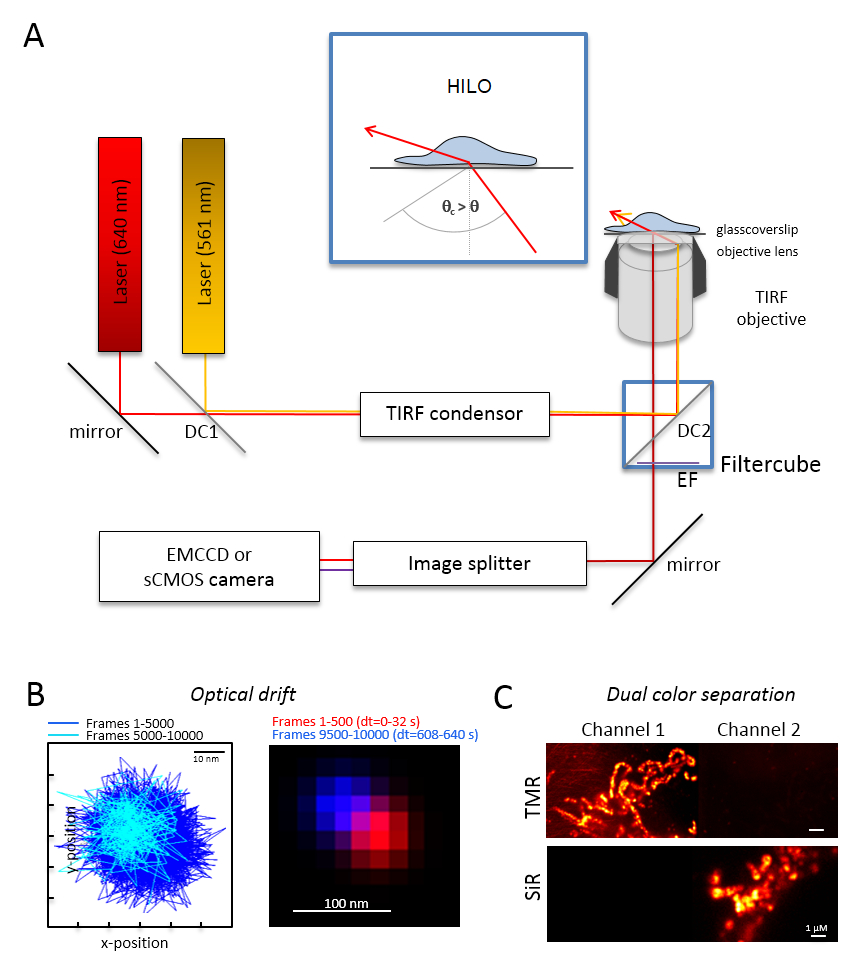
Nota: Un sistema di microscopia di base per l'imaging di doppio-colore singola molecola si basa su un microscopio invertito: è dotata di due laser accoppiato tramite una polarizzazione multi-mode-ottico mantenendo le fibre monomodali in una singola riflessione interna totale (TIR) condensatore, un obiettivo a immersione in olio progettato per TIRF, un filtri di emissione polyband, uno splitter di immagine e una fotocamera altamente sensibili (Figura 1). Un condensatore TIR, è necessario che per la regolazione continua dell'angolo incidente permette di passare tra la epi-, fortemente inclinato e foglio laminato ottico (illuminazione sottile ed inclinata (HILO)29) e la modalità di eccitazione TIRF con ottimizzato profondità di penetrazione. Immagini sono acquisite con un sistema di sensore raffreddato altamente sensibili, ad esempio, un retro-illuminato elettrone moltiplicando addebitato fotocamera dispositivo accoppiato (EMCCD) (efficienza quantica QE > 90%) o una fotocamera sCMOS (QE > 80 – 90%).- Determinare la deriva optical imaging perline fluorescenti (Vedi punto 2.2) alle stesse condizioni come quelle che saranno successivamente utilizzati per l'esperimento, per esempio, quando 10.000 fotogrammi vengono registrate nell'esperimento, registra anche 10.000 fotogrammi con il campione della perla. Per la determinazione della deriva ottica, confrontare la posizione delle perle nel primo fotogramma e l'ultimo fotogramma acquisito (Figura 1B). Se necessario, dopo la serie di immagini per ottica deriva30 di correggere e/o utilizzare ambienti stabili deriva.
- Dotare il cubo di filtro con il divisore di fascio dicroico appropriato, ad esempio, per fluorescenza arancia e rosso, più i filtri di adeguata emissione per fluorescenza arancio e fluorescenza rossa. Dotare lo splitter di immagine con i filtri adatti. Per verificare la possibile perdita dei segnali da un canale in altro canale singolo colore campioni registrati in entrambi i canali (Figura 1).

Figura 1 : Layout ottico per microscopia rilevamento e localizzazione multi-colore (TALM) con emettitori di arancione e rossi. (A) installazione di microscopio invertito con almeno due laser di eccitazione, un condensatore TIRF, un obiettivo adatto TIRF, uno splitter di immagine e una telecamera sensibile. Inserto: per eccitare gli organelli all'interno delle cellule, l'angolo del fascio incidente deve essere impostato più piccola di quanto l'angolo critico per TIRF raggiungere altamente inclinata e laminato illuminazione ottica foglio (HILO). DC1: Specchio dicroico 1; DC2: Specchio dicroico 2. EF: filtro di emissione. (B) prova su ottica deriva da posizioni di una perlina fluorescente per 10.000 fotogrammi con lo stesso frame rate a partire dai seguenti esperimenti di imaging (qui: 15 Hz). Collegato posizioni dei primi 500 telai e l'ultima 500 fotogrammi mostrano la deriva. Inoltre, un'immagine unita con la posizione del primo e l'ultimo frame in rosso e blu mostrano una minima deriva. La deriva è che la distanza tra il centro dei segnali diviso per il tempo di registrazione totale, qui 125 pm/s. (C) Check sulla netta separazione dei segnali, qui TMR e SiR. Per entrambi i canali, sono state generate immagini somma cumulativa di 3.000 fotogrammi (TMR nel canale 1) e SiR nel canale 2. SiRHTL è stato fissato a Tom20-HaloTag e TMRHTL a OxPhos complesso V-HaloTag. I colori sono falsi colori. Scala bar = 100 nm (B) e 1 µm (C). Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
-
Immagini generate allineamento fisico dell'immagine splitter
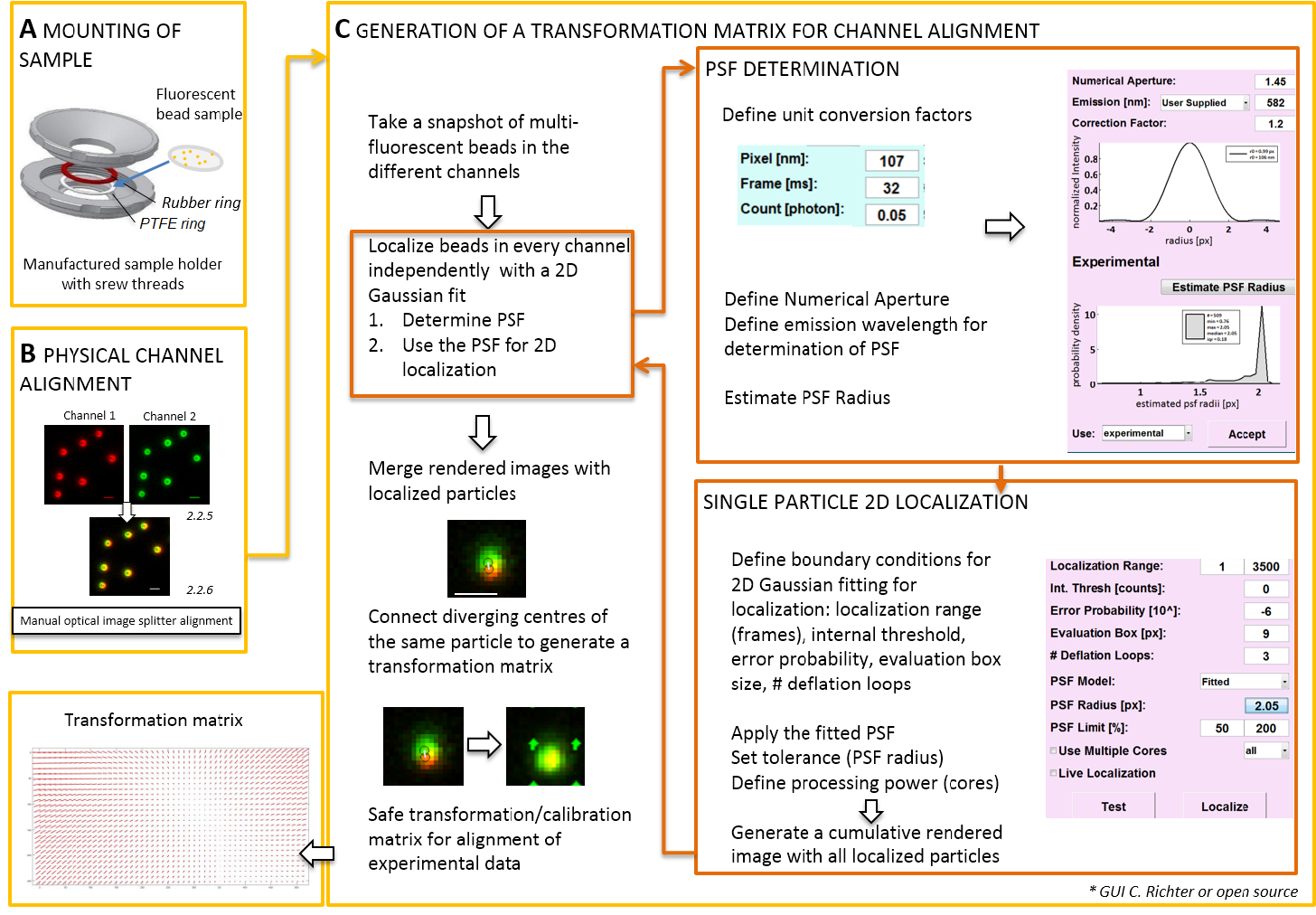
Nota: Per il montaggio del campione preparato su un vetrino coprioggetti, un self-made supporto del campione può essere utilizzato (Figura 2A). Per evitare la polvere, ecc. che rientrano nel campione, collocare il coperchio della piastra di coltura senza bloccare sopra la camera, quando montato. Il supporto del campione stesso può essere utilizzato per montare il vetrino coprioggetto con perline fluorescenti o cellule; Quando le cellule sono immaginate, aggiungere 0,5 – 0,8 mL di mezzo di imaging. L'immagine splitter divide l'immagine in due o più spettralmente separati canali e loro side-by-side proietta la stessa fotocamera. Questo processo potenzialmente introduce distorsioni sistematiche tra i canali a causa di distinti percorsi ottici attraversati e ostacola l'analisi diretta di colocalizzazione. Pertanto, in primo luogo eseguire allineamento fisico e secondo, post-correzione allineamento con una matrice di trasformazione. Per entrambi i processi di allineamento, perline fluorescenti devono essere distribuiti omogeneamente in tutto il campo visivo.- Montare il campione preparato con le perline fluorescenti in supporto del campione tra il politetrafluoroetilene (PTFE)-anello e l'anello di gomma rossa (Figura 2A).
- Avviare il microscopio, tutti i componenti hardware e tutti i software necessari per microscopia.
- Pulire l'obiettivo e la parte inferiore del coprivetrino con un panno di tessuto panno imbevuto di isopropanolo. Poi asciugare entrambi gli elementi con una salvietta fresca. Mettere una goccia di olio per immersione sulla pupilla della lente dell'obiettivo.
- Posizionare il portacampioni con il campione di tallone sul palco microscopio in modo che la parte inferiore del coprivetrino contatti l'olio. Concentrarsi sui branelli di trasmissione della luce o una linea laser.
- Regolare la potenza dei due laser di eccitazione per raggiungere intensità di segnale simile nei due canali di fluorescenza. Cerca una località con molti distinti segnali fluorescenti.
- Generare una visualizzazione unita dei canali fluorescente utilizzando il software di controllo della telecamera. Quindi utilizzare le viti a immagine splitter per inclinare manualmente gli specchi interni dello splitter immagine per ottenere la migliore sovrapposizione dei segnali dai due canali fluorescenti (Figura 2B).
Nota: attenzione! Non superare la gamma dinamica della fotocamera.
-
Allineamento dei canali spettralmente separati dal software eseguendo la trasformazione spaziale
Nota: La parte seguente mostra la post-correzione allineamento e la procedura di localizzazione con il nostro plugin software (disponibile su richiesta).- Avviare il microscopio TIRF software di controllo e scegliere di visualizzare singoli canali in modalità streaming. Prendere una fluorescenza di emozionante immagine snapshot in tutti i canali (Figura 2).
- Utilizzare questa immagine snapshot per produrre la matrice di trasformazione (Vedi Figura 2).
Nota: La matrice di trasformazione viene utilizzata per una trasformazione spaziale, in genere uno affine, che corregge per la traduzione (divergenza dei segnali da una singola fonte di punto tra i due canali). - Avviare il plugin di analisi del software (possono essere ottenute su richiesta dal nostro laboratorio, Vedi Figura 2).
- Caricare le immagini registrate in precedenza doppio colore (Vedi punto 2.2) di perline fluorescenti nel software. Scegliere l'orientamento usata dei canali fluorescenti. Selezionare 'Sì' quando chiesto 'calibrare immagini', quindi selezionare l'istantanea precedentemente assunto.
- Aprire "Gestione unità" per definire i fattori di conversione unità (dimensione dei pixel, frame rate, fattore di conversione del fotone).
- Aprire il "MANAGER di localizzazione". Determinare che il punto di diffusione prima funzione (PSF). Premere il pulsante: "Raggio di PSF". Nella finestra "PSF Estimator" che si apre, definire l'apertura numerica e l'emissione massima. Avviare "stima PSF raggio" facendo clic su. Accettare lo sperimentali ottenuti FPF. definire la casella di valutazione, il numero di cicli di deflazione, e quanti core del computer sono utilizzati per il calcolo. Premere "localizzare" per avviare la distribuzione di intensità delle singole particelle di montaggio di una funzione gaussiana simmetrica 2D (Figura 2).
- "Accettare" la sperimentali ottenuti FPF. definire la casella di valutazione, il numero di cicli di deflazione, e quanti core del computer sono utilizzati per il calcolo. Premere "localizzare" per avviare la distribuzione di intensità delle singole particelle di montaggio di una funzione gaussiana simmetrica 2D (Figura 2).
- Aprire il "gestore di taratura". Nell'immagine renderizzata unita dei due canali, sono mostrati i segnali originali e i centri localizzati. Scegliere la modalità "affine". Connettersi manualmente coppie corrispondenti di centri localizzate nei due canali che hanno avuto origine dalla stessa perlina fluorescente tracciando una linea di connessione.
- Collegare i segnali corrispondenti distribuiti in tutto il campo visivo. Dopo questo, premere "accetta". Salvare la calibrazione.
Nota: La trasformazione spaziale è campionata a ogni branello fluorescente e interpolata in mezzo. La funzione di trasformazione estratti rappresenta una Δr(x,y) di campo di spostamento che viene utilizzato per correggere successivamente le localizzazioni sperimentale dual-colore singola molecola, in modo che essi sovrapposizione all'interno della loro precisione di localizzazione. La matrice di trasformazione spaziale è in genere uno affine che corregge per la conversione, ridimensionamento e rotazione tra canali con precisione nanometrica, e si può dedurre da questo mapping uno a uno manuale (Figura 2).

Figura 2 : Flusso di lavoro per l'allineamento di doppio colore. (A) il coprioggetto con le perline fluorescenti è montato in un supporto del campione tra un PTFE e un anello di gomma. Quindi la parte superiore e inferiore della camera sono avvitati tra loro. (B) allineamento fisico delle visualizzazioni canale generati dal divisore di immagine. Registrato segnali fluorescenti dai branelli (0,1 µm) in due canali (verde e rosso, falsi colori) vengono unite. Le viti corrispondenti alle splitter ottico di immagine sono attivate manualmente fino ad ottenere la migliore sovrapposizione dei segnali diversi (colore giallo, pannello inferiore). (C) generazione di una matrice di trasformazione per l'allineamento del canale post-processive. Per la precisa localizzazione di una particella, è necessario determinare che il punto di diffusione di funzione (PSF) in dipendenza dalla lunghezza d'onda di emissione e l'apertura numerica dell'obiettivo. Il centro di un PSF può essere determinato dal relativo profilo di intensità analizzato da una gaussiana bidimensionale simmetrica in forma. La localizzazione il picco del segnale risultante è poi proiettata sui segnali originali, sfocati. In un'immagine unita, i centri localizzati dei segnali da due canali sono collegati per generare una matrice di trasformazione che viene successivamente utilizzata per l'allineamento post-processive dei dati sperimentali. Scala bar = 1 µm (B, C). Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
-
Formazione immagine di singola molecola delle proteine di membrana mitocondriale
Nota: Tutti gli esperimenti sono effettuati a temperatura ambiente. Le cellule T o cellule non aderenti devono essere immobilizzate in agarosio prima dell'imaging di31.- Montare il campione con le cellule aderenti il coprivetrino tra la gomma e PTFE anelli (Figura 3A). Riempire la camera con 0,5 – 0,8 mL di mezzo di imaging.
- Ripetere i passaggi 2.2.2–2.2.5.
- Regolare il microscopio TIRF angolo di illuminazione controllo software per creare un angolo di incidenza è minore dell'angolo critico per la modalità di TIR eccitare la regione specifica di interesse tramite un HILO foglio32 (modalità di HILO, Figura 1A).
Attenzione: Evitare il contatto visivo diretto con il raggio laser! - Impostare il guadagno di EM e scegliere un tempo di esposizione adatto per l'esperimento che raccoglie sufficienti fotoni per ogni frame.
- Impostare la potenza del laser per raggiungere un alto segnale/rumore (S/N) rapporto (Figura 3B), dal momento che la precisione di localizzazione corrisponde direttamente al S/N33 (Figura 3).
- Trovare una zona nella periferia delle cellule con i mitocondri non sovrapposte, di forma allungati e segnali di singola molecola (Figura 3D; Complementare dei Video 1). Se nessun segnale di singola molecola sono visibile, attendere lo sbiancamento provoca la comparsa di segnali di singola molecola (Figura 3E).
- Registrare fino a quando il numero di segnali è troppo basso per ragionevole continuazione (solitamente 1.000 – 10.000 fotogrammi in base al comportamento d'imbianchimento della tintura fluorescente, Figura 3F).
- Avviare l'imaging elaborazione software e cerca strutture mitocondriali generando un'immagine somma cumulativa di rendering di almeno 1.000 fotogrammi registrati (Figura 3).
Nota: Il più veloce possibile frame rate è dettato dalla zona di lettura. Il campo di vista per un canale è ridotto da un divisore di doppio-colore immagine (512 x 512 pixel) a 256 x 512 pixel e di un Quad-colore a 256 x 256 pixel. Così, per l'utilizzo di un'immagine splitter per due colori, questo è 30 Hz. impostare la modalità di trasferimento del telaio per ottenere il minor tempo possibile lettura. - Avviare dati raw plugin e carico di analisi software. Selezionare le immagini di orientamento e carico di canale. Utilizzare la matrice di trasformazione dal passaggio 2.3.9 quando chiesto "Calibra immagini". Canali vengono visualizzati separatamente.
- Aprire "Gestione unità" come prima per definire i fattori di conversione di unità per ogni canale. Aprire il "MANAGER di localizzazione" per ogni canale. Quindi definire la casella di valutazione, numero di cicli di deflazione, aggiungere il PSF teorico per le condizioni utilizzato e impostare il numero di core del computer vengono utilizzato per il calcolo. Infine, premere il tasto "localizzare" per ottenere particelle singole localizzate (Figura 3 H; Complementare dei Video 2).
- Nota che il programma infine genererà un'immagine superresolution cumulativo Mostra tutte localizzate particelle (Figura 3I).
- Eseguire l'analisi, ad esempio, di software open source o il nostro software disponibili su richiesta.
- Traccia le singole molecole in entrambi i canali localizzati, ad esempio, con il tracciante multipla10
Nota: Passaggio 2.4.13 necessita di conoscenza preliminare (sperimentale) circa la diffusibilità delle proteine di interesse per impostare correttamente le condizioni al contorno. Di solito, trovare le corrette condizioni di contorno è un processo iterativo.

Figura 3 : Passaggi durante la microscopia localizzazione singola molecola. (A), A coprire i vetrini con il campione viene montato tra la parte superiore e inferiore (grigio) del supporto del campione fatti in casa (progettato da J. Bereiter-Hahn). Un anello di gomma (rosso) e un anello in PTFE (bianco) sigillare il sistema da sopra e sotto il vetrino coprioggetto, quando le parti di supporto del campione sono bullone insieme. (B) rapporto segnale-rumore del segnale TMR. (C) calcolato istogramma di precisione di localizzazione per localizzate in tutte le particelle. (D) scelta di una regione ragionevole per l'imaging, qui, la periferia di cellule con i mitocondri chiaramente separate. (E) registrazione ed elaborazione di immagini: un singolo fotogramma con segnali distinti singola molecola è mostrato (qui, sono state registrate singole molecole di CV-HaloTag/TMRHTL ). Intensità di TMR (F) sopra il tempo di registrazione. (G) immagine somma cumulativa di 3.000 fotogrammi, non trasformati. (H) particelle di CV-HaloTag/TMRHTL localizzata con una funzione gaussiana 2D da un singolo fotogramma. (io) somma immagine mostrando tutte le particelle di CV-HaloTag/TMRHTL localizzate da 3.000 telai cumulativo, resa. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
Risultati
Analisi di imaging e colocalizzazione di multi-colore può contribuire a determinare la localizzazione sub-organellari delle proteine. Abbiamo dimostrato questo in precedenza con la fosfatasi citosolica e tensin omologo, PINK1, che ha diverse sedi sub-mitocondriali dovute alla sua lavorazione da proteasi mitocondriale17. PINK1 è un fattore importante, garantendo la funzionalità mitocondriale34,35. Per det...
Discussione
Qui, una tecnica per la localizzazione di singola molecola di colore doppio delle proteine della membrana cellulare è stata presentata. Seguendo il protocollo, le proteine di membrana sono fuse a proteine che reagiscono con le tinture della rodamina TMR e SiR coniugato ai loro rispettivi substrati con etichetta automatica. Rodamina coloranti sono luminose e fotostabili e pertanto consentono di imaging ripetitivo1. Per performance di successo, alcuni condizioni e argomenti critici devono essere te...
Divulgazioni
Gli autori non hanno nulla a rivelare.
Riconoscimenti
Gli autori vorrei ringraziare il gruppo di biofisica e Jacob Piehler presso l'Università di Osnabrück per supporto continuo, Wladislaw Kohl per assistenza tecnica e preparazione del materiale e il Consiglio di CellNanOs per la fornitura di microscopi per l'uso. Il progetto è stato finanziato il 944 SFB.
Materiali
| Name | Company | Catalog Number | Comments |
| (2-(4-(2-hydroxyethyl)-1-piperazinyl)-ethanesulfonic acid, 1 M) (HEPES) | Biochrom | #1104E | |
| DC1: Dichroid beam splitter | Chroma | 640 dcxr | NC506031 |
| DC2: Polychroic Mirror, beamsplitter | Chroma | zt405/488/561/640rpc | discontinued |
| Dulbecco´s Phosphate-Buffered Saline (PBS) 1x (w/o Ca & Mg) | Sigma-Aldrich & Co. | #RNBF8311 | |
| Earle´s MEM without phenol red, without L-Glutamine and without NaHCO3 containing 1% FBS, 0.1% HEPES, 0.1% NEAA, 0.1% Alanyl-L-Glutamine and 34.78% sodium hydrogen carbonate (NaHCO3 0.75g/l) | Imaging medium | ||
| Earle´s minimum essential medium (MEM) with phenol red, containing 1% Fetal Bovine Serum Superior (FBS), 0.1% HEPES (2-(4-(2-hydroxyethyl)-1-piperazinyl)-ethanesulfonic acid, 1 M), and 0.1% non-essential amino acids (NEAA) | Growth medium | ||
| EF: Emission filter quadbandpass | AHF analysentechnik | F72-866 | Brightline HC 446 nm/523 nm/600 nm/677 nm |
| EMCCD camera | Andor | Andor iXON 897 | EMCCD camera |
| Emission filter QuadView filter cubes, orange | AHF analysentechnik | F39-637 | bandpass 582 - 619 nm |
| Emission filter QuadView filter cubes, red | Chroma | bandpass 655 - 725 nm (HQ 690/70) | |
| FBS (Fetal bovine serum) superior | Biochrom | S0615 | |
| Fluorescent beads: TetraSpeck™ Microspheres, 0.1 µm, fluorescent blue/green/orange/dark red | Thermo Fisher Scientific | T7279 | fluorescent microspheres |
| Glutamine | Biochrom | #0951C | |
| HeLa cells | DSMZ | ACC-57 | Cervical carcinoma cells from patient Henrietta Lacks |
| Hela cells CI::paGFP, stable | Muster et al., PLOSOne 2010 | ||
| Hela cells CV g::Halo7-Tag, stable | Appelhans et al., NanoLett 2012 | ||
| Hela cells Tom20::Halo7-Tag, stable | Appelhans et al., NanoLett 2012 | ||
| Image splitter | Photometrics | Dual-View QV2 | image splitter emission |
| Imaging processing software | ImageJ2 / Fiji | freeware | |
| Immersion Oil - ImmersolTM 518 F (ne = 1.518, ve = 45) | Carl Zeiss Jena GmbH | 444960-0000-000 | |
| Inverted epifluorescence microscope | Olympus IX-71/73/83 | ||
| Laser 561 nm, 200 mW | CrystaLaser | CL-561-200 | 561 nm emission |
| Laser 642 nm, 140 mW | Omicron | Luxx-642-140 | 642 nm emission |
| MATLAB | MathWorks | version R2013a | |
| MEM with Earle's Balanced Salt Solution 2.2 g/L NaHCO3, stable glutamine w/o PR | Biochrom | FG-0385 | |
| MEM with Earle's Balanced Salt Solution with 2.2 g/L NaHCO3, stable glutamine, Phenolred | Biochrom | FG-0325 | |
| MitoTracker® Deep Red FM | Thermo Fisher Scientific | M22426 | dye |
| MitoTracker® Green FM | Thermo Fisher Scientific | M7514 | dye |
| Multi-mode-optical polarization maintaining monomode fiber | Pointsource/Qioptiq | KineFLEX | |
| NHS-PEG-MAL, Rapp Polymer | Rapp Polymere GmbH Tübingen | coverslip coating | |
| non-essential amino acids (NEAA) | Biochrom | #0802E | |
| PEG 800 (Polyethylene glycol) 10 % | Carl Roth GmbH | Art No. 0263.1 | coverslip coating |
| Penicillin/Streptomycin | Biochrom | #0122E | |
| Plasmid for PINK1-Halo7-Tag expression | Beinlich et al., ACS Chemical Biology 2015 | ||
| Poly-L-lysine (1.2 mg/ml) | Sigma-Aldrich & Co. | Cat. No.P9155 | coverslip coating |
| RGD Peptide (Ac-CGRGDS-COOH) | Coring System Diagnostix GmbH, Gernsheim | coverslip coating / Intergrin receptor motif | |
| Silicon Rhodamine linked to HaloTag®-Ligand (SiRHTL) | personal gift from Kai Johnson | dye | |
| Software analysis plugin | self-written C. P. Richter, Biophysik Osnabrück | SLIMFAST 16g | |
| Tetramethylrhodamine / SNAP-Cell® TMR-Star linked to SNAP-Ligand (TMRstar) | New England Biolab® | S9105S | dye |
| Tetramethylrhodamine linked to HaloTag®-Ligand (TMRHTL) | Promega | G8251 | dye |
| TIRF condensor | Olympus | Cell^TIRF MITICO System | TIRF condensor |
| TIRF microscope controlling software | Olympus cellSens 1.12 | ||
| TIRF objective | Olympus | 150x oil objective (N.A. 1.45; Olympus UAPO) | |
| Trypsin/EDTA 10x | Biochrom | #0266 | |
| Water H2O 99,5 % Rotipuran® Low organic | Carl Roth GmbH | Art. No. HN57.1 |
Riferimenti
- Appelhans, T., Richter, C., Wilkens, V., Hess, S., Piehler, J., Busch, K. Nanoscale organization of mitochondrial microcompartments revealed by combining tracking and localization microscopy. Nano Letters. 12 (2), 610-616 (2012).
- Appelhans, T., Busch, K. Single Molecule Tracking and Localization of Mitochondrial Protein Complexes in Live Cells. Methods Mol Biol. 1567, 273-291 (2017).
- Rust, M. J., Bates, M., Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods. 3 (10), 793-795 (2006).
- Gould, T. J., Verkhusha, V. V., Hess, S. T. Imaging biological structures with fluorescence photoactivation localization microscopy. Nat Protoc. 4 (3), 291-308 (2009).
- Pennacchietti, F., Gould, T. J., Hess, S. T. The Role of Probe Photophysics in Localization-Based Superresolution Microscopy. Biophys J. 113 (9), 2037-2054 (2017).
- Wegel, E., Göhler, A., Lagerholm, B. C., Wainman, A. Imaging cellular structures in super-resolution with SIM, STED and Localisation Microscopy: A practical comparison. Scientific reports. , (2016).
- Pellett, P., et al. Two-color STED microscopy in living cells. Biomedical Optics Express. 2 (8), (2011).
- Ishigaki, M., et al. STED super-resolution imaging of mitochondria labeled with TMRM in living cells. Mitochondrion. 28, 79 (2016).
- Liss, V., Barlag, B., Nietschke, M., Hensel, M. Self-labelling enzymes as universal tags for fluorescence microscopy, super-resolution microscopy and electron microscopy. Scientific Reports. 5, 17740 (2015).
- Sergé, A., Bertaux, N., Rigneault, H., Marguet, D. Dynamic multiple-target tracing to probe spatiotemporal cartography of cell membranes. Nat Methods. 5 (8), (2008).
- Appelhans, T., Busch, K. B. Dynamic imaging of mitochondrial membrane proteins in specific sub-organelle membrane locations. Biophysical reviews. 9 (4), 345-352 (2017).
- Wilmes, S., et al. Triple-color super-resolution imaging of live cells: resolving submicroscopic receptor organization in the plasma membrane. Angewandte Chemie. 51 (20), 4868-4871 (2012).
- Niewidok, B., et al. Single-molecule imaging reveals dynamic biphasic partition of RNA-binding proteins in stress granules. J Cell Biol. , (2018).
- Wurm, C. A., Jakobs, S. Differential protein distributions define two sub-compartments of the mitochondrial inner membrane in yeast. FEBS Lett. 580 (24), 5628-5634 (2006).
- Schmidt, R., Wurm, C. A., Punge, A., Egner, A., Jakobs, S., Hell, S. W. Mitochondrial cristae revealed with focused light. Nano Lett. 9 (6), 2508-2510 (2009).
- Kukat, C., Wurm, C. A., Spahr, H., Falkenberg, M., Larsson, N. G., Jakobs, S. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proceedings of the National Academy of Sciences of the United States of America. 108 (33), 13534-13539 (2011).
- Beinlich, F., Drees, C., Piehler, J., Busch, K. Shuttling of PINK1 between Mitochondrial Microcompartments Resolved by Triple-Color Superresolution Microscopy. ACS chemical biology. 10 (9), 1970-1976 (2015).
- Shim, S., et al. Super-resolution fluorescence imaging of organelles in live cells with photoswitchable membrane probes. Proceedings of the National Academy of Sciences. 109 (35), 13978-13983 (2012).
- Sbalzarini, I., Mezzacasa, A., Helenius, A., Koumoutsakos, P. Effects of Organelle Shape on Fluorescence Recovery after Photobleaching. Biophysical Journal. 89 (3), 1482-1492 (2005).
- Reits, E., Neefjes, J. From fixed to FRAP: measuring protein mobility and activity in living cells. Nature Cell Biology. 3 (6), E145-E147 (2001).
- Goehring, N., Chowdhury, D., Hyman, A., Grill, S. FRAP Analysis of Membrane-Associated Proteins: Lateral Diffusion and Membrane-Cytoplasmic Exchange. Biophysical Journal. 99 (8), 2443-2452 (2010).
- Bacia, K., Haustein, E., Schwille, P. Fluorescence correlation spectroscopy: principles and applications. Cold Spring Harbor protocols. 2014 (7), 709-725 (2014).
- Sukhorukov, V., Dikov, D., Busch, K., Strecker, V., Wittig, I., Bereiter-Hahn, J. Determination of protein mobility in mitochondrial membranes of living cells. Biochimica et biophysica acta. 1798 (11), 2022-2032 (2010).
- Kim, T. K., Eberwine, J. H. Mammalian cell transfection: the present and the future. Anal Bioanal Chem. 397 (8), 3173-3178 (2010).
- Graham, F. L., van der Eb, A. J. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology. 52 (2), 456-467 (1973).
- Wedeking, T., et al. Spatiotemporally Controlled Reorganization of Signaling Complexes in the Plasma Membrane of Living Cells. Small. 11 (44), 5912-5918 (2015).
- Mironov, S. L., Ivannikov, M. V., Johansson, M. [Ca2+]i signaling between mitochondria and endoplasmic reticulum in neurons is regulated by microtubules. From mitochondrial permeability transition pore to Ca2+-induced Ca2+ release. J Biol Chem. 280 (1), 715-721 (2005).
- Poot, M., et al. Analysis of mitochondrial morphology and function with novel fixable fluorescent stains. J Histochem Cytochem. 44 (12), 1363-1372 (1996).
- Tokunaga, M., Imamoto, N., Sakata-Sogawa, K. Highly inclined thin illumination enables clear single-molecule imaging in cells (vol 5, pg 159). Nature Methods. 5 (5), 455 (2008).
- Elmokadem, A., Yu, J. Optimal Drift Correction for Superresolution Localization Microscopy with Bayesian Inference. Biophys J. 109 (9), 1772-1780 (2015).
- Barlag, B., et al. Single molecule super-resolution imaging of proteins in living Salmonella enterica using self-labelling enzymes. Sci Rep. 6, 31601 (2016).
- Tokunaga, M., Imamoto, N., Sakata-Sogawa, K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nature Methods. 5 (2), 159-161 (2008).
- Mortensen, K. I., Churchman, L. S., Spudich, J. A., Flyvbjerg, H. Optimized localization analysis for single-molecule tracking and super-resolution microscopy. Nat Methods. 7 (5), 377-381 (2010).
- Jin, S., Youle, R. J. PINK1- and Parkin-Mediated Mitophagy at a Glance. Journal of Cell Science. 125, 795-799 (2013).
- Yamano, K., Youle, R. J. PINK1 is degraded through the N-end rule pathway. Autophagy. 9 (11), 1758-1758 (2013).
- Wiedemann, N., et al. Machinery for protein sorting and assembly in the mitochondrial outer membrane. Nature. 424 (6948), 565-571 (2003).
- Thompson, R., Larson, D., Webb, W. Precise Nanometer Localization Analysis for Individual Fluorescent Probes. Biophysical Journal. 82 (5), 2775-2783 (2002).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati