
Method Article
Label-Free Imaging of Single Proteins Secreted from Living Cells via iSCAT Microscopy
In This Article
Summary
We present a protocol for the real-time optical detection of single unlabeled proteins as they are secreted from living cells. This is based on interferometric scattering (iSCAT) microscopy, which can be applied to a variety of different biological systems and configurations.
Abstract
We demonstrate interferometric scattering (iSCAT) microscopy, a method capable of detecting single unlabeled proteins secreted from individual living cells in real time. In this protocol, we cover the fundamental steps to realize an iSCAT microscope and complement it with additional imaging channels to monitor the viability of a cell under study. Following this, we use the method for real-time detection of single proteins as they are secreted from a living cell which we demonstrate with an immortalized B-cell line (Laz388). Necessary steps concerning the preparation of microscope and sample as well as the analysis of the recorded data are discussed. The video protocol demonstrates that iSCAT microscopy offers a straightforward method to study secretion at the single-molecule level.
Introduction
Secreted proteins play a significant role in various physiological processes1. Because of this, they are routinely studied as a collective ensemble (proteomics) or as individual entities2,3. Proteomics traditionally investigates the entire set of proteins present in a particular biological system by way of e.g., enzyme-linked immunosorbent assays (ELISA), flow cytometry, or mass spectrometry4,5,6. Single proteins, on the other hand, are generally detected using a variety of techniques that are based on fluorescence7,8, plasmonics9,10, or cryogenic electron11 microscopies. All of these techniques use complex instruments, labeling, or both and often lack dynamics information as they only deliver long-term information about the system under study.
Here we use iSCAT12,13 microscopy to sense individual secretory proteins with sub-second temporal resolution14. Importantly, the technique detects the weak scattered signal intrinsic to every protein12,14. The amount of light that a small bioparticle scatters scales with its polarizability. Assuming that the shape of a protein can be approximated by an effective scattering sphere14,15,16, and that different proteins have very similar refractive indices, the measured signal can be directly connected to the molecular weight (MW) of the protein. The empirical calibration of iSCAT contrast versus molecular weight by reference measurements allows one to distinguish proteins of different sizes. iSCAT experiments can readily be complemented by fluorescence microscopy17,18, immunosorbent reagents, as well as fluorescent or scattering labels to allow for a specific detection of any protein of interest14,17,19.
In principle, iSCAT functions by amplifying a protein’s weak scattered light via interferometric mixing with a secondary reference wave. The detected intensity ( ) in an iSCAT microscope is described by
) in an iSCAT microscope is described by

where  is the incident intensity,
is the incident intensity,  is a coefficient for the contribution of the reference wave,
is a coefficient for the contribution of the reference wave,  signifies the scattering strength of the nano-object under study, and
signifies the scattering strength of the nano-object under study, and  is the phase shift between the scattered and reference waves14. Either the transmitted or back-reflected incident light is typically used as a reference wave, where in each case
is the phase shift between the scattered and reference waves14. Either the transmitted or back-reflected incident light is typically used as a reference wave, where in each case  accounts for the transmissivity or reflectivity of the sample chamber, respectively. The term
accounts for the transmissivity or reflectivity of the sample chamber, respectively. The term  is proportional to the protein’s scattering cross section and can be neglected compared to the cross term. Thus, setting
is proportional to the protein’s scattering cross section and can be neglected compared to the cross term. Thus, setting  for complete destructive interference, the detected light is given by
for complete destructive interference, the detected light is given by  where
where  is the reference intensity and
is the reference intensity and  is the interference intensity.
is the interference intensity.
iSCAT microscopy offers an excellent method to study biological processes at the single-molecule level. As an example, we investigate Laz388 cells — an Epstein-Barr virus (EBV) transformed B lymphocyte cell line20,21 — as they secrete proteins such as IgG antibodies16. However, the method is general and can be applied to a variety of other biological systems. iSCAT is inherently unspecific and can detect any protein or nanoparticle or it can be extended with common surface functionalization methods for specific or multiplexed detection. Its simplicity and ability to be combined with other optical techniques, such as fluorescence microscopy, make iSCAT a valuable complementary tool in cell biology.
Protocol
CAUTION: Please read all relevant material safety data sheets (MSDS) before using any chemicals, observe all appropriate safety practices, and wear personal protective equipment (laser safety goggles, eye protection, gloves, laboratory coats) as needed.
1. Building the iSCAT Microscope16,18
NOTE: The iSCAT microscope typically consists of a modified inverted microscope setup. In brief, a laser is focused onto the back focal plane of a high numerical aperture (NA) objective and an imaging lens is used to focus the particle’s back-scattered light onto a camera chip. In general, this wide-field microscope can be built from scratch or based on an existing inverted microscope. This protocol covers the essential steps to realize the setup, while changes in the used hardware are possible. A more detailed description of the assembly of an iSCAT microscope can be found in the work of Arroyo et al.18.
CAUTION: An iSCAT microscope involves a Class IIIB to Class IV laser light source. Appropriate eye protection is necessary when assembling and aligning the microscope optics. During the microscope assembly, make sure that the laser beam path remains straight and is not deflected as new optical components are added.
- Set up the illumination path of the microscope.
- Using a damped optical table and a rigid metal block, build a microscope sample stage18 that incorporates a high numerical aperture (NA) objective (100X/1.46 NA) and a translation unit that allows for lateral sample translation as well as change of focus position for the objective.
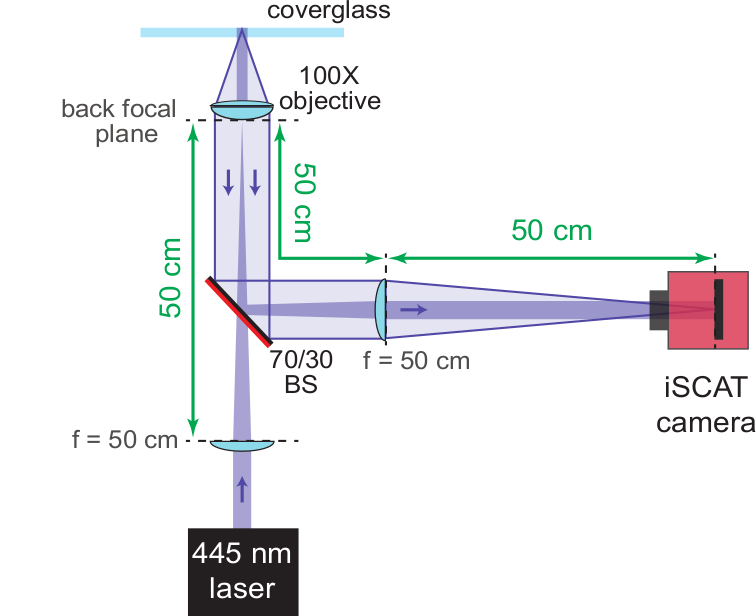
NOTE: Operation of an iSCAT microscope at the limit of detecting single proteins is highly susceptible to external vibrations. A centrosymmetric piezo stage that supports the sample from all sides is recommended to limit acoustic excitations of the sample that would otherwise compromise focal and lateral stability. Figure 1 shows a suitable sample stage, including the piezo translation unit and the objective. In addition, it is recommended that massive and stable mounts are used for all optical components discussed in the following steps. Such components are readily available from commercial optics suppliers. - Use a 50 cm focal length singlet lens (wide-field lens) and a 45° (vertical) coupling mirror to focus the light of a diode laser at wavelength 445 nm onto the back focal plane of the objective. This creates a collimated beam at the objective’s forward focus and will become the iSCAT illumination source. If necessary, filter the laser spatially prior to the 50 cm lens via a 30 µm pinhole or single mode fiber.
- Apply a droplet of immersion oil to the objective and place a glass coverslip in the sample plane of the microscope stage. This will result in a beam that reflects back down through the imaging objective.
NOTE: The reflection arises from the air-glass interface at the upper surface of the sample coverslip and will serve as the basis for the iSCAT reference beam. Residual dirt or dust on the surface of the coverslip will give rise to scattering point sources, which aid in the correct focusing of the imaging objective in the next steps.
CAUTION: The majority of the laser light transmits through the coverslip and travels straight up from the objective. Place an opaque specular diffuser (e.g., a paper card) above the coverslip to minimize the risk of injury from the laser light.
- Using a damped optical table and a rigid metal block, build a microscope sample stage18 that incorporates a high numerical aperture (NA) objective (100X/1.46 NA) and a translation unit that allows for lateral sample translation as well as change of focus position for the objective.

Figure 1: iSCAT sample stage. The photograph shows the massive aluminum block on which the piezo translation unit (black) is mounted as well as the centered 100x objective. The 3-axis piezo stage allows for precise positioning of the sample in the focal plane of the objective. Coarse focusing is carried out by rotating a threaded tube on which the objective is mounted (not depicted). The block is positioned on the optical table with four steel pedestals above the 45° coupling mirror. Please click here to view a larger version of this figure.
{kind=link}
- Set up the imaging path of the microscope.
- Introduce an antireflection (AR)-coated beam splitter (70% reflection, 30% transmission) at a 45° angle relative to the incident beam, and approximately 10 cm after the wide-field lens. Point the AR coating towards the laser source. This transmits the incident beam, and reflects the reference and scattered beams at a 90° angle to the incident beam.
NOTE: AR-coated and/or wedged beam splitters are recommended as significant ghosting and fringe effects can occur when using beam splitting cubes, or uncoated planar beam splitters. See the Discussion for more details. If desired, any unwanted reflected beam arising from the back side of the beam splitter may be blocked by use of an iris diaphragm. - A thick beam splitter will introduce a significant beam displacement so that the laser might not enter the objective straight anymore. If necessary, realign the laser beam path before the beam splitter to ensure the correct propagation through the objective.
NOTE: The beam splitter might also be added before the wide field lens and the microscope stage is positioned (in step 1.1.2.), so that the beam will not be displaced later. - At this point, focus the imaging arm of the interferometer and ensure that the sample plane and camera are parfocal. Place a concave f = -45 cm lens at a position 5 cm after the wide-field lens in the incident beam path. This will result in a collimated beam entering the back aperture of the objective.
- With a screen placed in the reflected arm of the interferometer, move the objective in the vertical direction to find the coarse focal position. The objective is in focus when the beam hitting the screen is collimated.
NOTE: Figure 2 shows a schematic of this process. - Remove both the f = -45 cm lens and the screen when coarse focusing is complete.
NOTE: Instead of using a negative focal length lens, the wide-field lens itself may be placed on a movable mount and shifted out of the beam path for this step. However, to achieve the most stable microscope configuration, it is recommended to hold the wide-field lens in a fixed position. - Add a second f = 50 cm singlet lens to focus the scattered light and to collimate the reflected light onto the sensor of a CMOS camera. Ensure that the lens is placed 50 cm from the back focal plane of the objective so as to re-collimate the reference beam and focus the scattered light.
- Place the CMOS chip 50 cm away from the f = 50 cm lens and position the beam directly onto the middle of the chip.
NOTE: The following parameters are typically used for imaging. The output power of the laser (wavelength 445 nm) is set to 100 mW. Pinhole and beam splitter attenuate the transmitted light so that the effective power entering the objective is about 9 mW. The beam diameter at the sample position amounts to 6 µm. With the used imaging lens, the effective magnification of the system is about 300x. The size of the image on the CMOS chip is set to 128 × 128 pixels within the illuminated area, resulting in a field of view of approximately 5 × 5 µm2. Figure 3 shows a schematic of the fully assembled iSCAT microscope.
- Introduce an antireflection (AR)-coated beam splitter (70% reflection, 30% transmission) at a 45° angle relative to the incident beam, and approximately 10 cm after the wide-field lens. Point the AR coating towards the laser source. This transmits the incident beam, and reflects the reference and scattered beams at a 90° angle to the incident beam.

Figure 2: Coarse focusing of the iSCAT microscope. The schematic shows the arrangement of the optics to help bring the system into focus. The AR-coated back side of the beam splitter (70/30 BS) is marked in red. Important distances are provided in green. The focal lengths (f) of the lenses used are denoted. Components in the blue dashed box are added in steps 1.2.6 - 1.2.7. The concave lens (used to re-collimate the converging iSCAT beam) and the screen are removed later. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: iSCAT microscope. The schematic shows the completely assembled iSCAT microscope. The AR-coated back side of the beam splitter (70/30 BS) is marked in red. Important distances are provided in green. The focal lengths (f) of the lenses used are denoted. Please click here to view a larger version of this figure.
{kind=link}
- Set up additional imaging channels.
NOTE: This section adds another imaging path to the microscope that allows for the observation of a large area surrounding the iSCAT laser via bright-field microscopy, and to monitor cell viability via fluorescence microscopy.- Couple the output of an LED light source (approximately 500 nm < λ < 580 nm) into a long working distance 20X/0.4 NA objective, and install mechanical components above the sample chamber that allow for focusing and lateral positioning of the LED output onto the sample.
- Ensure that the LED’s output spectrum covers the excitation range of the cell death marker (propidium iodide (PI)) and does not interfere with its fluorescence (λ > 600 nm). Use optical filters if necessary.
- Move the upper objective laterally so that the upper (wide-field) and lower (iSCAT) objectives are collinear. This is determined by placing a screen under the lower objective and maximizing the intensity of transmitted LED light on the screen. Place a λ = 550 nm short-pass dichroic mirror (SPDM) to split the transmitted LED light from the iSCAT laser path.
- Split this beam into two channels with an 8% reflective/92% transmissive beam splitter (BS). The 92% path is the fluorescence channel and the 8% path is used for bright-field imaging.
- Image the bright-field channel onto a CMOS camera using an f = 5 cm achromatic doublet lens.
- Image the fluorescence channel onto a separate CMOS camera using an f = 5 cm achromatic doublet lens and a λ = 600 nm long-pass filter to block the excitation light. Figure 4a shows a schematic of the fully assembled microscope including all imaging channels.
- Couple the output of an LED light source (approximately 500 nm < λ < 580 nm) into a long working distance 20X/0.4 NA objective, and install mechanical components above the sample chamber that allow for focusing and lateral positioning of the LED output onto the sample.

Figure 4: iSCAT microscopy of proteins secreted by single cells. (a) Schematic of the microscope described in the protocol. See section 1 for more information. Abbreviations: LED, light-emitting diode; SPF, short-pass filter; obj, objective; SPDM, short-pass dichroic mirror; BS, beam splitter; LPF, long-pass filter; BF, bright-field; fluor, fluorescence; C1-C3, camera 1-3. (b) Bright-field image of a single Laz388 cell about 4 µm away from the iSCAT field of view (depicted by a white square). Image taken by camera C3, scale bar: 10 µm. (c) Fluorescence image of the same region shown in (b) with the position of the cell marked by a white circle. The absence of fluorescence indicates that the cell is viable. Image taken by camera C2, scale bar: 10 µm. (d) Raw iSCAT camera image snapshot with 80 µs exposure time. Image taken by camera C1. (e) iSCAT image of the same region after spatiotemporal background subtraction as described in the discussion section. The image was integrated over 1000 sequential raw frames (d) with a final frame time of 400 ms and reveals the surface roughness of the glass coverslip. (f) Corresponding differential iSCAT image that shows the binding event of 2 proteins onto the coverslip. The image was constructed by subtracting two consecutive filtered images (e). Scale bars in (d), (e), and (f): 1 µm. This figure has been adapted from McDonald, M.P. et al.16. Copyright 2018 American Chemical Society. Please click here to view a larger version of this figure.
{kind=link}
- Set up the computer and software.
- Connect all cameras to a computer. Install respective driver packages and obtain/write software for their control.
NOTE: Suitable hardware is necessary for high-speed acquisitions. At a minimum, a multi-core processor, 16 GB of RAM, a frame grabber card, and a solid-state disc for data storage are recommended. - Observe the iSCAT image on the CMOS camera and ensure that it is in focus by finding a residual dust or dirt particle on the glass coverslip. Verify that the particle’s image is a circularly symmetric point spread function (PSF).
NOTE: The main reason for a non-circularly symmetric PSF is that the laser beam does not enter the objective straight but at a small angle with respect to the optical axis. This is corrected by adjusting the angle and position of the incident beam with the 45° coupling mirror. - Compare the camera images of the bright-field and the fluorescence channels. Ensure that both are in focus and display the same area. Verify that the position of the iSCAT laser is approximately in the center of the image and take note of its position for later reference. See Figure 4b – 4f for typical camera images.
NOTE: Use a cell sample or fluorescent beads to find the focus of the two channels. Temporarily remove the fluorescence long-pass filter to adjust the system. The focus for conventional imaging of the cells needs to be slightly higher than the iSCAT focal plane. To compensate for this without moving the objective, displace the cameras from their positions in the focus of the two respective f = 5 cm lenses. - Set the necessary camera parameters. Use a fixed frame rate and disable software gain and correction tools.
NOTE: The following parameters are used: The iSCAT camera is set to 5000 frames per second (fps) with an exposure time of 80 µs. As mentioned above, the image size is 128 × 128 pixels. Both bright-field and fluorescence cameras operate at full frame size (1280 × 1024 pixels). Bright-field imaging is carried out with a 20 ms exposure time. The fluorescence camera is set to 750 ms exposure time, and 5 consecutive frames are accumulated to form one final image. Bright-field and fluorescence images are acquired at fixed 20 s time intervals.
- Connect all cameras to a computer. Install respective driver packages and obtain/write software for their control.
2. Preparation of the Experiment
- Prepare the stock microscopy medium.
- Add 25 mL of HEPES Buffer Solution (1 mol/L) to 975 mL of RPMI 1640 medium to get a final 1 L of 25 mmol/L HEPES solution. Alternatively, use a buffer medium with HEPES already included.
NOTE: HEPES is used to maintain the pH value of the medium during a measurement in ambient conditions (e.g., outside an incubator and without constant CO2 supply). - Take an aliquot of the solution needed for an experiment and let it warm up to room temperature. 2 mL of medium is sufficient. Keep the remaining stock solution at 4 °C.
- Add 25 mL of HEPES Buffer Solution (1 mol/L) to 975 mL of RPMI 1640 medium to get a final 1 L of 25 mmol/L HEPES solution. Alternatively, use a buffer medium with HEPES already included.
- Prepare the microscope cuvette.
NOTE: The following steps describe the procedure for a custom-built sample holder consisting of an aluminum base plate and an acrylic cuvette dish that both fixes the coverslip and couples it to the piezoelectric 3D positioner. Commercially available sterile culture dishes with a glass bottom may also be used.
NOTE: Figure 5 shows photographs of the custom-built sample holder.- Take a new microscopy coverslip and rinse it with deionized water (DI-water) and ethanol. Air dry the slide with nitrogen or pressurized air.
- Clean the coverslip in an oxygen plasma atmosphere (0.3 mbar gas pressure) for 10 min at 500 W RF power. This removes all organic impurities from the surface.
- Clean the acrylic cuvette dish by immersing it in 0.2 mol/L NaOH solution for approximately 10 min. Rinse with DI water.
- Assemble the sample holder and cover it with a plastic Petri dish until needed in the experiment.

Figure 5: Custom-built sample holder. (a) Sample holder components: (1) acrylic cuvette dish; (2) aluminum base plate; (3) holding screws; (4) silicone O-ring; (5) coverslip. (b) Fully assembled sample holder. Please click here to view a larger version of this figure.
{kind=link}
- Prepare the microscope.
- Turn on the iSCAT illumination laser, the bright-field/fluorescence illumination LED, cameras, and the acquisition computer/software. Block the laser beam at a position before the objective.
- Ensure that the 100X/1.46 NA objective is clean. If not, use lens cleaning wipes and ethanol to clean the objective in accordance with manufacturer guidelines.
- Apply a drop of immersion oil on the microscope objective.
- Take the sample holder (assembled in section 2.2) and carefully mount it on the piezo stage of the iSCAT microscope so that the sample coverslip is centered on the microscope objective. Be attentive and careful so as not to damage the objective lens. Fasten the unit to the piezoelectric positioner with thumb screws while ensuring that the manufacturer specified maximum torque is not exceeded.
- Add 1 mL of stock microscopy medium (prepared in section 2.1.) into the cuvette.
- Add 2 drops of propidium iodide stain to the medium as a cell death marker16,22.
- Unblock the laser and bring the system into focus. First, verify that the objective is positioned at the correct distance from the coverslip by repeating steps 1.2.3. – 1.2.5. (Figure 2). Then, fine tune the focus with the z-axis of the piezo stage.
- Confirm that all settings for the light sources, cameras, and software are set correctly. This includes parameters such as laser power, LED intensity, camera frame rates, camera exposure times, or software saving paths.
NOTE: Saving videos at high frame rates can produce large file sizes. Ensure enough free disc space on the computer. - Block the laser beam again. The microscope is now ready for an experiment.
- Prepare the cells.
NOTE: Laz388 cells20 are cultured in RPMI 1640 medium supplemented with 10% fetal calf serum (FCS), amino acids, pyruvate, and antibiotics. The cells are incubated at 37 °C and 5% CO2 and are split and provided with fresh medium every 2-3 days23.- Take the cell culture flask from the incubator and aspirate medium containing approximately 1 x 106 cells. To determine the correct volume, quantify the concentration of the cell culture by use of a hemocytometer.
- Mix the cell solution with 10 mL of RPMI 1640 medium at room temperature and centrifuge the sample at 300 x g for 7 min.
- Carefully extract and discard the supernatant while ensuring that the pellet of concentrated cells remains undisturbed.
- Repeat steps 2.4.2. – 2.4.3. with the concentrated pellet of cells.
- Re-suspend the cells in 0.5 mL stock microscopy medium (prepared in section 2.1.) and immediately use them in an experiment.
3. iSCAT Microscopy of Secreting Cells
- Ensure that the laser beam is blocked to prevent the cells from being directly exposed to the iSCAT laser light.
- Inject the cells into the sample cuvette.
- Inject approximately 3 µL of the cell sample (prepared in section 2.4.) slightly off-center into the sample cuvette. Gently touch the pipette tip to the coverslip and slowly inject the cell solution. Allow the cells to settle on the coverslip.
NOTE: Use small volume pipette tips (10 µL) or long, flexible gel-loading tips. - Ensure that the density of cells is below approximately 1 cell per 500 µm² so that single-cell measurements are not influenced by multiple cells in the area surrounding the iSCAT laser.
- If the number of cells is too low, repeat step 3.2.1. until a sufficient number is available.
- If the coverage of cells is too dense, use an injection of approximately 20 µL of additional microscopy medium to disperse the cells across the coverslip.
- Inject approximately 3 µL of the cell sample (prepared in section 2.4.) slightly off-center into the sample cuvette. Gently touch the pipette tip to the coverslip and slowly inject the cell solution. Allow the cells to settle on the coverslip.
- Using the piezo positioner, move the sample laterally to position a cell close (approximately 10 µm) to the iSCAT field of view. Ensure that the cell does not enter the iSCAT field of view as direct exposure to the 445 nm laser light might be harmful for the cell.
- Use bright-field and fluorescence images to locate and verify the cell’s viability25.
NOTE: A viable cell has a round shape in the bright-field image and is not fluorescent, whereas cell death is indicated by strong fluorescence signals arising from the presence of propidium iodide inside the cell22. - Unblock the iSCAT laser beam and ensure that the coverslip surface is still in focus. Enclose the isolation table to minimize drift and acoustical coupling from the ambient surroundings.
NOTE: The latter is accomplished with heavy optical curtains or acrylic panels surrounding the optical table. - Start the measurement by acquiring images from the iSCAT, bright-field, and fluorescence cameras. Automate and control the process through software to maximize experimental efficiency. Periodically check the viability of the cell and the focus of the system.
NOTE: Depending on the laser intensity, optical components, and exposure time settings of the camera, the iSCAT laser might interfere with the fluorescence camera. If this behavior is observed, consider shuttering the iSCAT laser temporarily during fluorescence image acquisitions.
4. Data Analysis
NOTE: Experimental data is inherently noisy, and iSCAT images are no different. There are several sources of noise in a typical iSCAT measurement, including wavefront distortions in the incident light source, surface roughness of the coverslip, and camera noise. The section below presents some ways in which these noise sources are remedied via post processing. Additionally, lateral mechanical instabilities of the setup lead to noisy data and must be addressed accordingly, as described in the discussion section below. The described analyses are performed with custom MATLAB scripts.
- Minimize camera noise by filtering the raw data with a two-dimensional Fourier filter that excludes high spatial frequencies. The size of the filter needs to be adjusted to fit the specific experimental configuration (mostly determined by the system’s numerical aperture).
NOTE: Features in the image with higher spatial frequencies than the optical system originate from extraneous sources (such as camera read-out noise) and can be neglected. - Convert the images from raw camera counts to iSCAT contrast.
NOTE: The signal detected by the camera is . iSCAT contrast is defined as
. iSCAT contrast is defined as  where
where  is the intensity of the reference light, in this case the part reflected by the coverslip, and
is the intensity of the reference light, in this case the part reflected by the coverslip, and  is the interference between and the scattered intensity (
is the interference between and the scattered intensity ( ).
).
- Separate the signal into and by computing the temporal mean of particular frames in which the particles of interest are not present. The resulting image provides the reference signal .
NOTE: Alternatively, an active background subtraction step may be performed as described in the discussion below. - Calculate contrast according to
 12,14,16.
12,14,16.
- Separate the signal into
- Create a rolling differential image by subtracting each consecutive frame from its successor.
NOTE: Residual signals from the surface roughness of the coverslip and wavefront distortions are effectively removed in this step as they are constant within consecutive frames. The rolling differential removes these residual signals, leaving only the protein bindings that occur from one frame to the next. This dynamic background subtraction is beneficial as it is not sensitive to long-term sample drifts. - Apply a peak-seeking algorithm to detect and index single particles for each frame and determine their specific contrast and position.
- Use the information collected in step 4.4 to create histograms of protein binding events and relate their extracted contrasts to protein mass through a calibration curve compiled from known protein samples14,24.
Results
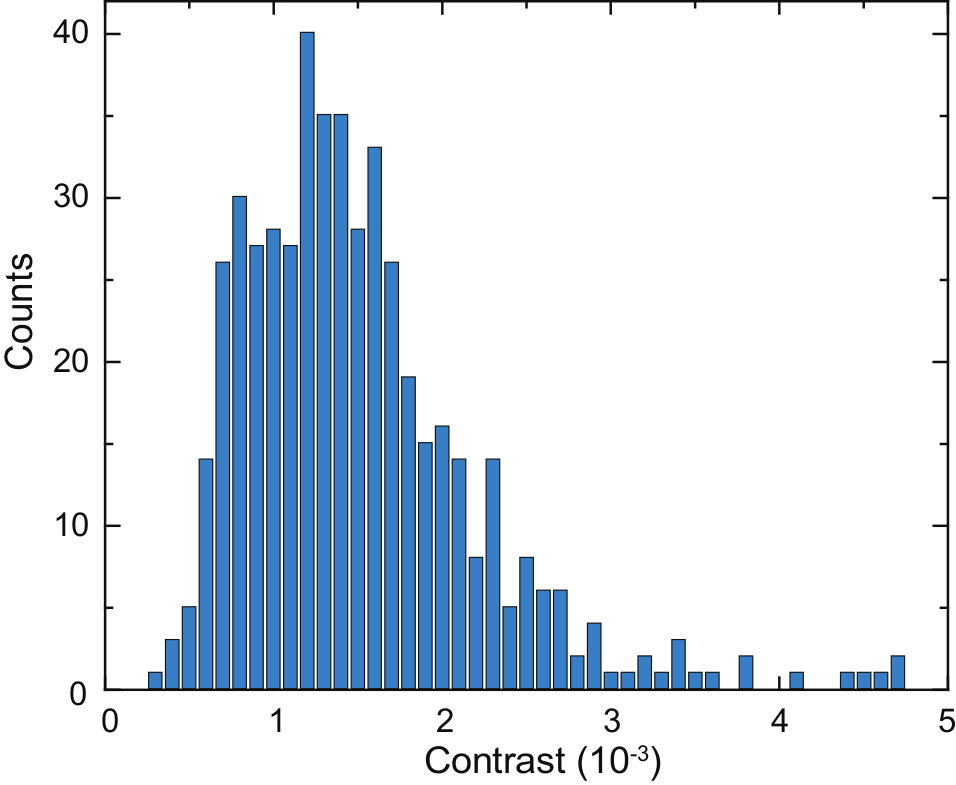
A schematic of an iSCAT microscope is shown in Figure 4a. Representative bright-field, fluorescence, and raw iSCAT images are shown in Figure 4b, 4c, and 4d, respectively16. Figure 4e and 4f show the results of background removal and differential post processing; two adsorbed proteins are visible as diffraction-limited spots in Figure 4f. Figure 6 shows a histogram of the detected proteins over the course of 125 s. These data were obtained by applying a peak-seeking algorithm to the captured images to count the binding events and catalog their contrast16. A total number of 503 proteins were detected.
Next, secreted species are identified by comparison with reference measurements carried out on purified protein solutions, or through additional measurements with functionalized glass surfaces14,16. The iSCAT data, thus, directly visualize cellular secretion dynamics on a subsecond scale16. As an example, we have previously found that IgG antibodies are a major fraction of the Laz388 secretome and are released from the cell at a rate of ca. 100 molecules per second16. Additionally, other particles spanning a range of 100 kDa - 1000 kDa are secreted by the cells16. The described method can be further employed e.g., to investigate the spatial concentration gradient of secretions surrounding a cell16, or to determine the temporal dynamics of cellular lysis16.

Figure 6: Quantification of secreted proteins by a single Laz388 cell. The histogram shows detected proteins during a time period of 125 s. Contrast values are accumulated in 1 x 10-4 contrast bins (blue bars). A total of 503 individual proteins were counted during this measurement. The experiment was repeated 10 times with similar results. This figure has been adapted from McDonald, M.P. et al.16. Copyright 2018 American Chemical Society. Please click here to view a larger version of this figure.
{kind=link}
Discussion
One of the most crucial aspects to obtaining useful iSCAT data is the ability to find the correct focal position at the coverslip surface, and, furthermore, to hold this position for long periods of time. Failing to do so will result in broadened PSFs, weak iSCAT signals, and drift-associated artifacts in dynamics analyses. It turns out that finding the focal plane on a clean, bare coverslip surface is not an easy task as surface features are not visible against the large reference beam background (see Figure 4d).
Raw iSCAT images are often obscured by background signals that arise from wavefront impurities in the excitation source, and can hinder one's ability to find the correct imaging plane. Active wavefront subtraction is a useful way to circumvent this issue and subsequently monitor the iSCAT focus during a measurement16. One way to accomplish this is through spatial sample modulation. In brief, a function generator applies a 50 Hz square wave to the external control port of the piezo stage, resulting in a spatial sample modulation at the applied frequency (290 nm amplitude). Synchronous camera acquisitions are triggered from the same source, and, when combined through lock-in principles, result in a wavefront-compensated image14,16. The resulting image typically shows the surface roughness of the coverslip (Figure 4e). Small features remaining on the glass after cleaning can be used to bring the microscope into focus. Parameters used for this active background subtraction step may be changed according to the frame rate, exposure time, or hardware.
As mentioned above, the use of a high-quality beam splitter in the iSCAT setup (step 1.2.1.) is recommended, as imaging artefacts like ghosting or interference arising from thin planar beam splitters will influence the image and disturb the measurement. Figure 7 shows a comparison between a high-quality and low-quality beam splitter. Both raw iSCAT images show the same area on the coverslip containing some residual particles. The same iSCAT setup was used to capture both images, only the beam splitter was exchanged. Figure 7a shows the image formed on the camera by use of a thicker (5 mm), AR-coated, and wedged beam splitter. Due to the wedged design, the reflected beam from the back surface of the beam splitter is anti-parallel to the reflection arising from the front surface and is not entering the objective. No interference artefacts occur. Figure 7b shows the same field of view on the sample but this time a thinner (1 mm) planar beam splitter was used. The two reflections from front and back surfaces of the beam splitter are parallel and propagate to the camera. Interference artefacts are clearly visible.

Figure 7: Comparison of iSCAT images produced with high- and low-quality beam splitters. (a) Resulting raw iSCAT image by use of a 5 mm thick, AR-coated, and wedged beam splitter. (b) Resulting raw iSCAT image of the same area by use of a 1 mm thick planar beam splitter. Both beam splitters have the same splitting ratio (50% reflection, 50% transmission). Interference artefacts arising from Fresnel reflections are clearly observed in the image produced with the 1 mm thick planar beam splitter. Scale bars: 2 µm. Please click here to view a larger version of this figure.
{kind=link}
In this protocol we describe a wide-field illumination scheme for iSCAT as it is fast, easy to realize and allows for parallel sensing over a large area14. Another common approach is to use acousto-optic deflectors (AODs) and scan a confocal beam across the sample12,17. This approach avoids the need for high-quality wavefronts but is more experimentally complex than conventional wide-field imaging. Furthermore, the speed of confocal illumination is limited by that of AODs. Depending on the desired experimental parameters, either confocal or wide-field illumination schemes can, in principle, be utilized to detect single proteins secreted from living cells.
As discussed throughout the protocol, it is imperative to minimize lateral mechanical fluctuations in the sample stage of the microscope. Even nanometer deviations in the position of the sample can lead to variations in consecutive camera frames and induce significant extraneous noise in the differential image. It is therefore recommended to use a mechanically stable microscope stage and a damped optical table (step 1.1.1.) and to cover the setup with optical curtains or panels during an experiment (step 3.5.).
An active focus stabilization scheme could also be considered for long-term measurements. In this approach, a second laser is incorporated into the microscope in a total internal reflection (TIR) arrangement, and subsequently imaged onto a quadrant photodiode. Changes in the system's focus translate into lateral displacements of the TIR laser spot on the quadrant diode, which can then be used in an active feedback loop to control the z-axis of the piezo stage26. Long-term vertical drift effects are thus eliminated.
Several modifications and extensions can be applied to the presented technique to address specific experimental needs. For example, commercial microscope stage incubators are available that could readily be incorporated into the iSCAT microscope for long-term imaging of cells. Other techniques can also be implemented to complement iSCAT imaging, such as confocal or TIR fluorescence microscopies17. To adapt on the system under study, iSCAT secretion measurements can be carried out in other cell media such as DMEM or DPBS, however, the pH indicator phenol red should be avoided as it can disturb the experiment due to absorption of the laser light. Additionally, supplements like fetal calf serum (FCS) or human platelet lysate (hPL) contain proteins that may interfere with iSCAT detection. Depending on the desired sensitivity of the experiment, these supplements should be excluded from the microscopy medium.
iSCAT relies on an analyte's ability to scatter light—a property that is intrinsic to all proteins—and is thus inherently nonspecific. Nevertheless, some degree of specificity is possible as iSCAT signals scale linearly with protein mass14,27,28. This allows for the calibration of an iSCAT system using standard protein samples, such as bovine serum albumin (BSA) and fibrinogen14,27,28. In fact, very recently, Young et al.28 have extended on the work of Piliarik & Sandoghdar14 and have shown that iSCAT can be used to determine the molecular weight of proteins as small as streptavidin (53 kDa) with a mass resolution of 19 kDa and an accuracy of about 5 kDa. Several conventional approaches can further complement iSCAT by providing an extra level of specificity. As an example, enzyme-linked immunosorbent assays (ELISA), and/or other surface modifications, restrict protein binding events so that only the target protein is detected16.
In this protocol, we described how iSCAT microscopy can be used to investigate cellular secretions at the single protein level with subsecond temporal resolution16. The technique is general and can be implemented on any commercial or home-built microscope. In contrast to single-molecule fluorescence approaches, the method does not suffer from photobleaching or blinking effects but it still achieves single-protein sensitivity. These features make iSCAT a powerful tool in the field of biosensing and microscopy. Future applications will focus on elucidating complex cellular interactions such as immunological response to a stimulus or cellular communication.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was supported by the Max Planck Society, an Alexander-von-Humboldt Professorship, and the Deutsche Forschungsgemeinschaft (CRC 1181). We thank Stefanie Schaffer at Universitätsklinikum Erlangen for providing Laz388 cells and for useful discussions. We thank Simone Ihloff and Maksim Schwab at MPL for technical support.
Materials
| Name | Company | Catalog Number | Comments |
| Item/Device | |||
| 100x / 1.46 NA objective | Zeiss | 420792-9800-000 | alpha Plan Apochromat oil immersion |
| 20x / 0.4 NA objective | Leica | 566049 | N Plan |
| Piezo Stage | PI | P-517k020 | 3-axis stage with 100x100x10µm range |
| Diode laser (445 nm) | Lasertack | PD-01236 | |
| Optics/Optomechanics | Thorlabs/Newport | - | lenses, mirrors, posts, mounts |
| Pinhole | Thorlabs | P30H | |
| LED light source | Thorlabs | MCWHL5 | |
| Shortpass filter (580 nm) | Omega Optical | 580SP | to modify the spectrum of the LED for fluorescence excitation |
| Longpass filter (500 nm) | Thorlabs | FEL0500 | to modify the spectrum of the LED for fluorescence excitation |
| 70R/30T beam splitter | Newport | 20Q20BS.1 | |
| Economy beam splitter | Thorlabs | EBS1 | used for the comparison of fringe effects |
| Wedged plate beam splitter | Thorlabs | BSW26 | used for the comparison of fringe effects |
| Shortpass dichroic mirror (550 nm) | Edmund Optics | 66249 | |
| 8R/92T beam splitter | Thorlabs | BP108 | |
| CMOS camera | Photonfocus | MV1-D1024E-160-CL | for iSCAT aquisition |
| CMOS cameras | Mightex | SCE-B013-U | for bright field / fluorescence aquisition |
| Longpass filter (600 nm) | Thorlabs | FELH0600 | |
| Computer | Fujitsu Siemens | - | Core i7 Processor, 16 GB RAM, SSD |
| Acquisition Software | LabVIEW | - | LabVIEW 2016 Suite |
| Analysis Software | Matlab | - | Matlab 2014 Suite |
| Plasma Cleaner | Diener | Diener pico | |
| Incubator | Binder | Model CB | |
| Centrifuge | Eppendorf | 5810R | |
| Reagent/Material | |||
| RPMI 1640 medium | Gibco | 11835063 | without phenol red |
| HEPES Buffer Solution (1M) | Sigma Aldrich | 59205C | |
| Cover slides | Marienfeld | 107052 | |
| Glass bottom culture dishes | ibidi | 81158 | |
| Fluorescent Microspheres | Invitrogen | F8821 | used for calibration |
| Immersol immersion oil | Zeiss | 444960 | |
| Propidium iodide stain | Invitrogen | R37108 | |
| Small pipette tips | Eppendorf | 30075005 | |
| Flexible pipette tips | Eppendorf | 5242956003 | |
| Ethanol, 99.8% | Fisher Scientific | E/0650DF/15 | |
| Sodium hydroxide, pellets | Sigma Aldrich | 221465 | for preparing 0.2M NaOH solution |
References
- Hathout, Y. Approaches to the study of the cell secretome. Expert Review of Proteomics. 4 (2), 239-248 (2007).
- Pandey, A., Mann, M. Proteomics to study genes and genomes. Nature. 405 (6788), 837-846 (2000).
- Makridakis, M., Vlahou, A. Secretome proteomics for discovery of cancer biomarkers. Journal of Proteomics. 73 (12), 2291-2305 (2010).
- Bantscheff, M., Schirle, M., Sweetman, G., Rick, J., Kuster, B. Quantitative mass spectrometry in proteomics: a critical review. Analytical and Bioanalytical Chemistry. 389 (4), 1017-1031 (2007).
- MacBeath, G. Protein microarrays and proteomics. Nature Genetics. 32, 526-532 (2002).
- Seder, R. A., Darrah, P. A., Roederer, M. T-cell quality in memory and protection: implications for vaccine design. Nature Reviews Immunology. 8 (4), 247-258 (2008).
- Moerner, W. E., Fromm, D. P. Methods of single-molecule fluorescence spectroscopy and microscopy. Review of Scientific Instruments. 74 (8), 3597-3619 (2003).
- Borisov, S. M., Wolfbeis, O. S. Optical biosensors. Chemical Reviews. 108 (2), 423-461 (2008).
- Dantham, V. R., Holler, S., Barbre, C., Keng, D., Kolchenko, V., Arnold, S. Label-Free Detection of Single Protein Using a Nanoplasmonic-Photonic Hybrid Microcavity. Nano Letters. 13 (7), 3347-3351 (2013).
- Zijlstra, P., Paulo, P. M. R., Orrit, M. Optical detection of single non-absorbing molecules using the surface plasmon resonance of a gold nanorod. Nature Nanotechnology. 7 (6), 379-382 (2012).
- Rickgauer, J. P., Grigorieff, N., Denk, W. Single-protein detection in crowded molecular environments in cryo-EM images. eLife. 6, e25648 (2017).
- Lindfors, K., Kalkbrenner, T., Stoller, P., Sandoghdar, V. Detection and Spectroscopy of Gold Nanoparticles Using Supercontinuum White Light Confocal Microscopy. Physical Review Letters. 93 (3), 037401 (2004).
- Jacobsen, V., Stoller, P., Brunner, C., Vogel, V., Sandoghdar, V. Interferometric optical detection and tracking of very small gold nanoparticles at a water-glass interface. Optics Express. 14 (1), 405 (2006).
- Piliarik, M., Sandoghdar, V. Direct optical sensing of single unlabelled proteins and super-resolution imaging of their binding sites. Nature Communications. 5, 4495 (2014).
- Roux, K. H. Immunoglobulin Structure and Function as Revealed by Electron Microscopy. International Archives of Allergy and Immunology. 120 (2), 85-99 (1999).
- McDonald, M. P., et al. Visualizing Single-Cell Secretion Dynamics with Single-Protein Sensitivity. Nano Letters. 18 (1), 513-519 (2018).
- Kukura, P., Ewers, H., Müller, C., Renn, A., Helenius, A., Sandoghdar, V. High-speed nanoscopic tracking of the position and orientation of a single virus. Nature Methods. 6 (12), 923-927 (2009).
- Ortega Arroyo, J., Cole, D., Kukura, P. Interferometric scattering microscopy and its combination with single-molecule fluorescence imaging. Nature Protocols. 11 (4), 617-633 (2016).
- Spindler, S., et al. Visualization of lipids and proteins at high spatial and temporal resolution via interferometric scattering (iSCAT) microscopy. Journal of Physics D: Applied Physics. 49 (27), 274002 (2016).
- Lazarus, H., et al. Characterization of a unique cell line (LAZ 221) from human acute lymphocytic ("null" cell) leukemia. Cancer Research. 38 (5), 1362-1367 (1978).
- Mackensen, A., et al. Evidence for in situ amplification of cytotoxic T-lymphocytes with antitumor activity in a human regressive melanoma. Cancer Research. 53 (15), 3569-3573 (1993).
- Crowley, L. C., Scott, A. P., Marfell, B. J., Boughaba, J. A., Chojnowski, G., Waterhouse, N. J. Measuring cell death by propidium iodide uptake and flow cytometry. Cold Spring Harbor Protocols. 2016 (7), 647-651 (2016).
- Freshney, R. I. Primary Culture. Culture of Animal Cells. , (2005).
- Dahmardeh, M., et al. . Unpublished data. , (2018).
- Majno, G., Joris, I. Apoptosis, oncosis, and necrosis. An overview of cell death. American Journal of Pathology. 146 (1), 3-15 (1995).
- Bellve, K., Standley, C., Lifshitz, L., Fogarty, K. Design and Implementation of 3D Focus Stabilization for Fluorescence Microscopy. Biophysical Journal. 106 (2), 606a (2014).
- Liebel, M., Hugall, J. T., Van Hulst, N. F. Ultrasensitive Label-Free Nanosensing and High-Speed Tracking of Single Proteins. Nano Letters. 17 (2), 1277-1281 (2017).
- Young, G., et al. Quantitative mass imaging of single biological macromolecules. Science. 360 (6387), 423-427 (2018).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved