
Method Article
Spontaneous Formation and Rearrangement of Artificial Lipid Nanotube Networks as a Bottom-Up Model for Endoplasmic Reticulum
In This Article
Summary
Solid-supported, protein-free, double phospholipid bilayer membranes (DLBM) can be transformed into complex and dynamic lipid nanotube networks and can serve as 2D bottom-up models of the endoplasmic reticulum.
Abstract
We present a convenient method to form a bottom-up structural organelle model for the endoplasmic reticulum (ER). The model consists of highly dense lipidic nanotubes that are, in terms of morphology and dynamics, reminiscent of ER. The networks are derived from phospholipid double bilayer membrane patches adhering to a transparent Al2O3 substrate. The adhesion is mediated by Ca2+ in the ambient buffer. Subsequent depletion of Ca2+ by means of BAPTA/EDTA causes retraction of the membrane, resulting in spontaneous lipid nanotube network formation. The method only comprises phospholipids and microfabricated surfaces for simple formation of an ER model and does not require the addition of proteins or chemical energy (e.g., GTP or ATP). In contrast to the 3D morphology of the cellular endoplasmic reticulum, the model is two-dimensional (albeit the nanotube dimensions, geometry, structure, and dynamics are maintained). This unique in vitro ER model consists of only a few components, is easy to construct, and can be observed under a light microscope. The resulting structure can be further decorated for additional functionality, such as the addition of ER-associated proteins or particles to study transport phenomena among the tubes. The artificial networks described here are suitable structural models for the cellular ER, whose unique characteristic morphology has been shown to be related to its biological function, whereas details regarding formation of the tubular domain and rearrangements within are still not completely understood. We note that this method uses Al2O3 thin-film-coated microscopy coverslips, which are commercially available but require special orders. Therefore, it is advisable to have access to a microfabrication facility for preparation.
Introduction
The ER carries out crucial tasks in the biological cell including protein folding, lipid synthesis, and calcium regulation1,2. The ER morphology is intrinsic to the functions it performs. It combines planar stacks and dense-dynamic tubular domains, which continuously interact with the cytoskeleton and undergo constant motion and rearrangement. Some of the remodeling that ER structures undergo include the continuous transformation between planar sheets and tubes, vesicle formation from or fusion to the ER lumen, elongation of pre-existing tubes, tube retraction, fusion, and breakage3. The peculiar structure of the tubular networks is energetically unfavorable. The pathways and mechanisms by which the ER generates and maintains this organization as well as how this relates to its function is not yet fully understood4,5.
It is known that the ER malfunctions when it loses its homeostatic state, resulting in ER stress, a condition caused by an increase in protein synthesis, accumulation of misfolded proteins, or changes in Ca2+ and oxidative balance. ER stress in turn causes deformation of the natural morphology of the organelle, specifically by disturbing the network organization6,7. As a response, the cell activates a repair mechanism to return to a homeostatic state. Failure in repair can lead to ER-induced cell apoptosis, which contributes to several metabolic and degenerative diseases such as Alzheimer's disease, type 2 diabetes, Parkinson's disease, amyotrophic lateral sclerosis, and several others7,8. Current research targets the organization of the tubular ER networks, and several studies are focusing on reconstituting the ER in vitro2. A few existing models2,9,10 require proteins to initiate and maintain membrane curvature3,11 and help the organelle reach its shape. Clearly, model systems that mirror some of the key structural and organizational features of the ER and provide access to advanced experimental studies are in great demand.
We present here procedures for the preparation of a facile, protein/chemical energy-free, dynamic in vitro model for the ER, providing a basic platform to study ER morphology and associated functions4. In this method, an ER model is fabricated with a bottom-up approach using only a few elements, in which the molecules of interest can be integrated to add complexity. The network represents ER structure and dynamics. Furthermore, reversible transformation between the planar membrane and the tubes, vesicle formation from the tubes, tube fusion, sliding and retraction can all be observed. In addition to serving as a bottom-up model for the incompletely understood cellular ER, the lipid route to nanotube networks described in this protocol can be applicable for researchers studying self-assembly, nanofluidics, single-molecule and colloid transport phenomena, Marangoni flow, and other related fields. The only molecular building blocks used in our method are phospholipids. The protocol requires little laboratory work and basic equipment and is accessible for the incorporation of additional elements.
Protocol
1. Preparation of Phospholipid Vesicle Suspension
NOTE: For all materials referred to as "clean" in this protocol, thoroughly wash them with isopropanol followed by deionized water and blow-dry them with nitrogen. Note that a treatment of glass substrates with strongly oxidizing acidic agents (Piranha solution), which is typically applied in preparation protocols for supported lipid films on solid substrates, should not be performed on Al2O3-coated carriers.
- Place in a clean 10 mL round-bottom or inverted pear-shaped glass flask: Soy L-α phosphatidyl choline (PC, 69% w/w), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE, 30% w/w), and a lipid-conjugated fluorophore of choice [e.g., Texas Red 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine triethylammonium salt (TR-DHPE, 1% w/w)] in chloroform; for a total amount of 3000 µg of lipids in 300 µL of chloroform, resulting in a final concentration of 10 mg/mL.
NOTE: Use clean, glass, gas-tight syringes with polytetrafluoroethylene plungers when handling compounds containing chloroform.
CAUTION: Chloroform is toxic and highly volatile and should always be handled under a fume hood with associated personal protective equipment. - Connect the flask to a rotary evaporator, position with a tilt of 45°, and rotate at 24 rpm inside a water bath at 23 °C for 6 h with reduced air pressure to slowly and completely remove the chloroform. Start reducing the pressure right after initiating the rotation by steps of 20 kPa every 2 min until it reaches 20 kPa (150 Torr, 80% vacuum).
NOTE: The formation of a homogenous lipid film of uniform thickness in the preparation vessel is the most important requirement for the rotavap procedure. Lipid preparations are sensitive to rotation velocity, rapid pressure changes, and final pressure value; therefore, strictly follow the slow reduction steps as well as the end pressure and rotation velocity. Position the flask with a tilt of 45° to guarantee that the dehydrated lipid cake is formed evenly as a film on the wall of the flask. Too fast of a rotation leads to turbulences and too slow of a rotation leads (due to gravity) to the accumulation of a thick layer of fluid at the bottom of the flask. During the subsequent overnight swelling process, a very inhomogenous lipid mass is produced that does not respond well to the final sonication step, and the resulting fractions are of different compositions. Pressure and time within the range specified in the method ensures slow desolvation. With chloroform as the solvent, too rapid of a drop in pressure cools down the mixture, resulting in increased viscosity and aggregate and uneven film formation. The long time period of 6 h is recommended in order to remove solvent to the greatest extent possible, as the organic solvent partitions into the lipid material upon rehydration. - After 6 h, stop the rotation and increase the air pressure again, gradually, by steps of 20 kPa every 2 min until reaching 100 kPa. Remove the flask from the rotary evaporator and add 3 mL of PBS and 30 µL of glycerol. Gently swirl the flask to dissolve the glycerol. Use an air-tight glass stopper to seal the flask containing the lipids.
NOTE: The glycerol is used to prevent the complete dehydration of the lipid film and enables bilayer separation12. It should be heated before use to decrease its viscosity, which facilitates the handling of this compound. The warmed glycerol does still not immediately mix with the PBS buffer. Gentle swirling is required until the glycerol is completely dissolved. - Store the flask in the refrigerator at 4 °C overnight for rehydration and swelling of the lipid films.
- On the following day, sonicate the lipids with an ultrasonic water bath at room temperature (RT, ~21 °C) and at 35 kHz frequency until achieving a uniform, slightly turbid lipid suspension.
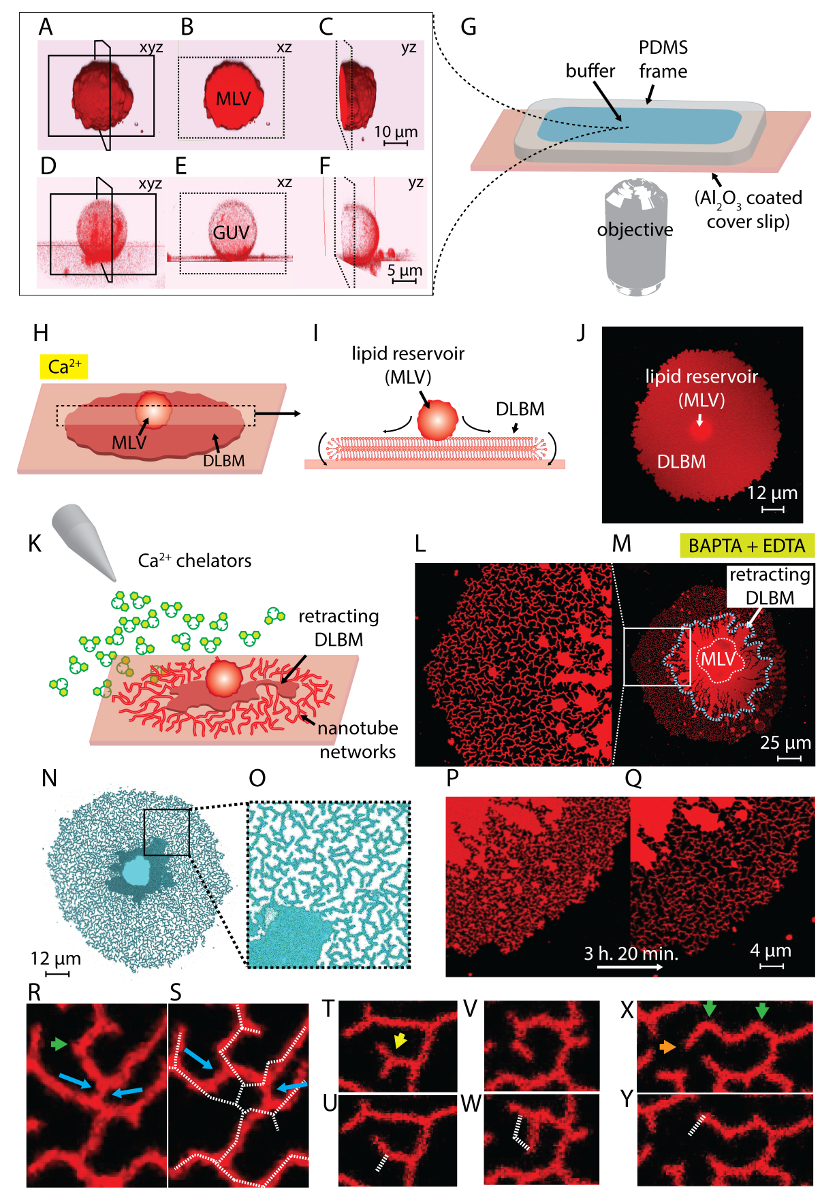
NOTE: Sonication can take around 10-30 s. Prolonged sonication (~1 min) produces heat and is detrimental to vesicle formation. - Steps 1.1-1.5 yield a suspension containing two types of vesicular structures: multilamellar vesicles (MLV) and giant unilamellar vesicles (GUV) (Figure 1A-1F).
- For storage, divide the lipid suspension into 100 µL aliquots, using a total of 30 microcentrifuge tubes, and store them in a freezer at -20 °C.
NOTE: Flash freezing with liquid nitrogen is not necessary and not used before storage. The protocol can be paused here. Leaving lipid suspensions in the 4 °C refrigerator for prolonged times causes lipid lysis, which affects the membrane composition.
2. Preparation of Substrates
NOTE: The following protocol is performed at a cleanroom classified as ISO 8 in the ISO 14644-1 standard specification. Atomic layer deposition (ALD) is used to fabricate Al2O3 substrates. The specified process parameters are instrument-dependent and may vary between different models of equipment. They can be used as initial parameters to develop the process.
- Set the temperature of the ALD reactor to 200 °C.
- Load the glass surfaces (e.g., glass coverslips) into the sample chamber together with a silicon wafer, which will be used later as a reference surface to determine thickness of the deposition by ellipsometry.
NOTE: Glass substrates were used out-of-the-box and were not solvent-cleaned before deposition. They were only flushed with nitrogen gas to remove particles. - Evacuate the loading chamber to 400 Pa (3 torr) and transfer the samples into the main reaction chamber, and evacuate it to 200 Pa.
NOTE: The temperature of the reactor must be maintained at 200 °C for proper deposition. Temperature fluctuations after sample loading must therefore be equilibrated before initiating the deposition. Chamber pressure is set to be higher than reactor pressure to avoid any precursors to spread outside the chamber. - Start depositing the atomic film. One cycle consists of a 150 ms pulse trimethyl aluminum exposure, followed by a 1 s purge, and subsequently, a H2O exposure of 200 ms duration followed by a 1 s purge.
NOTE: All settings including chamber and reactor pressure, length of the cycles, and purges are automated to achieve a defined rate of deposition. These parameters may vary among the various models of equipment. The pre-configured recipes are often prepared by the vendor or tool responsible in the clean room and communicated to the user as deposited film thickness per unit of time. - To reach 10 nm of Al2O3 on the substrate, repeat the process for 100 cycles. The number of cycles depends on the deposition rate, which may vary among different recipes or equipment.
- To remove the samples from the reactor, first vent the chamber until its pressure reaches atmospheric pressure, then remove the samples.
- Store the samples in air tight containers at RT until use.
NOTE: No further cleaning is recommended before use. The protocol can be paused here.
NOTE: The samples should ideally be used immediately after deposition. The optimal storage requires positioning the surfaces inside polypropylene wafer carriers, followed by enveloping the carriers in clean room-compatible plastic bags, which are to be nitrogen-flushed before vacuum sealing. The purpose is to avoid exposing the surface to air-borne contaminants. If necessary, the surfaces can be kept in air tight containers at RT at maximum for 5 days. Longer storage is not recommended. For users who do not have easy access to a clean room nearby, and purchase or obtain the surfaces from abroad, re-oxidizing the substrates by means of oxygen plasma or ozone treatment may be an alternative solution13.
3. Transformation of Molecular Phospholipid Films to Tubular Networks
- Thaw the lipid suspension and transfer a 4 µL droplet of suspension onto a clean glass microscope slide/coverslip.
- Desiccate the droplet for 20 min. The droplet will collapse into a flat circular film of lipids after desiccation, which is visible to the eye.
- Rehydrate the lipid film with 1 mL of HEPES buffer (see Table of Materials) for 3 min.
NOTE: The volume of rehydration buffer affects the density of vesicle suspension (the number of vesicles per unit volume), which is subsequently transferred to the observation chamber. Depending on the volume of the observation chamber and the desired vesicle density, the rehydration volume can be tuned to 0.5-1 mL. Clean borosilicate slides tend to support droplets of several hundred microliters up to 1.5 mL without problems. Since the coverslip does not need to be moved, this does not lead to a technical problem. On more hydrophobic surfaces, such as SU-8 polymer-coated slides, even 1.5 mL can be deposited12.
NOTE: Lipids should be freshly prepared, as exposure of the rehydrated lipid film for more than 20 min at RT leads to evaporation of the buffer and partial dehydration of the previously rehydrated vesicles, which leads to a poorly defined composition. - Prepare the observation chamber: to allow for buffer exchange by means of an automatic pipette, which is required to initiate the ER transformation, an observation chamber with an open top was used. This chamber consists of a polydimethylsiloxane (PDMS) frame with dimensions 1.5 x 1.5 x 0.5 cm, adhered onto the Al2O3 deposited coverslip. A scheme of the mounted observation chamber is represented in Figure 1G. The following steps were performed to fabricate the PDMS frame and assemble the observation chamber:
- Prepare a KOH solution by mixing 100 g of KOH with 100 mL of isopropanol in a beaker in an ice bath. Stir for 10 h or longer until the KOH is completely dissolved using a magnetic stirrer and magnetic stir plate.
CAUTION: KOH solution is corrosive and can lead to skin burns, so always handle with proper personal protective equipment.
NOTE: The solubility of KOH in isopropanol is not as high as in water. The dissolution is exothermic. Crushing the KOH pellets prior to dissolving and continuous stirring is advisable. - Submerge a glass Petri dish (d = 6 cm) into the KOH solution at RT and keep it overnight.
- The following day, remove the glass dish from the solution, immerse it in a container with deionized water for 5 min, rinse it several times with water, and place it inside a drying oven at 80 °C for 1 h. Blow the surface briefly with a stream of nitrogen to ensure that particles are removed.
- To passivate the surface and prevent bonding to PDMS, transfer 200 µL of dimethyldichlorosilane with a plastic syringe into a clean plastic container such as a weighting boat.
- Store the glass Petri dish together with the silane for 1 h in an evacuated desiccator (low vacuum, ~20 kPa).
CAUTION: Dimethyldichlorosilane is toxic and should always be handled under a fume hood with associated personal protective equipment. - Wait 15 min before collecting the Petri dish in order for the remaining dimethyldichlorosilane vapor to dissipate. The Petri dish is now silanized, and the surface is hydrophobic.
NOTE: A quick way to test the success of this step is to place a water droplet onto the silanized Petri dish. The contact angle of the droplet with the surface must visibly increase compared to untreated glass. - In a 250 mL plastic container (transparent plastic cup fresh from the package), mix 10 g of silicone elastomer base with 1 g of silicone elastomer curing agent (10:1). Stir using a plastic stirrer/plastic spatula for 5 min.
NOTE: Air bubbles are formed upon stirring, and the PDMS will look pale-white. - Desiccate the mixture to degas at < 20 kPa until all expanding air bubbles have collapsed (higher vacuum accelerates the process). Pour the degassed mixture into the silanized Petri dish.
- Cure at 65 °C for 2 h in an oven.
NOTE: It is possible to double the curing speed by increasing the temperature to > 95 °C. The increasing curing temperature results in an increase in stiffness of the material. - Cool down the Petri dish filled with the cured PDMS to RT and remove the PDMS slab with a spatula.
- With a scalpel, cut the frame into the dimensions and geometry appropriate for the available opening in the microscope stage. Dimensions of 1.5 (length) x 1.5 (width) x 0.5 (height) cm are suitable for most setups.
- Bring the smooth side (the bottom side that was in contact with the Petri dish) of the PDMS frame into contact with the active side of the surface where the Al2O3 film resides, and gently apply pressure to push the frame and surface against each other to make them adhere.
NOTE: The adhesion between the PDMS frame and Al2O3 substrate is weak. The presence of air bubbles at the contact interface may cause de-attachment and, consequently, the leakage of the buffer and related content. The PDMS frame can be used several times if immediately after and before each use it is rinsed with isopropanol, followed by rinsing with DI water and blow-drying with nitrogen. The silanized Petri dish can also be reused.
- Prepare a KOH solution by mixing 100 g of KOH with 100 mL of isopropanol in a beaker in an ice bath. Stir for 10 h or longer until the KOH is completely dissolved using a magnetic stirrer and magnetic stir plate.
- Fill the observation chamber with Ca2+-HEPES buffer (see Table of Materials).
NOTE: The surface should be used immediately after unsealing the package. Contact with air leads to adsorption of contaminants, which decreases the activity of the surface gradually. The chamber should be filled with buffer immediately after assembly. Do not fill the entire chamber volume to allow for addition of the rehydrated lipids in the subsequent step. - Place the chamber onto the confocal microscope stage. Transfer the rehydrated lipid material, now a suspension containing giant vesicles, into the chamber with a plastic Pasteur pipette (Figure 1A-1G).
- Wait 10-20 min to let the vesicles adhere onto the substrate and spread across the surface (Figure 1H-1J).
NOTE: The spreading starts immediately after deposition of lipids on the surface. The rate of spreading might slightly vary depending on the lipid composition, Al2O3 deposition technique (ADL, RF-sputtering, chemical vapor deposition, etc.), freshness of the substrate, and divalent cation concentration in the buffer. Ensure that buffer exchange is performed before rupturing of the spreading patches14. - After observing multiple lipid spreads, slowly remove the ambient buffer via an automatic pipette, such that only a thin buffer film remains on the bottom.
NOTE: Rapid removal of buffer perturbs the lipid structures on the surface. - Proceed to the ambient buffer exchange by slowly filling the observation chamber with chelator-HEPES buffer (see Table of Materials) using an automatic pipette (Figure 1K).
NOTE: Abrupt addition of buffer perturbs the lipid structures on the surface. - This final step yields dynamic nanotubular networks, formed as a result of the chelator-induced depinning and retraction of the DLBM to the MLV4 (Figure 1L-1Y).
4. Microscopy Observation
- Acquire the images with an inverted laser scanning confocal microscope using a 40X oil (1.3 NA) immersion objective with a scanning frequency of 400 Hz. Employ a white light laser source to excite the Texas Red DHPE at 595 nm. Collect the emission from 605 to 700 nm by using a hybrid photon detector.
NOTE: Alternatively, an epi-fluorescence microscope can be used for imaging. Depending on the light sources available, select an appropriate lipid-dye conjugate.
Results
The lipid suspension obtained in step 1 of the protocol and used throughout the experiments contains two main types of vesicles: MLVs and GUVs. Figure 1A-1F shows laser scanning confocal micrographs of the vesicles in the initial sample constructed in 3D. Figure 1A-1C shows an MLV (lipid deposit) in the xyz, xz, and yz planes, respectively. Figure 1D-1F shows similar views of a giant unilamellar vesicle (GUV). The inner part of the GUVs, which lack multilamellarity, is hollow; hence, the lipid material for spreading is significantly limited. Therefore, the only useful lipid reservoirs for this method are MLVs.
When the lipid suspension is transferred to the observation chamber containing the Ca2+-HEPES buffer (Figure 1G), the MLVs (Figure 1A-1C) start to settle on the Al2O3 surface. Upon contact, the vesicles adhere to the surface, and a circular flat double lipid bilayer membrane (DLBM) starts to spread from each MLV onto the solid support (Figure 1H-1J). The MLV acts as a reservoir that provides the lipids for the continuously expanding DLBM. The distal (upper) bilayer membrane, with respect to the solid support, is connected to the proximal (lower) bilayer membrane along the circumference of the circular edge, performing a rolling motion (Figure 1I). The two bilayers are positioned flatly on top of each other, only having a thin liquid film encapsulated between them. During spreading, the proximal membrane continuously adheres to the support surface underneath, while the distal membrane is laterally pulled at the edges by the expanding proximal membrane edge. The spreading of the DLBM is mediated by Ca2+, which acts as a fusogenic agent between lipid head groups from the proximal membrane and solid substrate4.
If the spreading continues for prolonged periods, the membrane tension increases, leading to ruptures (Figure 2A and 2B). After that point, membrane retraction, which is necessary to create the ER tubular morphology, can no longer be induced. Therefore, it is important to recognize the ruptured membranes. Since the lipids in our experiments are fluorescently labelled, the rupturing can be directly observed. A key indicator of rupturing is the significant drop in fluorescence intensity in the ruptured region (Figure 2A and 2B)14. The rupturing is a result of the increased tension and subsequent pore formation in the distal membrane. Under a fluorescent microscope, the ruptured regions will therefore exhibit half the emission intensity (single proximal bilayer) of the unruptured regions (double bilayer)14 (Figure 2B). During pore formation, the membrane material, which was initially positioned at the ruptured regions, migrates to the edges of the spreading patch. This in turn causes a growth of the overall patch area. Therefore, a rapid expansion of the contour of the circular patch can also be observed during rupturing.
To avoid the extensive growth leading to the rupture of the patches, immediately after the circular patch area reaches 100-200 µm, the Ca2+-HEPES buffer is gently removed with an automatic pipette until a thin film of liquid remains on the surface. The Chelator-HEPES buffer is then gently added to the chamber to initiate the retraction (Figure 1K). A complete dehydration of the sample (Figure 2C) or rapid exchange of buffers (Figure 2D and 2E) causes disturbance, rupturing, or deformation of the patches. The addition of chelators gradually removes Ca2+ from the space between the surface and membrane. The Chelator-HEPES buffer gradually accesses the inter-bilayer-substrate space, starting from the periphery of the lipid membrane patch. Therefore, the removal of the pinning sites starts from the edges of the circular patch and propagates inwards (Figure 1K-1Q). As a result of depinning, the lipid membrane starts to detach and retract from the edges inwards, progressing towards the MLV at the center. The retraction process leads to a new interface of dynamically developing lipidic tubular networks4 (Figure 1L-1Y). The persistent regions of pinning, which do not allow the membrane to detach completely, remain on the surface and nucleate tabulation, leading to long branched network of nanotubes. (Figure 1L-1Y). Continuous de-pinning and further retraction of lipid nanotubes are observed over time as result of the gradual chelation process. This coarsening and rearrangement of the network branches plays a key role in the dynamic behavior of the tubular networks, which resemble the smooth ER.
Figure 1L-1Y shows the micrographs of the nanotube networks obtained in the protocol. Figure 1L is a close-up of the region in Figure 1M marked in the white frame. The continuous bright-red regions in Figure 1L and 1M are the retracting fraction of the DLBM (marked with a blue dashed line in Figure 1M). The micrograph of a tubular network in Figure 1N and 1O is inverted to increase contrast. Figure 1P and 1Q depicts the reduction of the tubular density on a membrane region over the course of 3 h and 20 min. The decrease of tubular density occurs due to the gradual depinning followed by retraction of the DLBM from the surface over the experimental time period. Over time, the number of points freed from pinning increases, leading to rearrangements and a reduction of the area covered by tubes (Figure 1P and 1Q). The tubular rearrangements are motivated by surface free energy minimization of a lipid nanotube suspended between two fixed points. It is well-established that the most efficient way of minimizing the surface energy of a nanotube is to reduce its length15. Therefore, when the fusogenic regions, which initially hold the nanotubes on the surface, are de-pinned, the nanotubes slide and arrange themselves spontaneously, adopting a minimal length. These rearrangements cause a gradually reduced coverage of the surface by the nanotubes (Figure 1P and 1Q).
We cannot visualize the Ca2+-mediated pinning points, but we establish their locations as the points where tubes have terminal or sharp turns. Sharp turns are referred to as V-junctions15 or turning-points because of the sudden shift in direction of the tube's alignment (green arrows in Figure 1R and 1X). The end-point represents the terminus of the tube, which prevents the tube from retracting (orange arrows in Figure 1X). During re-organization, energetically favorable disposition of tubes identified as "Y-junctions" or "3-way junctions", appear. The Y-junction connects three tubes with approximately 120° angles between each tube, where the shortest total tube length can be secured. The Y-junctions, which do not possess an end-point and instead are positioned between multiple nanotubes, are not pinned. This is the only Y-junction type that can perform sliding (blue arrows, Figure 1R). As shown in Figure 1R and 1S, sliding of a Y-junction along a highly instable intersection results in the formation of two individual Y-junctions (blue arrows in Figure 1S). The dashed white line superimposed onto Figure 1S represents the contour of the tubular network fragment in Figure 1R. A fraction of the Y-junctions possesses end terminals (yellow arrow in Figure 1T) which, over time, eventually retract (Figure 1U). The transformation of a V-junction to a single, straight tube by depinning of the intersection point of the two line segments and by retraction of one of the tubes forming the V shape can be observed in Figure 1V and 1W and Figure 1X and 1Y, respectively.

Figure 1: Transformation of lipid deposits to ER-like tubular networks. (A-F) Laser scanning confocal micrographs of the vesicles in the initial sample, constructed in 3D. (A-C) Multilamellar lipid vesicle (MLV, lipid deposit) in xyz, xz, and yz planes, respectively. (D-F) Similar views of a giant unilamellar vesicle (GUV). The inner part of the GUVs is hollow, which makes the lipid material for spreading significantly limited. The useful lipid reservoirs for this method are therefore MLVs. (G) Illustration of the observation chamber mounted on an inverted microscope onto which the buffer and lipids are deposited. The chamber is composed of a PDMS frame adhered onto an Al2O3 coated coverslip, providing an open volume top. (H-J) Illustration of the spreading phenomena of the MLV in the presence of Ca2+. (H) Upon contact with Al2O3, the MLV spontaneously spreads in form of a circular, double lipid bilayer membrane (DLBM). (I) Schematic side view of the DLBM in the xz plane, where the peripheries perform rolling motion. MLV (d = 5-15 µm) and DLBM (thickness = 10 nm) are not drawn to scale. (J) A confocal micrograph of a spreading DLBM from top-view. (K) Describes the main step of this protocol, where the buffer is exchanged to one containing Ca2+ chelating agents, inhibiting the spreading, causing retraction of the DLBM to the MLV, and leading to formation of the lipid nanotubes. (L-Y) Micrographs of the nanotube networks obtained with the described method. (L) Close-up of the region in (M) marked in a frame. The continuous bright-red regions in (L and M) represent the DLBM (also marked with a blue dashed line in M). The micrograph of a tubular network in (N and O) is inverted to increase contrast. (P and Q) Depiction of the reduction of the tubular density on a membrane region over the course of 3 h and 20 min. (R-Y) Representative tubular re-arrangements. (R and S) Sliding, (T and U) transitioning of a Y-junction to V-junction by retraction of one end-point, and (V and W) de-pinning of a turning point, resulting in the eradication of a V-junction. (X and Y) Retraction of an end-point. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Potential negative results. (A and B) Rupturing of the distal membrane due to a long waiting time before the exchange of the buffers. The ruptured areas, where the proximal membrane becomes visible, appear as dark regions in comparison to the unruptured distal membrane regions. The inset to panel B shows the light intensity along the blue arrow on the micrograph. The intensity of the ruptured regions of the membrane (proximal/single bilayer becomes visible) corresponds to half the intensity of the unruptured membrane (distal/double bilayer). (C) Appearance of a dry lipid patch formed as a result of removal of all liquid from the observation chamber. (D and E) Disruption of the membrane by rapid exchange of buffers via automatic pipette. In (D), the MLV has split into 2 MLVs, leading to a non-circular, deformed lipid patch. In (E), the pattern of the flow (arrows), created by strong injection of the chelator-HEPES buffer, is reflected on the tubulated membrane structure. Please click here to view a larger version of this figure.
{kind=link}
Discussion
In the following discussion, the critical steps, possible modifications, and limitations of the protocol are described. The first critical step is the proper assembly of the observation chamber, given that the adhesion of the PDMS frame to the Al2O3 surface is intrinsically weak. In the case where the frame does not adhere to the substrate properly, the content will leak from the observation chamber and the experiment will come to a halt. The main factors that hinder proper sealing of the surface and frame are 1) a lack of thorough cleaning of the (re-used) PDMS frame and 2) air bubbles that occasionally get entrapped between frame and substrate. A newly prepared or thoroughly cleaned PDMS frame should be used. The frame should be rinsed before and after each use with isopropanol, followed by rinsing with DI water and blow-drying with nitrogen. The Al2O3 surfaces do not require any prior cleaning, since they are fabricated in a cleanroom environment and kept in sealed containers until use. Due to the amphoteric nature of Al2O3, it should not be exposed to strongly acidic or basic solutions. Other designs for the observation chamber can be employed, depending on accessibility of the individual setup. Important features of this chamber are free access to the liquid sample from the open top and the inertness of the frame material with respect to the used solutions and samples. The chamber dimensions are also a significant factor, as they should accommodate a volume from 0.5 to 1 mL. Since the surfaces used are typically standard-sized coverslips (24 x 60 mm), the volume of the chamber is mostly determined by the thickness of the frame. To our knowledge, spacers with the size and depth which can accommodate the sample volumes typically handled in this protocol are not commercially available. We therefore have dedicated a section in the protocol to details of the fabrication and assembly of a sample chamber frame.
The other critical step in this protocol is the buffer exchange. A challenge in this step is the timing needed to perform this exchange. The spreading of the MLV upon contact with the Al2O3 substrate is instant, and the continuous expansion of the DLBM leads to its rupturing, terminating the experiment (Figure 2A,B). Therefore, the spreading should be constantly monitored, and the buffer exchange must be performed in a timely manner. The exchange should not be performed too quickly after initialization of the spreading, in order to allow the membrane patches to reach an optimal size (100-200 µm in diameter). On the other hand, continuous adhesion on the surface causes high membrane tension, which leads to rupturing. Thus, all membrane patches eventually rupture if spreading is not interrupted. The timing of the rupturing differs for each patch, since it depends on the size and internal structure of the MLV and accessibility of the lipids therein. Hence, the moment of the exchange should be arranged to a timepoint at which the un-ruptured patches with optimal sizes represent a majority of the entire population. Another challenge in the buffer exchange step is the rate of removal and addition of the buffers. Performing this substitution too rapidly has a detrimental influence on the final membrane structures (Figure 2C-E). The excessive extraction of the Ca2+-HEPES buffer without leaving a thin liquid film on the substrate, results in dried and irreversibly deformed membrane patches (Figure 2C). Even if a proper amount of liquid is maintained on the surface, abrupt addition of the Chelator-HEPES buffer also causes perturbation of the membrane structures. Figure 2D,E shows the typical appearance of hydrodynamically disrupted membrane patches. The overall morphological disruption does not necessarily influence the dynamic properties of the final structures (i.e., the tubular rearrangements in remaining areas will still occur). However, it will become difficult to observe the material transformation on the deformed structures. For example, in Figure 2D, it would be difficult to determine the direction towards which MLV the DLBM retracts.
One possible modification of the protocol is the lipid composition used. The main focus has been on phospholipids that dominate the ER composition in mammals and yeast16 (e.g., phosphatidylcholine (PC), phosphatidylethanolamine (PE), and phosphatidylinositol (PI). The original experiments were performed using PC and PI mixtures4. In the results presented, a mixture of PC and DOPE and a derivative of PE was used. However, not all arbitrary lipid compositions have been found to create the tubular structures obtained through this protocol. A few of the other experimentally investigated lipid mixtures involve total heart extract, soy bean extract polar, E. coli extract polar, mixtures of PC with stearoyl-2-hydroxy-sn-glycero-3-phosphoinositol (Lyso-PI) in varying ratios, and mixtures of PC-PE-PI-Posphatidyl serine (PS) in varying ratios. Since tubular membrane structures possess high curvatures and require special arrangement of individual lipid molecules, it is expected that the observed phenomenon is lipid composition-specific.
Another modification applied in this protocol is the method for surface fabrication. Here, the ALD was used to fabricate the Al2O3 coated coverslips. This differs from the originally reported deposition method, reactive sputtering4. While this indicates that an alternative surface fabrication method can still lead to ER-like tubulation, one important limitation appears to be the specificity of the surface material. The mode of spreading and strength of adhesion is highly dependent on properties of the surface material, which influence factors such as electrostatic interactions, wettability, hydrophobicity, and roughness of the surface. Al2O3 surfaces provide optimal adhesion strength, and lipid films can both attach strongly enough to spread as a double lipid bilayer membrane and detach to form tubular networks upon removal of Ca2+ ions. We previously tested the same experiment with SiO2, in which the multilamellar vesicles spread as a double lipid bilayer membrane, but no tubular network formation was observed upon addition of chelators17. De-attachment and tube formation are observed only on Al2O3 or plasma etched Al18. Our investigations revealed that the contributing parameter leading to such a phenomenon was the zeta potential of the surfaces, for which Al and Al2O3 were close to zero (mV) and SiO2 significantly negative. The zeta potential of borosilicate is similar to SiO219; therefore, adhesion of lipid films on borosilicate is equally strong and irreversible. In fact, multilamellar lipid reservoir contact with borosilicate surfaces typically leads to immediate rupture and formation of single lipid bilayers20. The Al2O3 surfaces required for this protocol are not readily or commercially available. They can, however, be custom-ordered from specialty glass and substrate manufacturers. Access to cleanroom facilities with thin-film fabrication equipment is highly recommended.
The other existing bottom-up methods to fabricate ER-like tubular networks2,10 involve proteins as well as input of chemical energy (e.g., GTP and ATP). Rapoport and colleagues2 reported the formation of ER-networks on glass coverslips in vitro by mixing the membrane-bending proteins present in ER, with phospholipids and GTP. The work by Bachand et al.10 shows how such dynamic tubular networks can be created by using molecular motors and ATP as the energy source. This protocol presented does not require membrane proteins or hydrolysis of organic compounds for energy. The only essential components are the solid substrate and the phospholipids. Purification and extraction of proteins are not required. This protocol provides, in terms of simplicity of the constitutive molecules, the most basic ER model.
With this basic, lipid-based ER model established, building up complexity by adding ER-associated components is of interest, since it enables investigation of individual impacts on the system. Similar to the actual ER networks, tubes in the model are dynamic. The conjugation to and migration of the labeled membrane proteins, or fluorescent particles throughout the tubular network, may give information about the direction of membrane movement. Encapsulation and monitoring of the fluorescent liquids inside the DLBM and tubes during the transformation and a possible mapping of the intratubular content transport may serve as another focus. Finally, a transition from the 2D ER model resulting from this protocol towards a 3D smooth ER model may be adopted through encapsulation of the networks in hydrogel architectures.
Disclosures
The authors have nothing to disclose.
Acknowledgements
We thank Prof. Aldo Jesorka from the Chalmers University of Technology in Sweden for his invaluable comments on the manuscript. This work was made possible through financial support obtained from the Research Council of Norway (Forskningsrådet) Project Grant 274433, UiO: Life Sciences Convergence Environment, the Swedish Research Council (Vetenskapsrådet) Project Grant 2015-04561, as well as the start-up funding provided by the Centre for Molecular Medicine Norway & Faculty of Mathematics and Natural Sciences at the University of Oslo.
Materials
| Name | Company | Catalog Number | Comments |
| Pear-shape flask 10 mL | Lenz Laborglasinstrumente | 3.0314.13 | In which the lipid mixture is prepared |
| Hamilton 5 mL glass syringe (P/N) | Hamilton | P/N81520 | For transfer of the chloroform to beaker |
| Custom large hub needle Gauge 22 S | Hamilton | 7748-18 | Removable needle for syringe specified in row 3 |
| Hamilton 250 µL glass syringe | Hamilton | 7639-01 | Used for transfer of lipids in chloroform to the flask |
| Large hub Gauge 22 S | Hamilton | 7780-03 | Removable needle for syringe specified in row 5 |
| Hamilton 50 µL glass syringe | Hamilton | 7637-01 | Used for transfer of fluorophore-conjugated lipids to the flask |
| Small hub Gauge 22 S | Hamilton | 7770-01 | Removable needle for syringe specified in row 7 |
| Chloroform anhydrous (≥99%) | Sigma-Aldrich | 288306 | Used to complete the lipid mixture to a total of 300 µL |
| Soy L-α Phosphatidyl choline lipid (Soy PC) | Avanti Polar Lipids Inc | 441601 | phospholipid species contributing to 69% of the total composition/mixture |
| 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) | Avanti Polar Lipids Inc | 850725 | phospholipid species contributing to 30% of the lipid composition/mixture |
| L-α Phosphatidyl inositol lipid (Soy PI) | Avanti Polar Lipids Inc | 840044 | alterative phospholipid species contributing to 30% of the lipid composition/mixture (from the original article Bilal and Gözen, Biomaterials Science, 2017) |
| Texas Red 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine,triethylammonium salt (Texas Red DHPE) | Invitrogen (Thermo Fisher Scientific) | T1395MP | Fluorescent-lipid conjugate, 1% of the lipid composition/mixture |
| Digital Dry Baths/Block Heaters | Thermo Fischer | 88870006 | To warm glycerol in order to decrease its viscosity |
| Glycerol for molecular biology (≥99%) | Sigma Life Science | G5516 | For lipid preparation |
| PBS buffer (pH=7.8); ingredients below in rows 17-21 | Used to prepare the lipid suspension | ||
| TRIZMA base, primary standard and buffer (≥99%) | Sigma Life Science | T1503 | Used to prepare PBS buffer |
| Potassium phosphate tribasic, reagent grade (≥98%) (K3PO4) | Sigma-Aldrich | P5629 | PBS buffer ingredient |
| Magnesium sulfate heptahydrate, BioUltra (≥99,5%) KT (MgSO47H2O) | Sigma Life Science | 63138 | PBS buffer ingredient |
| Potassium phosphate monobasic, anhydrous, free flowing, Redi-Dri, ACS (KH2PO4) | Sigma-Aldrich | 795488 | PBS buffer ingredient |
| Ethylenediaminetetraacetic acid disodium salt dihydrate ACS reagent, 99.0-101.0% (Na2EDTA) | Sigma-Aldrich | E4884 | PBS and Chelator-HEPES buffer ingredient |
| Ultrasonic cleaner USC-TH | VWR | 142-0084 | Ultrasonication of rehydrated lipids |
| Rotary evaporator - Büchi rotary evaporator Model R-200 | Sigma | Z626797 | For evaporation of chloroform |
| Pressure meter - Vacuum regulator IRV-100 | SMC | IRV10/20 | For controlling the pressure value during lipid dehydration |
| HEPES-buffer (pH=7.8); ingredients below in rows 26-27 | Used for rehydration of lipids. Content: 10 mM HEPES with 100 m NaCl diluted in ultrapure deionized water | ||
| HEPES ≥99.5% (titration) | Sigma Life Science | H3375 | HEPES-buffer ingredient |
| Sodium chloride for molecular biology, DNase, RNase, and protease, none detected, ≥98% (titration) (NaCl) | Sigma Life Science | S3014 | HEPES-buffer ingredient |
| Calcium-HEPES buffer (pH=7.8); effective ingredient below in row 29 | Used for spreading of lipids. Content: 10 mM HEPES, 100 mM NaCl, 4 mM CaCl2 diluted in ultrapure deionized water | ||
| Calcium chloride anhydrous, BioReagent, suitable for insect cell culture, suitable for plant cell culture, ≥96.0% (CaCl2) | Sigma Life Science | C5670 | To prepare Calcium-HEPES buffer |
| Chelator-HEPES buffer (pH=7.8); effective ingredient below in row 31 | Used to promote the formation of tubular networks. Content: 10 mM HEPES, 100 mM NaCl, 10 mM EDTA and 7 mM BAPTA diluted in ultrapure deionized water | ||
| 1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrasodium salt ≥95% (HPLC) (BAPTA-Na4) | Sigma Life Science | 14513 | Chelator-HEPES buffer ingredient |
| Sodium Hydroxide | Sigma | 30620 | Basic solution used to adjust the pH of the buffers |
| pH meter accumet™ AE150 pH | Fisher Scientific | 1544693 | Used to measure the pH of all buffers |
| Glass petri dish | VWR | HECH41042012 | 6 cm, used for making the PDMS sheet |
| Potassium hydroxide ACS reagent, ≥85%, pellets (KOH) | Sigma-Aldrich | 221473 | To make the KOH solution for cleaning glass petri dish for the fabrication of the PDMS sheet |
| Isopropanol prima ren 99.5% | Antibac AS | 600079 | KOH solution ingredient |
| Heating and drying oven - venticell | MMM Medcenter Einrichtungen GmbH | MC000714 | For drying of the glass petri dish after silanization and to cure PDMS |
| Dichlorodimethylsilane ≥99.5% | Sigma-Aldrich | 440272 | Used for silanization of glass petri dish in which PDMS sheet is prepared |
| Vacuum pump | Cole-Parmer | EW-79202-05 | Connected to desiccator |
| Sylgard 184 silicone elastomer curing agent | Dow corning | 24236-10 | Kit to make PDMS solution |
| Sylgard 184 Silicone elastomer base | |||
| Disposable scalpel | Swann-Morton | 11798343 | Used to cut the PDMS |
| Cover slips | Menzel -Gläser | MEZ102460 | 24x60 mm. Used to deposit thin film of Al2O3 |
| Atomic layer deposition system | Beneq | TFS200 (model number) | Atomic Layer deposition system used to deposit thin film of Al2O3 in microscope cover glass |
| Ellipsometer | J.A. Woollan Co. | Alpha-SE (model name) | System used to charcaterize the thickness of the film deposited on glass surface |
| Laser scanning confocal microscope | Leica Microsystems | Leica TCS SP8 X | Microscope used for visualization of the experiment |
| Objective 40x, 1.3 NA | Leica Microsystems | 1550635 | Used for visualization of the experiment |
| White light laser source | Leica Microsystems | Leica TCS SP8 X | For excitation of the membrane fluorophore |
References
- Chen, S., Novick, P., Ferro-Novick, S. ER structure and function. Current Opinion in Cell Biology. 25 (4), 428-433 (2013).
- Powers, R. E., Wang, S., Liu, T. Y., Rapoport, T. A. Reconstitution of the tubular endoplasmic reticulum network with purified components. Nature. 543 (7644), 257-260 (2017).
- Pendin, D., McNew, J. A., Daga, A. Balancing ER dynamics: Shaping, bending, severing, and mending membranes. Current Opinion in Cell Biology. 23 (4), 435-442 (2011).
- Bilal, T., Gözen, I. Formation and dynamics of endoplasmic reticulum-like lipid nanotube networks. Biomaterials Science. 5 (7), 1256-1264 (2017).
- Shibata, Y., et al. Mechanisms determining the morphology of the peripheral ER. Cell. 143 (5), 774-788 (2010).
- Ozcan, L., Tabas, I. Role of endoplasmic reticulum stress in metabolic disease and other disorders. Annual Review of Medicine. 63, 317-328 (2012).
- Yamanaka, T., Nukina, N. ER dynamics and derangement in neurological diseases. Frontiers in Neuroscience. 12, (2018).
- Taalab, Y. M., et al. Mechanisms of disordered neurodegenerative function: Concepts and facts about the different roles of the protein kinase RNA-like endoplasmic reticulum kinase (PERK). Reviews in the Neurosciences. , (2018).
- Shemesh, T., et al. A model for the generation and interconversion of ER morphologies. Proceedings of the National Academy of Sciences of the United States of America. 111 (49), 5243-5251 (2014).
- Bouxsein, N. F., Carroll-Portillo, A., Bachand, M., Sasaki, D. Y., Bachand, G. D. A continuous network of lipid nanotubes fabricated from the gliding motility of kinesin powered microtubule filaments. Langmuir. 29 (9), 2992-2999 (2013).
- Sackmann, E. Endoplasmatic reticulum shaping by generic mechanisms and protein-induced spontaneous curvature. Advances in Colloid and Interface Science. 208, 153-160 (2014).
- Jesorka, A., et al. Generation of phospholipid vesicle-nanotube networks and transport of molecules therein. Nature Protocols. 6, 791 (2011).
- Hook, D. A., Olhausen, J. A., Krim, J., Dugger, M. T. Evaluation of Oxygen Plasma and UV Ozone Methods for Cleaning of Occluded Areas in MEMS Devices. Journal of Microelectromechanical Systems. 19 (6), 1292-1298 (2010).
- Gözen, I., et al. Fractal avalanche ruptures in biological membranes. Nature Materials. 9 (11), 908-912 (2010).
- Lobovkina, T., Dommersnes, P., Joanny, J. -. F., Hurtig, J., Orwar, O. Zipper Dynamics of Surfactant Nanotube Y Junctions. Phys Rev Lett. 97, (2006).
- van Meer, G., Voelker, D. R., Feigenson, G. W. Membrane lipids: where they are and how they behave. Nature Reviews Molecular Cell Biology. 9 (2), 112-124 (2008).
- Gözen, I., et al. Repair of large area pores in supported double bilayers. Soft Matter. 9 (10), 2787-2792 (2013).
- Gözen, I., et al. Thermal migration of molecular lipid films as a contactless fabrication strategy for lipid nanotube networks. Lab on a Chip. 13 (19), 3822-3826 (2013).
- Sides, P. J., Hoggard, J. D. Measurement of the Zeta Potential of Planar Solid Surfaces by Means of a Rotating Disk. Langmuir. 20 (26), 11493-11498 (2004).
- Nissen, J., Jacobs, K., Rädler, J. O. Interface Dynamics of Lipid Membrane Spreading on Solid Surfaces. Physical Review Letters. 86 (9), 1904-1907 (2001).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved