
Method Article
Real-Time Monitoring of Aurora kinase A Activation using Conformational FRET Biosensors in Live Cells
In This Article
Summary
The activation of the multifunctional Ser/Thr kinase AURKA is hallmarked by its autophosphorylation on Thr288. Fluorescent probes relying on FRET can discriminate between its inactive and activated states. Here, we illustrate some strategies for probe engineering, together with a rapid FRET protocol to follow the kinase activation throughout mitosis.
Abstract
Epithelial cancers are often hallmarked by the overexpression of the Ser/Thr kinase Aurora A/AURKA. AURKA is a multifunctional protein that activates upon its autophosphorylation on Thr288. AURKA abundance peaks in mitosis, where it controls the stability and the fidelity of the mitotic spindle, and the overall efficiency of mitosis. Although well characterized at the structural level, a consistent monitoring of the activation of AURKA throughout the cell cycle is lacking. A possible solution consists in using genetically-encoded Förster’s Resonance Energy Transfer (FRET) biosensors to gain insight into the autophosphorylation of AURKA with sufficient spatiotemporal resolution. Here, we describe a protocol to engineer FRET biosensors detecting Thr288 autophosphorylation, and how to follow this modification during mitosis. First, we provide an overview of possible donor/acceptor FRET pairs, and we show possible cloning and insertion methods of AURKA FRET biosensors in mammalian cells. Then, we provide a step-by-step analysis for rapid FRET measurements by fluorescence lifetime imaging microscopy (FLIM) on a custom-built setup. However, this protocol is also applicable to alternative commercial solutions available. We conclude by considering the most appropriate FRET controls for an AURKA-based biosensor, and by highlighting potential future improvements to further increase the sensitivity of this tool.
Introduction
Aurora kinase A/AURKA is a multifunctional serine/threonine kinase, active throughout the cell cycle and at different subcellular compartments1. Understanding its broad spatiotemporal activation is particularly important in cancer, as AURKA is often overexpressed in epithelial and hematological malignancies, with patients showing poor responsiveness to the currently available therapies2.
Structural studies revealed that AURKA undergoes two steps to convert from an inactive to an active kinase. First, the autophosphorylation of Thr288 changes the conformation of the kinetic pocket of the kinase and activates it3,4,5,6. This step increases the catalytic activity of AURKA in human cells and in Xenopus laevis3,6,7, priming the kinase for full activity. Once activated, the interaction of AURKA with the targeting protein for Xklp2 (TPX2) induces a second conformational change5. This further modification allows AURKA to reach full enzymatic activity towards its substrates in the cell5,8,9,10.
For nearly two decades, insights on the activation and activity of AURKA were obtained mainly through a combination of biochemical approaches. These include the detection of phosphorylated Thr288 in cells or in vivo as a hallmark of AURKA activation, crystallographic analyses, and in vitro or in cellulo kinase assays to probe the activity of AURKA1. However, the spatiotemporal resolution of these approaches is poor or absent, and novel solutions were needed to broaden the knowledge of the dynamics of these two events.
The development of fluorescent probes in the last few years facilitated the monitoring of AURKA in live cells, allowing the following of its activation with greater spatiotemporal resolution. The most specific sensors for AURKA developed so far rely on the FRET principle (Förster’s Resonance Energy Transfer)11 to discriminate between inactive and active AURKA. The first sensor developed was a substrate-based biosensor of AURKA kinase activity. Substrate-based biosensors are constituted by a short aminoacidic sequence targeted by a given kinase for phosphorylation, and inserted within a donor/acceptor FRET pair and a binding domain recognizing the phosphorylated residue, which helps the folding of the biosensor for an efficient FRET process12. In the case of AURKA, a 14-aminoacid fragment of KIF2C targeted by phosphorylation was inserted between a CFP-YFP donor/acceptor pair13. However, this sensor has some major drawbacks. First, the KIF2C sequence used in this probe can be targeted both by AURKA and the closely-related kinase AURKB, thereby decreasing the specificity of this biosensor. Second, the sensor relies on the endogenous kinase for phosphorylation. Therefore, FRET efficiency can be undetectable or not significant if the quantities of the kinase are limiting (e.g., in subcellular compartments or cell cycle phases). To overcome these limitations, a new class of AURKA sensor was created known as “conformational sensors”. In these probes, the full-length sequence of AURKA was inserted within a donor fluorophore at the N-terminus, and an acceptor fluorophore at the C-terminus. Inactive AURKA presents an “open” conformation, which brings the N- and C-termini of the kinase away from each other. With such distance between the two termini (> 10 nm), the donor/acceptor pair are in a non-permissive configuration for FRET. On the contrary, autophosphorylated AURKA adopts a “closed” conformation, with the two protein termini and the two fluorophores in proximity. This was shown to allow FRET between the donor and the acceptor, which can be measured using the variations in the donor lifetime14,15. Such probes present several advantages. First, they are genetically-encoded, and they can be used to replace the endogenous kinase in the cell. Second, they rescue the phenotypes induced by the knockdown of AURKA, indicating that they are functional in the cell. Third, they allow to follow the activation of the kinase at different subcellular compartments and throughout the cell cycle. The probes detected the activation of AURKA at locations where the kinase is known to be activated (i.e. centrosomes and the mitotic spindle), and also participated in discovering the activation of AURKA at the mitochondria16. Last, these sensors allowed high-content screenings based on FRET/FLIM, where the conformational changes of AURKA were used to identify novel pharmacological inhibitors17.
In the present work, we describe a procedure to visualize AURKA activation in cultured cells. First, we will make an insight on potential fluorophore pairs for FRET. The choice of the most suitable donor/acceptor pair will be made according to the available microscope setup, or a particular downstream application as multiplex FRET18,19. Then, we propose a pipeline to explore the behavior of the biosensor(s) chosen in a rapid FRET/FLIM microscope setup. This pipeline will extend from cell culture and synchronization procedures to FLIM acquisition and data analysis. Last, we will discuss the potential advantages of this protocol, as an analogous strategy for biosensor design could be applied to other kinases, and it may also be used with other FRET-based imaging systems.
Protocol
NOTE: U2OS cells used in this protocol were purchased from American Type Culture Collection (ATCC, HTB-96), and they were tested free from mycoplasma. Step 2.1 to 2.7 should be performed under a laminar flow hood to keep cells and reagents sterile.
1. Choosing the donor/acceptor FRET pair
- Refer to the literature for the choice of the most suitable donor/acceptor FRET pairs. Useful examples can be found in20,21,22,23,24, although the final choice must be made according to the characteristics of the FRET/FLIM setup (available laser lines, filters, etc.). The following are some considerations on how to select a donor/acceptor pair.
- Choosing the donor: refer to FP base (https://www.fpbase.org/) for a complete set of information on the available fluorescent proteins. This database is constantly updated with all the newly-developed fluorophores.
- Refer to the Fluorescent Biosensor Database (https://biosensordb.ucsd.edu/index.php) for more information on the biosensors already available in the literature, together with the respective fluorescent proteins used.
- As a general starting point, choose a bright donor fluorophore. Good candidates are cyan fluorescent proteins as mTFP1 or ECFP, or GFP variants as EGFP or mEGFP.
- Oligomers can affect protein localization and/or function25. Consider using monomeric mutants of CFP as mTurquoise226, or Aquamarine27,28. These variants also have good quantum yield and extinction coefficients, which make them good candidates as FRET donors.
- Give preference to fluorescent proteins (both as donor or acceptors) insensitive to environmental changes like intracellular pH23, or to photobleaching25, as FRET efficiency can be heavily affected by these parameters29. Nowadays, fluorophores like mTurquoise2 or mTFP1 are extensively used as donors, thanks to their good photostability22,25,26.
- Choosing the acceptor: cyan donors are often paired with Yellow Fluorescent Protein (YFP) variants, as mVenus, Citrine, and YPet20,21,22,30. However, it should be noted that these proteins have a much greater sensitivity to pH, and globally display a poor photostability.
- Consider using newly-developed, pH-insensitive yellow variants of YFP as pH-Lemon31, green fluorophores as mNeonGreen23 or red fluorophores as mScarlet-I23,32 as previously-validated fluorescent acceptors for mTurquoise2.
- Alternatively, consider using non-fluorescent/dark derivatives of YFP as ShadowG33 or ShadowY34, which were shown to behave as good acceptors for cyan-fluorescent donors in FRET/FLIM experiments.
- If using mEGFP as a donor, consider using monomeric red acceptors as mCherry.
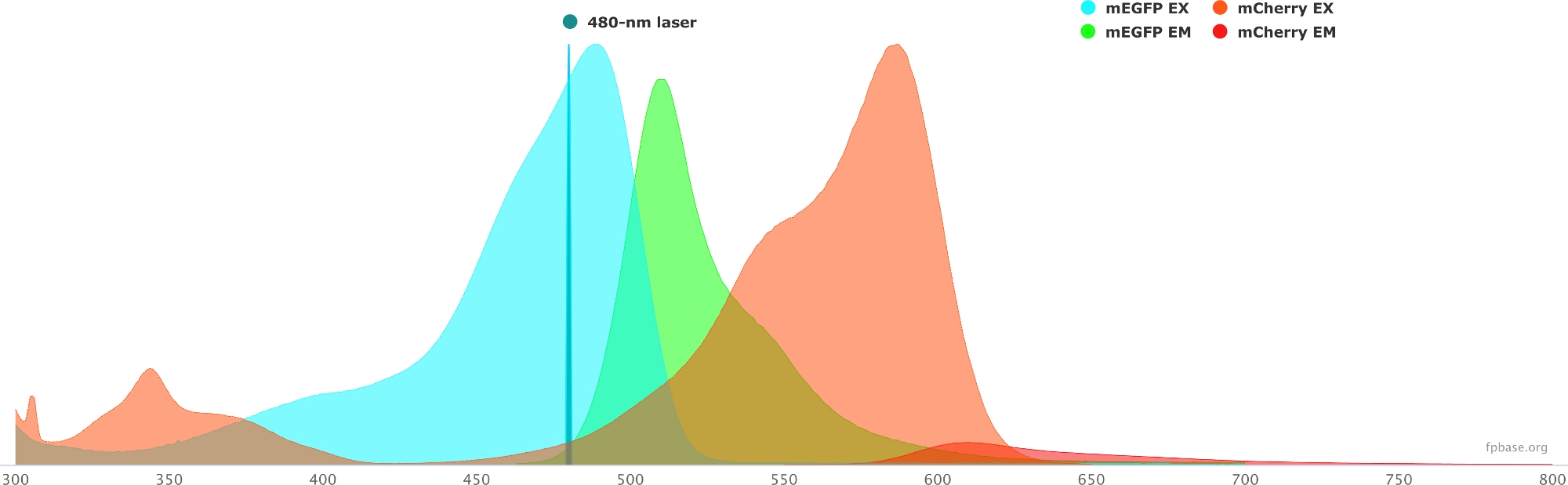
- Verify the spectral properties of the chosen donor/acceptor pair using the tools available on the FP base website. See Figure 1 for an example of the mEGFP/mCherry pair.
- On the FP base website, select the Tools drop-down menu and select Spectra viewer.
- In the drop-down menus, enter the name of the fluorophore pair to visualize (e.g., mEGFP and mCherry).
- Simulate the properties of the donor/acceptor pair with a specific light source by selecting a given laser in the drop-down menu. Alternatively, enter a specific laser wavelength by selecting Add laser. Click on the Normalize emission to this option to adjust the fluorophore spectra to the desired wavelength. Here, the wavelength used to excite GFP is at 480 ± 10 nm.
- Clone the selected donor/acceptor pair by adding one fluorophore at the N-terminus of the full-length sequence of AURKA, and one at the C-terminus. Follow the guidelines of the preferred cloning method to insert this construct in a mammalian expression vector of choice.
2. Cell culture, transfection and synchronization
- DAY 1. Prepare culture medium for U2OS cells: use Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS), 1% Penicillin-Streptomycin and 1% L-Glutamine (from here on, Complete growth media). Alternatively, DMEM with pre-supplemented L-Glutamine can also be used.
- If cells are frozen, thaw a vial at least 8 days prior to experiments (i.e., from point 2.5 on).
- Grow cells in an incubator dedicated to mammalian cell cultures at 37 °C and with 5% CO2. Clean and sterilize the incubator regularly to avoid contaminations.
- When the cells reach ~80% confluence:
- Wash them briefly with sterile 1x phosphate buffer saline (PBS) without Ca2+ and Mg2+.
- Trypsinize cells with sterile 0.05% Trypsin-EDTA according to manufacturer’s protocol and by placing cells in the incubator for 1-3 min.
- Inactivate trypsin by adding twice the volume of complete growth media; mix well.
- Centrifuge the cell suspension at 800 x g for 3-5 min.
- Count the cells using a hemocytometer and calculate the appropriate dilution for them to be at ~70-80% confluency in chamber slides the day after. Alternatively, similar supports for live cell imaging can also be used.
- Pipet the corresponding volume of cells in the chosen live cell imaging support, and place cells back in the incubator until the day after.
- DAY 2. Proceed with transfection. Follow the guidelines of the preferred transient transfection method(s) to obtain an optimal transfection efficiency (~50/80%). No specific transfection method is required. Note that the transfection efficiency may vary according to the cell line used. Incubate for 48 h.
NOTE: Produce stable clones containing each of the three vectors to bypass the need of transient transfections. At this stage, two types of controls should be planned. First, a “donor-only” control is required to verify that the presence of full-length AURKA does not perturb the lifetime of mTurquoise2 per se. Second, a biosensor carrying a kinase-dead mutation should be used as a negative control where FRET is abolished or significantly lowered. Alternatively to a kinase-dead mutant, a chemical inhibitor of AURKA activation as the ATP analog MLN8237 can be used as a negative control.- Plan ahead three transfection conditions, each of them in an independent well:
The “donor-only” vector (e.g., AURKA-mTurquoise2)
The “biosensor” (e.g., superYFP-AURKA-mTurquoise2)
A kinase-dead/”K162M” biosensor (e.g., superYFP-AURKA K162M-mTurquoise2) or alternatively, an inhibitor of AURKA activation (e.g., MLN8237) - Carry out three conditions for each independent donor/acceptor pair to compare.
- Consider doubling the number of transfected wells if comparing unsynchronized and G2/M-synchronized cells (see step 2.6).
- Plan ahead three transfection conditions, each of them in an independent well:
- DAY 3. Synchronize cells in G2/M. Add 100 ng/mL nocodazole dissolved in DMSO to each transfected well avoiding light exposure and incubate for 16 h (preferably overnight). If comparing unsynchronized and G2/M-synchronized cells, treat each transfection condition with nocodazole or with an equal volume of DMSO. For a better synchronization efficiency, prepare single-use aliquots of nocodazole in DMSO, store them at -20 °C and discard them after use.
NOTE: Cell synchronization efficiency can vary among cell lines. The optimal concentration of nocodazole and its incubation time should be experimentally determined by flow cytometry approaches prior to FRET/FLIM experiments. For statistically relevant FRET/FLIM analyses, we recommend a synchronization efficiency in G2/M of at least 50% of the overall cell population. - DAY 4. Nocodazole washout and FRET/FLIM imaging on mitotic cells
- Remove the culture medium with a pipette and replace it with pre-warmed, sterile PBS. Avoid light exposure if possible. Gently rock the plate.
- Repeat the washing procedure, always avoiding light exposure.
- Remove the second PBS washing and replace it with pre-warmed, sterile Leibovitz L-15 medium, supplemented with 20% Fetal Bovine Serum (FBS) and 1% Penicillin-Streptomycin (from here on, imaging media).
NOTE: Imaging media should be purchased without pH indicators (e.g., phenol red) and medium components as riboflavin. These substances are a source of autofluorescence that could perturb lifetime values. - Proceed with FRET/FLIM imaging. Minimize rapid changes in temperature and proceed to the imaging step (step 3) as fast as possible. Consider protecting the sample from light while transporting it to the microscope setup (i.e., by wrapping it in an aluminum foil or placing it in a box).
3. FRET/FLIM acquisitions
NOTE: FRET/FLIM acquisitions in this protocol were performed on a custom-built setup, described in35 and equipped with a control solution as in15,17 (Figure 2). The setup is now commercialized by Inscoper and it is made of a spinning-disk microscope with a white laser for pulsed excitation, and a high-rate time-gated intensifier in front of the camera. Temporal gates of 2 ns in a time window of 10 ns are sequentially used to obtain a stack of five time-gated images. These images are then used to calculate the pixel-by-pixel mean fluorescence lifetime according to the following equation: τ = ΣΔti • Ιi/ΣΙi, where Δti corresponds to the delay time of the of the ith gate while I indicates the pixel-by-pixel time-gated intensity image35,36. This method ensures rapid FLIM measurements: no fitting or binning steps are required, and lifetime can be calculated in an online mode, with minimal photon budget. The system also presents a user-friendly software interface. However, the same experiment can be performed under any other commercial microscope setup equipped for FLIM measurements.
- To ensure an optimal release of cells from the G2/M block into mitosis, perform experiments at 37 °C. If possible, perform FRET/FLIM experiments with microscope setups equipped with a thermostatic chamber.
- Turn on the thermostatic chamber of the microscope at least 30 min to 1 h before the experiment.
- Switch on the laser, the camera, the microscope setup and the imaging software (Figure 2).
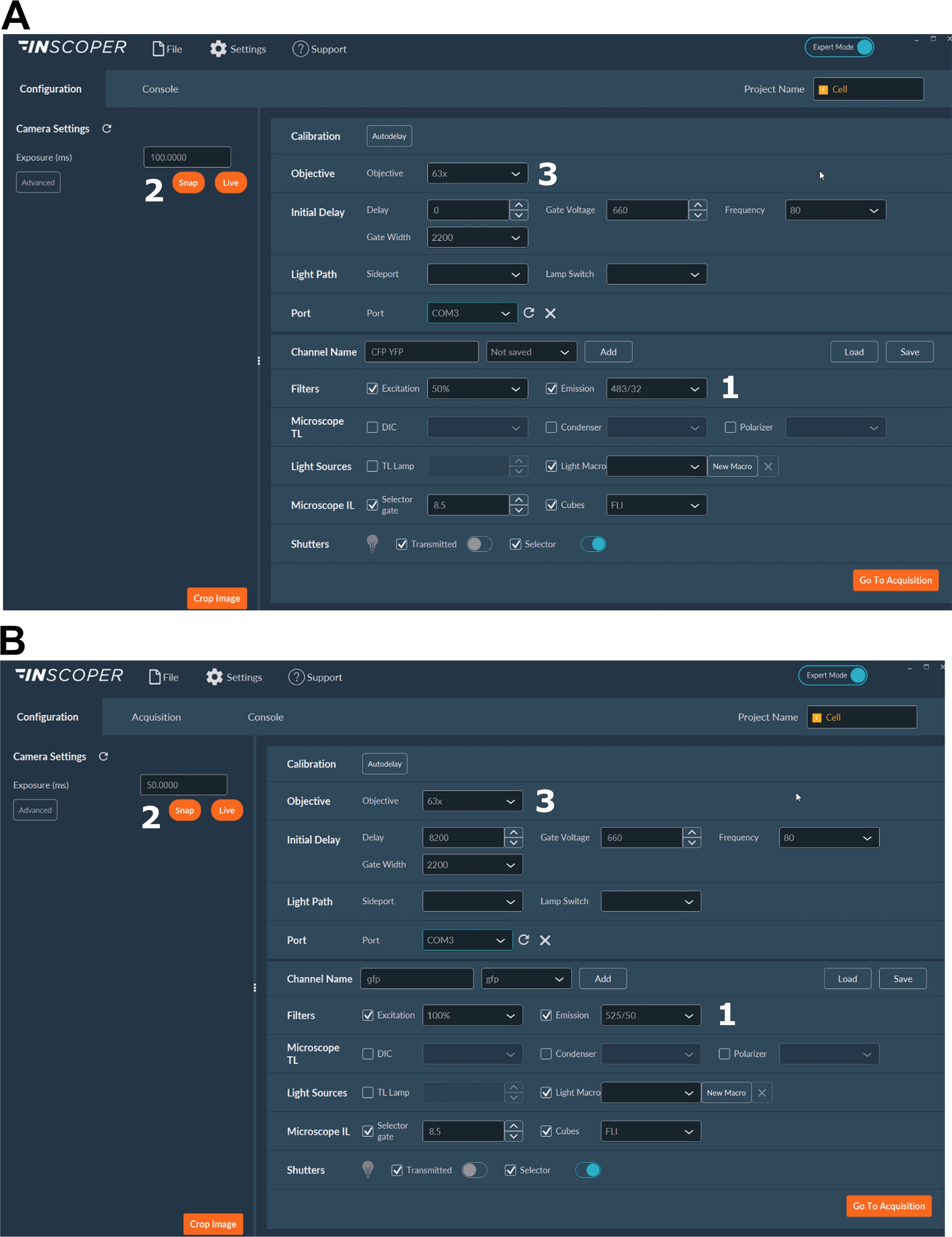
- Select the appropriate excitation and emission wavelengths for the donor fluorophore. Convenient wavelengths options are: λex 440/10 nm and λem 483/35 nm for mTurquoise2 (Figure 3A); λex 488/10 nm and λem 525/50 nm for GFP) (Figure 3B).
- Set the exposure time, generally between 30 and 100 ms (Figure 3). Beware that excessive laser power may result in induced phototoxic effects as photobleaching, which could in turn modify the fluorescence lifetime37. On the setup, validate the absence of photobleaching events by monitoring the fluorescence intensity of the first gate during time-lapse acquisitions. If variations in the fluorescence intensity are observable, discard the acquisition and adjust laser power.
NOTE: In the setup here, select an exposure time allowing at least 3000 gray levels in the first gate; otherwise the software will not calculate donor lifetime. This gray level value corresponds to the minimal photon budget necessary to obtain relevant lifetime values. - Before launching FRET/FLIM acquisitions, ensure that cells have entered mitosis by waiting until the appearance of the bipolar spindle (~20/30 min in U2OS cells). Since mitotic AURKA localizes mainly at this structure, verify mitotic progression by screening the formation of the spindle in cells directly under the microscope, with an external light source (Figure 1). Note that the time required for mitotic progression might vary according to the cell line used.
- If treatment with MLN8237 is planned, place the cells under the microscope and allow them to reach metaphase (approx. 20 min after nocodazole washout). Add 250 nM MLN8237 dissolved in DMSO both to cells expressing the donor-only construct and to cells expressing the biosensor.
- Control this condition on cells transfected as above and incubate with an equal volume of DMSO. For a better AURKA inhibition, prepare single-use aliquots of MLN8237 in DMSO, store them at -80 °C. Thaw them by placing the aliquots on ice, and discard them after use.
- Incubate for 10 min. After this period, the mitotic spindle will shrink and only a single, intense dot is left. A similar phenotype is observed when K162M mutants are used.
- For a better resolution of the mitotic spindle, use at least a 63x objective (Figure 3).
- Once found a cell in metaphase (see Figure 4 as an example of a cell in metaphase), adjust xyz coordinates to place it at the center of the field of view.
- For faster images, select one single z plane. Choose the plane where the mitotic spindle is more visible or intense.
- Start the recording. The acquisition time may vary according to the FLIM setup used (from few sec to min). The majority of the commercial setups available on the market will elaborate both the fluorescence micrograph and the pixel-by-pixel lifetime map. Save both images.
- Acquire at least 10 independent images from each transfection and/or treatment condition.
4. Calculation of ΔLifetime and comparison of FLIM values among donor/acceptor pairs
- Extract lifetime values from the whole pixel-by-pixel lifetime map (i.e., the entire mitotic spindle), or select regions of interest (ROIs) corresponding to specific subregions.
NOTE: According to the FRET/FLIM setup used, lifetime calculations may be performed directly on the acquisition software, or extracted with generic image processing solutions (e.g., Fiji/ImageJ: https://fiji.sc/). Software directly calculating lifetime values (also known as online mode) offer a more user-friendly solution, which is suitable for beginners and for microscopy users not fully familiar with FRET/FLIM. On the contrary, the extraction of lifetime values after acquisition often requires a fitting procedure. This option is less accessible to beginners, as some previous knowledge on mathematical models of fitting is necessary. - Once visualized or extracted, calculate the mean lifetime of the cells expressing the “donor-only” vector (e.g., AURKA-mTurquoise2), hereby mean donor lifetime.
- Subtract each independent lifetime value calculated in step 4.1 from the mean donor lifetime. Repeating this step for all cells in all conditions analyzed will give the ΔLifetime for every condition.
- Compare ΔLifetime values for the “donor-only”, the “biosensor” and the “K162M”, or the DMSO and the MLN8237 conditions.
NOTE: For the “donor-only” condition, ΔLifetime should result in values close to zero and corresponding to the experimental fluctuations of lifetime values. For the “biosensor” condition, ΔLifetime values should yield the net difference between the two constructs (see Figure 4 for an illustrated example). - Compare ΔLifetime values among different donor/acceptor pairs.
- Compare the “biosensor” conditions: do they show similar ΔLifetime?
- Do the “K162M” or the MLN8237 conditions show similar ΔLifetime among them? Is their ΔLifetime similar to the “donor-only” condition?
Results
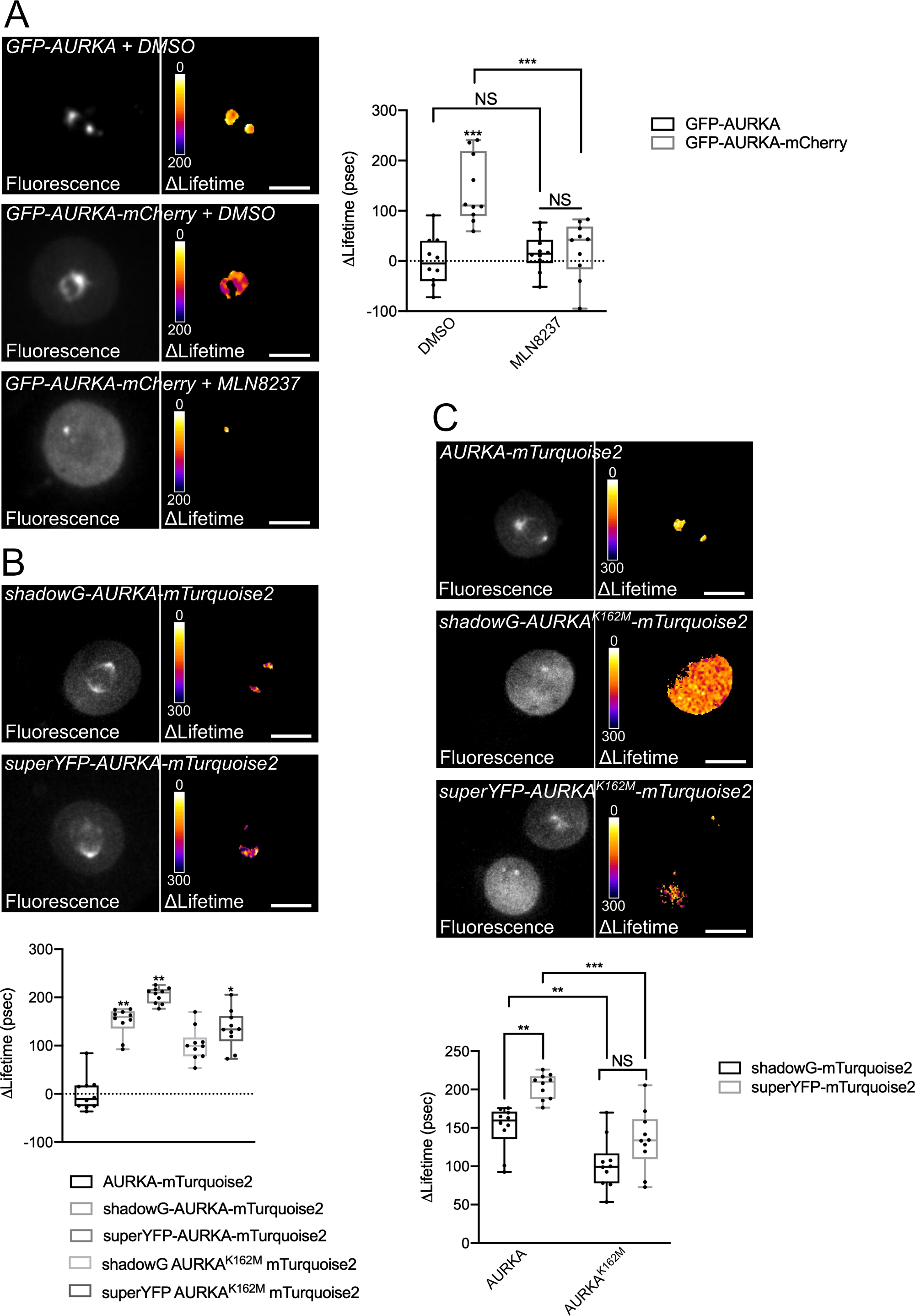
We followed the procedure described above to record the autophosphorylation of AURKA on Thr288 using two biosensors with different spectral properties. We compared the initial GFP-AURKa-mCherry probe14 with two biosensors with different spectral properties. These two probes rely on the fluorescent donor mTurquoise2 and on a non-fluorescent acceptor (ShadowG) in one case, or a yellow acceptor (superYFP) in a second case. We then inserted the full-length sequence of AURKA within each donor/acceptor pair. To have a negative control for AURKA activation, two strategies can be pursued. First, the use a small ATP-analog (MLN8237) interferes with the binding of ATP in the kinetic pocket of the kinase and prevents its activation38. Second, the mutation of Lys162 into Met (K162M), creates a kinase-dead version of each biosensor incapable of activating14,15,39. This mutation induces the disruption of a salt bridge normally established between Lys162 and Glu181, which results in a stable opening of the kinetic pocket of the kinase and triggers its overall inactivation40. As a negative control for FRET, we used an acceptor-devoid construct (GFP-AURKA or AURKA-mTurquoise2).
After synchronizing cells in G2/M and releasing them into mitosis, we measured the lifetime of all the transfected constructs at the mitotic spindle (Figure 4). Of note, this structure was considered as a whole, and no ROIs within the spindle were analyzed. We then calculated ΔLifetime for all conditions. As expected, the lifetime of GFP-AURKA or AURKA-mTurquoise2 (the “donor-only” conditions) was close to 0, indicating that the values measured for these constructs fluctuated around the mean value (Figure 4A,4B). Conversely, the ΔLifetime values for GFP-AURKA-mCherry were statistically different from the donor-only condition, with ΔLifetime increasing of ~130 ps (Figure 4A). Similar observations were made for shadowG-AURKA-mTurquoise2 and for superYFP-AURKA-mTurquoise2, with ΔLifetime increasing of ~150 and ~220 ps from the donor-only condition, respectively (Figure 4B,4C). These data can be easily visualized in single cells with a pseudocolor Lookup Table (LUT). In this case, values of ΔLifetime around 0 are pseudocolored yellow, while more significant differences are pseudocolored red/purple. Indeed, the pixel-by-pixel LUT was closer to yellow in cells expressing the donor-only constructs, while it was more in the red/purple spectrum in cells expressing either biosensor (Figure 4A,4B). This was also observed when the GFP-AURKA-mCherry biosensor was treated with the pharmacological inhibitor MLN8237.
We then analyzed the ΔLifetime of kinase-dead biosensors. These constructs showed intermediate ΔLifetime values: ΔLifetime was significantly higher when compared to the donor-only condition (Figure 4B,4C), but it was also significantly lower than their normal counterparts (Figure 4B,4D). The comparisons with cells treated with MLN8237 or expressing kinase-dead biosensors are necessary to estimate whether ΔLifetime variations for each donor/acceptor pair are solely linked to the activation of AURKA. In the case of GFP-AURKA-mCherry, ΔLifetime variations are abolished when an AURKA-specific inhibitor is used. Conversely, ΔLifetime variations are mostly, but not exclusively linked to AURKA activation in the case of shadowG-AURKA-mTurquoise2 and of superYFP-AURKA-mTurquoise2.

Figure 1: GFP (donor) and mCherry (acceptor) excitation and emission spectra.
Spectra were obtained and adapted from the FP base website (https://www.fpbase.org/), and adjusted to a 480 nm-laser excitation. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Image of the experimental workspace.
(1) The control solution; (2) white laser source; (3) CCD camera; (4) microscope setup; (5) external light source/lamp for ocular screening of the sample. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Representative images of the software for FLIM acquisition.
(A, B) (1) excitation and emission parameters for the donor (CFP in A, or GFP in B); (2) exposure time; (3) selection of the objective. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Representative images of AURKA FRET biosensors and their negative controls.
(A) (Micrographs) Fluorescence (green channel) and corresponding pixel-by-pixel ΔLifetime (donor only – biosensor) of U2OS cells expressing GFP-AURKA or GFP-AURKA-mCherry, synchronized at G2/M, released until the bipolar spindle is visible and then treated with DMSO or with MLN8237. ΔLifetime is illustrated with a pseudocolor scale (“Fire” lookup table). (Graph) Corresponding quantification and two-way ANOVA analysis for the indicated conditions. (B) (Micrographs) Fluorescence (cyan channel) and corresponding pixel-by-pixel ΔLifetime (donor only – biosensor) of U2OS cells expressing shadowG-AURKA-mTurquoise2 (upper panel) or superYFP-AURKA-mTurquoise2 (lower panel), synchronized at G2/M and released until the bipolar spindle is visible. ΔLifetime is illustrated with a pseudocolor scale (“Fire” lookup table). (Graph) Corresponding quantification and one-way ANOVA analysis of conditions represented in the above micrographs. (C) (Micrographs) Images of AURKA-mTurquoise2 (upper panel), shadowG-AURKA K162M-mTurquoise2 (middle panel) and superYFP-AURKA K162M-mTurquoise2 acquired and represented as in the micrographs. (Graph) Two-way ANOVA analysis for the indicated transfection conditions. The bar in boxplots represents the median; whiskers extend from the min to the max. n = 10 cells per condition of one representative experiment (of three). Individual values are represented as dots. Scale bar: 10 µm. *P < 0.05, **P < 0.01, ***P < 0.001 against each indicated condition in (A) the “AURKA-mTurquoise2” condition in (B), and against each indicated condition in (C). NS: not significant. Please click here to view a larger version of this figure.
{kind=link}
Discussion
Genetically-encoded FRET biosensors are reliable tools to measure the activation of single proteins or of entire signaling pathways41. Particularly, the AURKA FRET biosensor constitutes a preferential way to explore the activation of the kinase in time and space. However, some elements deserve special attention when designing or optimizing a FRET biosensor, not only in general terms but more specifically for AURKA.
First, the nature and the relative position of the donor/acceptor FRET pair can be adapted for specific functions of this kinase. AURKA is greatly enriched at the mitotic spindle during mitosis, but it is present throughout the cell cycle and at different subcellular locations (e.g., centrosomes, the nucleus, and mitochondria)1,2. If the biosensor is to be used in specific compartments like mitochondria, which can reach acidic pH, the choice of a pH-insensitive donor-acceptor FRET pair as mTurquoise2/shadowG should be made. Moreover, placing the FRET donor at the C-terminus could allow a better visualization of the biosensor at this subcellular compartment, and potentially even optimize FRET detection given that AURKA N-terminus was shown to partially cleave off at mitochondria16,42.
Second, a yet unexplored way of optimizing the AURKA FRET biosensor would require a more careful design of linkers between full-length AURKA and the donor/acceptor pair. Not only the distance between the fluorescent pair, but also the properties of the linker itself were shown to be key factors to improve FRET efficiency43,44,45,46. In this light, increasing the rigidity or the flexibility of the linker could either be detrimental to FRET efficiency, or further improve it.
Third, it is known that the overexpression of AURKA induces mitotic spindle abnormalities in a significant proportion of cells2. It would be interesting to compare ΔLifetime obtained by expressing the same FRET construct under a strong promoter like cytomegalovirus (CMV) – one of the most common promoters found in mammalian expression vectors – or under the AURKA minimal promoter sequence (CTTCCGG)14,47. This promoter was previously shown to rescue monopolar or multipolar spindles arising after the knock-down of the kinase, and its use did not induce cell cycle perturbations per se14,47. Although FLIM is insensitive to protein expression levels and relative concentrations in the cell11, benefiting from a thorough comparison of the two promoters on the same biosensor setup would broaden the understanding of the pool of activated AURKA at any given location. In addition, it would provide novel insights on how AURKA activation may change upon overexpression, which is relevant for epithelial and hematologic cancer paradigms.
Last, the downstream FRET application should also be taken into account. A future perspective in the field of AURKA would be to cumulate the kinase conformational biosensor with a substrate-based biosensor. Analyzing the FRET behavior of two biosensors simultaneously – a process known as multiplex FRET – requires a dark acceptor on the first biosensor to avoid spectral bleed through in the second donor channel. In the context of AURKA, this would open up the exciting new perspective of detecting the activation of the kinase with the first biosensor, and its enzymatic activity towards a given substrate with the second one. Recent developments in multiplexing now allow to cumulate up to three biosensors at a time48. Applying a similar method in the context of AURKA could represent a very promising strategy not only to test the activation-activity interplay of the kinase, but also to explore AURKA signaling cascades with unprecedented spatiotemporal resolution.
In conclusion, FRET/FLIM is a convenient way to deepen the knowledge on protein activity. On one hand, it allows to visualize the localization of a given protein in live cells, thanks to at least one fluorescent moiety. On the other hand, it can unravel protein conformational changes, which could be informative on protein activation and/or activity. Therefore, FRET/FLIM and conformational FRET biosensors have the potential of becoming widespread methods to follow signaling pathways in live cells, and with exquisite spatiotemporal resolution.
Disclosures
G.B. performed the experiments, wrote and reviewed the manuscript, and provided funding, M.T. reviewed the manuscript and provided support. M.T. is a scientific advisor and shareholder of the Inscoper company (France), which produces the solutions for rapid FLIM measurements shown in this manuscript. Inscoper partially supported the Open Access publication of the manuscript. Inscoper was not involved in experimental design, data handling, nor in the writing of the manuscript.
Acknowledgements
We thank the engineers of the Microscopy-Rennes Imaging Center (MRic, BIOSIT, Rennes, France) for advice and help, and particularly X. Pinson for critical reading of the manuscript. MRic is member of the national infrastructure France-BioImaging supported by the French National Research Agency (ANR-10-INBS-04). This work was supported by the Centre National de la Recherche Scientifique (CNRS), the Ligue Contre le Cancer Comités d’Ille et Vilaine, des Côtes d’Armor et du Finistère, and the Association pour la Recherche Contre le Cancer (ARC) to G.B.
Materials
| Name | Company | Catalog Number | Comments |
| Alisertib (MLN8237) | SelleckChem | S1133 | Use at a 250 nM final dilution |
| Dulbecco's Modified Eagle Medium (DMEM) | ThermoFischer Scientific Brand: Gibco | 41966052 | High glucose + L-glutamine + Sodium pyruvate |
| Fetal Bovine Serum (FBS) | ThermoFischer Scientific Brand: Gibco | 10270106 | |
| L15 | ThermoFischer Scientific Brand: Gibco | 21083027 | Leibovitz's L15 medium + L-glutamine, no Phenol red |
| LabTek | Nunc | 2515380 | |
| Nocodazole | Merck Brand: Sigma-Aldrich | M1404 | Use at a 100 ng/mL final dilution |
| Penicillin/Streptomycin | ThermoFischer Scientific Brand: Gibco | 15140122 | Penicillin-Streptomycin 10,000 U/mL (100x) |
| Phosphate Buffer Saline (PBS) | ThermoFischer Scientific Brand: Gibco | 14190169 | DPBS, no calcium, no magnesium |
| Trypsin/EDTA | ThermoFischer Scientific Brand: Gibco | 25300096 | Trypsin-EDTA 0.05%, Phenol Red (1x) |
References
- Bertolin, G., Tramier, M. Insights into the non-mitotic functions of Aurora kinase A: more than just cell division. Cellular and Molecular Life Sciences. , (2019).
- Nikonova, A. S., Astsaturov, I., Serebriiskii, I. G., Dunbrack, R. L., Golemis, E. A. Aurora A kinase (AURKA) in normal and pathological cell division. Cellular and Molecular Life Sciences. 70 (4), 661-687 (2013).
- Walter, A. O., Seghezzi, W., Korver, W., Sheung, J., Lees, E. The mitotic serine/threonine kinase Aurora2/AIK is regulated by phosphorylation and degradation. Oncogene. 19 (42), 4906-4916 (2000).
- Cheetham, G. M. T. Crystal Structure of Aurora-2, an Oncogenic Serine/Threonine Kinase. Journal of Biological Chemistry. 277 (45), 42419-42422 (2002).
- Bayliss, R., Sardon, T., Vernos, I., Conti, E. Structural basis of Aurora-A activation by TPX2 at the mitotic spindle. Molecular Cell. 12 (4), 851-862 (2003).
- Zhang, Y., et al. Identification of the auto-inhibitory domains of Aurora-A kinase. Biochemical and Biophysical Research Communications. 357 (2), 347-352 (2007).
- Littlepage, L. E., Wu, H., Andresson, T., Deanehan, J. K., Amundadottir, L. T., Ruderman, J. V. Identification of phosphorylated residues that affect the activity of the mitotic kinase Aurora-A. Proceedings of the National Academy of Sciences of the United States of America. 99 (24), 15440-15445 (2002).
- Kufer, T. A., et al. Human TPX2 is required for targeting Aurora-A kinase to the spindle. The Journal of Cell Biology. 158 (4), 617-623 (2002).
- Eyers, P. A., Erikson, E., Chen, L. G., Maller, J. L. A novel mechanism for activation of the protein kinase Aurora A. Current Biology. 13 (8), 691-697 (2003).
- Brunet, S., et al. Characterization of the TPX2 Domains Involved in Microtubule Nucleation and Spindle Assembly in Xenopus Egg Extracts. Molecular Biology of the Cell. 15 (12), 5318-5328 (2004).
- Padilla-Parra, S., Tramier, M. FRET microscopy in the living cell: Different approaches, strengths and weaknesses. BioEssays. 34 (5), 369-376 (2012).
- Aoki, K., Kamioka, Y., Matsuda, M. Fluorescence resonance energy transfer imaging of cell signaling from in vitro to in vivo: Basis of biosensor construction, live imaging, and image processing. Development, Growth & Differentiation. 55 (4), 515-522 (2013).
- Fuller, B. G., et al. Midzone activation of aurora B in anaphase produces an intracellular phosphorylation gradient. Nature. 453 (7198), 1132-1136 (2008).
- Bertolin, G., et al. A FRET biosensor reveals spatiotemporal activation and functions of aurora kinase A in living cells. Nature Communications. 7, 12674(2016).
- Bertolin, G., et al. Optimized FRET Pairs and Quantification Approaches To Detect the Activation of Aurora Kinase A at Mitosis. ACS Sensors. 4 (8), 2018-2027 (2019).
- Bertolin, G., et al. Aurora kinase A localises to mitochondria to control organelle dynamics and energy production. eLife. 7, (2018).
- Sizaire, F., Le Marchand, G., Pécréaux, J., Bouchareb, O., Tramier, M. Automated screening of AURKA activity based on a genetically encoded FRET biosensor using fluorescence lifetime imaging microscopy. Methods and Applications in Fluorescence. 8 (2), 024006(2020).
- Demeautis, C., et al. Multiplexing PKA and ERK1&2 kinases FRET biosensors in living cells using single excitation wavelength dual colour FLIM. Scientific Reports. 7, 41026(2017).
- Ringer, P., et al. Multiplexing molecular tension sensors reveals piconewton force gradient across talin-1. Nature Methods. 14 (11), 1090-1096 (2017).
- Nagai, T., Yamada, S., Tominaga, T., Ichikawa, M., Miyawaki, A. Expanded dynamic range of fluorescent indicators for Ca(2+) by circularly permuted yellow fluorescent proteins. Proceedings of the National Academy of Sciences of the United States of America. 101 (29), 10554-10559 (2004).
- Klarenbeek, J., Goedhart, J., van Batenburg, A., Groenewald, D., Jalink, K. Fourth-Generation Epac-Based FRET Sensors for cAMP Feature Exceptional Brightness, Photostability and Dynamic Range: Characterization of Dedicated Sensors for FLIM, for Ratiometry and with High Affinity. PLOS ONE. 10 (4), 0122513(2015).
- Fritz, R. D., et al. A Versatile Toolkit to Produce Sensitive FRET Biosensors to Visualize Signaling in Time and Space. Science Signaling. 6 (285), (2013).
- Mastop, M., et al. Characterization of a spectrally diverse set of fluorescent proteins as FRET acceptors for mTurquoise2. Scientific Reports. 7 (1), 11999(2017).
- vander Krogt, G. N. M., Ogink, J., Ponsioen, B., Jalink, K. A Comparison of Donor-Acceptor Pairs for Genetically Encoded FRET Sensors: Application to the Epac cAMP Sensor as an Example. PLoS ONE. 3 (4), 1916(2008).
- Cranfill, P. J., et al. Quantitative assessment of fluorescent proteins. Nature Methods. 13 (7), 557-562 (2016).
- Goedhart, J., et al. Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93. Nature Communications. 3 (1), (2012).
- Mérola, F., et al. Newly engineered cyan fluorescent proteins with enhanced performances for live cell FRET imaging. Biotechnology Journal. 9 (2), 180-191 (2014).
- Erard, M., et al. Minimum set of mutations needed to optimize cyan fluorescent proteins for live cell imaging. Molecular BioSystems. 9 (2), 258-267 (2013).
- Tramier, M., Zahid, M., Mevel, J. C., Masse, M. J., Coppey-Moisan, M. Sensitivity of CFP/YFP and GFP/mCherry pairs to donor photobleaching on FRET determination by fluorescence lifetime imaging microscopy in living cells. Microscopy Research and Technique. 69 (11), 933-939 (2006).
- Padilla-Parra, S., et al. Quantitative Comparison of Different Fluorescent Protein Couples for Fast FRET-FLIM Acquisition. Biophysical Journal. 97 (8), 2368-2376 (2009).
- Burgstaller, S., et al. pH-Lemon, a Fluorescent Protein-Based pH Reporter for Acidic Compartments. ACS Sensors. , (2019).
- Bindels, D. S., et al. mScarlet: a bright monomeric red fluorescent protein for cellular imaging. Nature Methods. 14 (1), 53-56 (2016).
- Murakoshi, H., Shibata, A. C. E., Nakahata, Y., Nabekura, J. A dark green fluorescent protein as an acceptor for measurement of Förster resonance energy transfer. Scientific Reports. 5, 15334(2015).
- Murakoshi, H., Shibata, A. C. E. ShadowY: a dark yellow fluorescent protein for FLIM-based FRET measurement. Scientific Reports. 7 (1), 6791(2017).
- Leray, A., Padilla-Parra, S., Roul, J., Héliot, L., Tramier, M. Spatio-Temporal Quantification of FRET in living cells by fast time-domain FLIM: a comparative study of non-fitting methods [corrected]. PloS One. 8 (7), 69335(2013).
- Padilla-Parra, S., Audugé, N., Coppey-Moisan, M., Tramier, M. Quantitative FRET analysis by fast acquisition time domain FLIM at high spatial resolution in living cells. Biophysical Journal. 95 (6), 2976-2988 (2008).
- Song, L., Hennink, E. J., Young, I. T., Tanke, H. J. Photobleaching kinetics of fluorescein in quantitative fluorescence microscopy. Biophysical Journal. 68 (6), 2588-2600 (1995).
- Manfredi, M. G., et al. Characterization of Alisertib (MLN8237), an investigational small-molecule inhibitor of aurora A kinase using novel in vivo pharmacodynamic assays. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research. 17 (24), 7614-7624 (2011).
- Katayama, H., et al. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nature Genetics. 36 (1), 55-62 (2004).
- Nowakowski, J., et al. Structures of the Cancer-Related Aurora-A, FAK, and EphA2 Protein Kinases from Nanovolume Crystallography. Structure. 10 (12), 1659-1667 (2002).
- Palmer, A. E., Qin, Y., Park, J. G., McCombs, J. E. Design and application of genetically encoded biosensors. Trends in Biotechnology. 29 (3), 144-152 (2011).
- Grant, R., et al. Constitutive regulation of mitochondrial morphology by Aurora A kinase depends on a predicted cryptic targeting sequence at the N-terminus. Open Biology. 8 (6), 170272(2018).
- Shimozono, S., Miyawaki, A. Engineering FRET Constructs Using CFP and YFP. Methods in Cell Biology. 85, 381-393 (2008).
- Komatsu, N., et al. Development of an optimized backbone of FRET biosensors for kinases and GTPases. Molecular Biology of the Cell. 22 (23), 4647-4656 (2011).
- Schifferer, M., Griesbeck, O. A Dynamic FRET Reporter of Gene Expression Improved by Functional Screening. Journal of the American Chemical Society. 134 (37), 15185-15188 (2012).
- Peroza, E. A., Boumezbeur, A. H., Zamboni, N. Rapid, randomized development of genetically encoded FRET sensors for small molecules. Analyst. 140 (13), 4540-4548 (2015).
- Reboutier, D., et al. Aurora A is involved in central spindle assembly through phosphorylation of Ser 19 in P150Glued. The Journal of Cell Biology. 201 (1), 65-79 (2013).
- Mo, G. C. H., Posner, C., Rodriguez, E. A., Sun, T., Zhang, J. A rationally enhanced red fluorescent protein expands the utility of FRET biosensors. Nature Communications. 11 (1), 1848(2020).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved