Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Combinaison de l’hybridation in situ par fluorescence multiplex avec l’immunohistochimie fluorescente sur des coupes de cerveau de souris fraîches, congelées ou fixes
Dans cet article
Résumé
Ce protocole décrit une méthode permettant de combiner l’hybridation in situ par fluorescence (FISH) et l’immunohistochimie par fluorescence (IHC) dans des coupes de cerveau de souris fraîches, congelées et fixes, dans le but d’obtenir un signal FISH et IHC de fluorescence multimarqueur. L’IHC a ciblé les protéines cytoplasmiques et membranaires.
Résumé
L’hybridation in situ fluorescente (FISH) est une technique moléculaire qui permet d’identifier la présence et la distribution spatiale de transcrits d’ARN spécifiques au sein des cellules. Le phénotypage neurochimique des neurones fonctionnellement identifiés nécessite généralement un marquage simultané avec plusieurs anticorps (protéine de ciblage) à l’aide de l’immunohistochimie (IHC) et une optimisation de l’hybridation in situ (ciblage de l’ARN), en tandem. Il est possible d’obtenir une « signature neurochimique » pour caractériser des neurones particuliers, mais les facteurs de complication incluent la nécessité de vérifier les cibles FISH et IHC avant de combiner les méthodes, et le nombre limité d’ARN et de protéines qui peuvent être ciblés simultanément dans la même section de tissu.
Nous décrivons ici un protocole, utilisant à la fois des préparations de cerveau de souris fraîches congelées et fixes, qui détecte plusieurs ARNm et protéines dans la même section du cerveau à l’aide de RNAscope FISH suivi d’un immunomarquage par fluorescence, respectivement. Nous utilisons la méthode combinée pour décrire le profil d’expression des ARNm de faible abondance (par exemple, le récepteur 1 de la galanine) et des ARNm de grande abondance (par exemple, le transporteur de glycine 2), dans les noyaux du tronc cérébral identifiés immunohistochimiquement.
Les considérations clés pour le marquage des protéines en aval de l’essai FISH vont au-delà de la préparation des tissus et de l’optimisation du marquage de la sonde FISH. Par exemple, nous avons constaté que la spécificité de la liaison et du marquage des anticorps peut être affectée de manière préjudiciable par l’étape de la protéase dans le test de sonde FISH. Les protéases catalysent le clivage hydrolytique des liaisons peptidiques, facilitant l’entrée de la sonde FISH dans les cellules, mais elles peuvent également digérer la protéine ciblée par le test IHC ultérieur, produisant une liaison hors cible. L’emplacement subcellulaire de la protéine ciblée est un autre facteur contribuant au succès de l’IHC après l’essai de la sonde FISH. Nous avons observé que la spécificité de l’IHC était conservée lorsque la protéine ciblée est liée à la membrane, alors que l’IHC ciblant la protéine cytoplasmique nécessitait un dépannage approfondi. Enfin, nous avons constaté que la manipulation des tissus congelés fixes montés sur lame était plus difficile que des tissus congelés frais, mais la qualité de l’IHC était globalement meilleure avec les tissus congelés fixes, lorsqu’ils étaient combinés avec RNAscope.
Introduction
Les protéines et les ARNm qui définissent neurochimiquement des sous-populations de neurones sont généralement identifiés par une combinaison d’immunohistochimie (IHC) et/ou d’hybridation in situ (ISH), respectivement. La combinaison de l’ISH avec les techniques IHC facilite la caractérisation des patrons de colocalisation propres aux neurones fonctionnels (codage neurochimique) en maximisant la capacité de marquage multiplex.
Les méthodes d’ISH fluorescentes (FISH), y compris l’ARNscope, ont une sensibilité et une spécificité plus élevées que les méthodes antérieures de détection de l’ARN telles que l’ISH radioactive et l’ISH chromogène non radioactive. FISH permet de visualiser des transcrits d’ARNm uniques sous forme de taches colorées ponctuées1. De plus, le test RNAscope permet de marquer un plus grand nombre de cibles d’ARN à la fois, à l’aide de différents marqueurs fluorophores. Malgré ces avantages, les limitations techniques peuvent affecter le nombre de fluorophores/chromogènes pouvant être utilisés dans une seule expérience. Il s’agit notamment de la disponibilité d’ensembles de filtres pour microscopes ; ces considérations sont aggravées lorsque l’identification neurochimique utilise la combinaison de la FISH et de l’IHC, par rapport à l’utilisation de chaque technique isolément, car les étapes inhérentes optimales à une méthode peuvent être préjudiciables à l’autre.
L’application antérieure de FISH combinée à l’IHC a démontré l’expression de cibles cellulaires spécifiques dans les lymphomes humains à cellules B2, les embryons de poussin3, les embryons de poisson-zèbre4, la rétine de souris5 et les cellules de l’oreille interne de souris6. Dans ces études, la préparation des tissus a consisté soit en une paraffine fixée au formol (FFPE)2,3,5, soit en une monture entière fraîche 4,6. D’autres études ont appliqué l’ARNscope chromogène à des préparations de cerveaux fixes de souris et de rats 7,8,9. En particulier, Baleriola et al.8 ont décrit deux préparations tissulaires différentes pour l’ISH-IHC combiné ; des sections de cerveau de souris fixes et des sections de cerveau humain FFPE. Dans une publication récente, nous avons combiné le FISH et l’IHC fluorescent sur des coupes fraîches congelées, afin de visualiser simultanément l’ARNm de faible abondance (récepteur de la galanine 1, GalR1), l’ARNm de haute abondance (transporteur de glycine 2, GlyT2) et la protéine vésiculaire transporteur d’acétylcholine (vAChT)10 dans la formation réticulaire du tronc cérébral.
Le noyau du tractus solitaire (NTS) est une région majeure du cerveau impliquée dans la fonction autonome. Située dans le cerveau postérieur, cette population hétérogène de neurones reçoit et intègre un grand nombre de signaux autonomes, dont ceux qui régulent la respiration. Le NTS abrite plusieurs populations neuronales, qui peuvent être phénotypiquement caractérisées par le modèle d’expression des cibles d’ARNm, y compris GalR1 et GlyT2 et des marqueurs protéiques de l’enzyme tyrosine hydroxylase (TH) et du facteur de transcription Paired-like homeobox 2b (Phox2b).
Le propriétaire du RNAscope recommande des préparations de tissus frais congelés, mais les tissus préparés par fixation par perfusion transcardique d’animaux entiers, ainsi que la cryoprotection à long terme (stockage à -20 °C) de coupes de tissus congelés fixes, sont courants dans de nombreux laboratoires. Par conséquent, nous avons cherché à établir des protocoles pour le FISH en combinaison avec l’IHC en utilisant des préparations de tissus frais congelés et congelés fixes. Ici, nous fournissons pour les coupes de cerveau fraîches congelées et congelées fixes : (1) un protocole pour la combinaison de FISH et d’IHC fluorescent (2) une description de la qualité de l’ARNm et du marquage protéique produit, lors de l’utilisation de chaque préparation, (3) une description de l’expression de GalR1 et GlyT2 dans le NTS.
Notre étude a révélé que, lorsqu’il est combiné avec la méthodologie RNAscope, le succès de l’IHC variait dans les préparations fraîches congelées et fixées et dépendait de la localisation des protéines cibles dans la cellule. Entre nos mains, le marquage des protéines liées à la membrane a toujours été couronné de succès. En revanche, l’IHC pour la protéine cytoplasmique a nécessité un dépannage même dans les cas où la protéine cytoplasmique était surexprimée chez un animal transgénique (Phox2b-GFP)11. Enfin, alors que GalR1 est exprimé dans les neurones non catécholaminergiques du NTS, l’expression de GlyT2 est absente du NTS.
Protocole
Un résumé des étapes de prétraitement des tissus se trouve à la figure 1. Toutes les procédures ont été effectuées conformément au Comité de protection et d’éthique des animaux de l’Université de Nouvelle-Galles du Sud, conformément aux directives relatives à l’utilisation et aux soins des animaux à des fins scientifiques (Australian National Health and Medical Research Council).
1. Préparation d’échantillons de tissus cérébraux frais congelés
- Perfusion transcardique
- Préparer un tampon phosphate (PB) hépariné (2500 U/L) de 0,1 M, pH 7,5. Fabriquez de la boue d’éthanol de glace carbonique en mélangeant de la glace carbonique avec de l’éthanol. Celui-ci aura une température d’environ −72 °C et sera utilisé pour la congélation immédiate du tissu récolté.
- Euthanasier des souris adultes C57BL/6 et Phox2b-GFP11 (identifiant MGI :5776545 de la base de données Mouse Genome Informatics) en les anesthésiant avec du pentobarbital de sodium (70 mg/kg, i.p.), à l’aide d’une aiguille de 27,5 pouces.
ATTENTION : Le pentobarbital est un barbiturique. Il est extrêmement toxique à fortes doses et peut entraîner la mort par arrêt respiratoire. Consultez les directives locales en matière de santé, de sécurité juridique et de sécurité des matériaux avant utilisation. - Exposez le cœur et canulez le ventricule gauche à l’aide d’une aiguille d’aspiration (calibre 23 pouces). Effectuer une perfusion transcardique avec un PB hépariné de 0,1 M jusqu’à ce que le sang s’éclaircisse (2-3 minutes) à un débit de 11-13 mL/min. Déterminer l’élimination du sang en surveillant la coloration du foie et de l’effusat de l’oreillette droite12.
- Isolez le cerveau de la cavité crânienne, incorporez-le immédiatement dans un composé à température de coupe optimale (OCT) dans un cryomoule ou une feuille d’aluminium et placez-le sur le bain d’éthanol de glace carbonique. Conservez le tissu incrusté congelé dans un récipient hermétique à - 80 °C jusqu’à 3 mois.
- Coupe de tissus frais congelés
- Réglez la température du cryostat à -20 °C. Laissez le tissu intégré à l’OCT et un mandrin de cryostat dans le cryostat pendant ~30 minutes pour permettre l’équilibrage à la nouvelle température.
REMARQUE : Gardez le tissu congelé en tout temps ; transporter les tissus du congélateur à -80 °C au cryostat sur de la glace carbonique. - Fixez le tissu au mandrin du cryostat pré-refroidi à l’aide d’un composé OCT. Dans ce protocole, des blocs de tissu ont été montés sur le mandrin dans le plan coronal.
REMARQUE : Coupez l’excès d’OCT du tissu, à l’aide d’une lame de rasoir, pour minimiser la quantité d’OCT coupée par le cryostat et ensuite transférée sur la lame de verre. - Découpez des sections coronales de 14 μm d’épaisseur et montez-les sur des lames de microscopie en verre chargées.
- Réchauffez les lames à température ambiante avant de monter les sections. Une fois la section montée, conservez les lames dans une boîte à lames dans le cryostat.
- Si plus d’une section doit être montée sur une diapositive, réchauffez la zone de la deuxième section en plaçant un doigt sur le côté opposé de la lame pendant 5 à 10 secondes pour faciliter l’adhérence de la section à la lame. Une section de tissu froid ne s’attachera pas à une lame froide. Les sections doivent adhérer aux glissières à plat ; Le pliage les fera tomber des glissières pendant les étapes de lavage.
- Si des fissures sont remarquées dans les sections, augmentez la température du cryostat de 1 à 5 °C pour éviter cela. Il est particulièrement important de placer des sections de tissus à proximité les unes des autres sur la même lame. Cela permettra d’éviter le gaspillage de sondes et de réactifs FISH pendant le test.
- Conservez les sections de mouchoirs montées sur des lames de verre dans un récipient hermétique à -80 °C jusqu’à 6 mois.
REMARQUE : Gardez les sections congelées en tout temps et évitez les cycles de congélation et de décongélation, afin d’éviter la dégradation de l’ARN. Transportez la boîte de lames de l’intérieur du cryostat au congélateur à -80 °C sur de la glace carbonique.
- Réglez la température du cryostat à -20 °C. Laissez le tissu intégré à l’OCT et un mandrin de cryostat dans le cryostat pendant ~30 minutes pour permettre l’équilibrage à la nouvelle température.
- Fixation de tissus frais congelés
- Le jour où le test de la sonde FISH doit être effectué, préparer du paraformaldéhyde (PFA) à 4 % dans 0,1 M PB, pH 7,5 (solution de PFA à 4 %). Filtrer en passant à travers du papier filtre (Grade 1 : 11 μm, Table des matériaux) dans un entonnoir Buchner ou un filtre à creuset.
ATTENTION : Le PFA est nocif et toxique par contact cutané ou par inhalation. Toutes les procédures avec une solution de PFA doivent être effectuées dans une hotte. Les déchets de solution de PFA doivent être éliminés avec soin, conformément aux protocoles de sécurité de l’établissement. - Refroidir la solution de PFA à 4 % à 4 °C. Transportez le mouchoir monté sur lame du congélateur à -80 °C dans de la glace carbonique et plongez-le immédiatement dans le fixateur pré-refroidi pendant 15 minutes.
REMARQUE : Il est important que cette étape de fixation ne dépasse pas 15 minutes, car une fixation excessive entraînera un marquage de fond non spécifique.
- Le jour où le test de la sonde FISH doit être effectué, préparer du paraformaldéhyde (PFA) à 4 % dans 0,1 M PB, pH 7,5 (solution de PFA à 4 %). Filtrer en passant à travers du papier filtre (Grade 1 : 11 μm, Table des matériaux) dans un entonnoir Buchner ou un filtre à creuset.
- Déshydratation de tissus frais congelés
- Déshydrater les coupes de tissus en immergeant les lames dans des concentrations graduées d’éthanol. Dans un bocal Coplin, immergez-les d’abord dans 50 %, puis 70 % et enfin dans de l’éthanol absolu, pendant 5 minutes chacun à température ambiante. Répétez l’incubation finale à l’éthanol absolu une deuxième fois.
- Séchez les lames à l’air libre et tracez le contour du groupe de sections à l’aide d’un stylo barrière hydrophobe, en veillant à ce que la zone interne soit réduite au minimum.
REMARQUE : Assurez-vous que la lame de verre est complètement sèche avant de tirer la barrière hydrophobe. La barrière hydrophobe doit entourer complètement les sections de tissu sans espaces et doit être sèche avant tout traitement ultérieur.
2. Préparation d’échantillons de tissus cérébraux congelés fixes
- Fixation de la perfusion transcardique
- Euthanasier les souris par anesthésie avec du pentobarbital sodique (70 mg/kg, i.p.) suivi d’une perfusion transcardique, d’abord avec 0,1 M PB puis une solution de PFA à 4 %. Fixer avec 10 minutes de perfusion à 11-13 mL/min.
- Isolez le cerveau de la cavité crânienne après perfusion-fixation et immergez-le pendant une nuit dans une solution de PFA à 4 %, à 4 °C.
- Coupe tissulaire de tissus fixés
- Rincer le cerveau dans une solution saline tamponnée au phosphate (PBS) stérile de 0,1 M avant de retirer les couches méningées, à l’aide d’un microscope à dissection, à l’aide d’une pince fine.
- Découpez le cerveau avec précision en blocs (séparez le tronc cérébral du cerveau antérieur avant la section du vibratome) à l’aide d’une matrice cérébrale (Table des matériaux). Plus précisément, coupez le tronc cérébral caudalement au niveau de la décussation pyramidale et disséquez le cervelet. De même, coupez immédiatement le cerveau antérieur rostral au chiasma optique.
- Fixez le tissu sur un mandrin de microtome vibrant à l’aide de cyanoacrylate et incorporez-le dans une solution de gélose à 2 %.
- Découper des coupes de tissu de 30 μm d’épaisseur à l’aide d’un microtome vibrant et conserver les coupes coupées dans une solution cryoprotectrice (saccharose sans RNase à 30 %, 30 % d’éthylène glycol, 1 % de polyvinylpyrrolidone (PVP-40), dans 0,1 M PB, pH 7,4). Les coupes de tissus peuvent être conservées dans un cryoprotecteur à -20 °C jusqu’à 6 mois.
- Préparation des sections fixes avant le FISH
- Le jour du FISH, lavez les sections flottantes trois fois, pendant 10 minutes par lavage, pour éliminer la solution cryoprotectrice. Pour le lavage, placer les sections dans du PBS de 0,1 M dans une plaque de culture cellulaire à 12 puits et agiter sur un agitateur à plate-forme rotative (90 - 100 tr/min).
- Après les lavages, utilisez un pinceau pour monter des sections sur des lames de microscopie en verre et séchez à l’air libre pendant au moins 2 heures.
REMARQUE : Les sections doivent adhérer à plat sur les lames car tout pli prononcé les fera se détacher pendant les lavages. - À l’aide d’un stylo barrière hydrophobe, tracez une barrière autour des sections pour limiter les réactifs FISH aux sections. Encore une fois, il est important de minimiser la surface interne du contour tracé avec le stylo barrière.
POINT DE RUPTURE POSSIBLE : Les sections peuvent être entreposées à température ambiante, pendant la nuit, pour poursuivre l’analyse le lendemain.
3. Dosage FISH
REMARQUE : Le reste du protocole s’applique à la fois aux tissus frais congelés et aux tissus congelés fixes.
- Préparer les réactifs et les instruments pour les étapes d’hybridation et d’amplification.
- Réglez un incubateur de paillasse et un bain-marie à 40 °C.
- Préparez une chambre humidifiée et à l’abri de la lumière pour l’incubation des lames. L’humidification empêche le dessèchement des tissus - les lames sont solidement placées au-dessus d’un réservoir humide. Idéalement, la chambre est en polystyrène robuste, elle est étanche à la lumière et à l’air pour maintenir une atmosphère saturée de vapeur d’eau. La fermeture de la chambre repose sur un frottement minimal pour éviter tout mouvement. Nous avons utilisé une boîte à lames doublée de lingettes de laboratoire humides (Table of Materials) en bas. Placez la boîte à lames à l’intérieur de l’incubateur pour la préchauffer à 40 °C.
- Réchauffez le tampon de lavage 50x (table des matériaux) et les sondes à 40 °C pendant 10 minutes, à l’aide du bain-marie, puis refroidissez-les à température ambiante.
- Préparez 1 L de tampon de lavage 1x à partir de la concentration de bouillon 50x.
- Préparer le mélange de sondes (Table des matériaux) : la sonde C1 est prête à l’emploi à la concentration de base tandis que les sondes C2 et C3 sont expédiées en concentration 50x et nécessitent une dilution avec le diluant fourni dans le kit.
REMARQUE : Les mélanges de sondes peuvent être conservés à 4 °C jusqu’à 6 mois.
- Traitement par protéase
- Incuber les sections avec la protéase III (tableau des matériaux) à température ambiante pendant 30 minutes.
REMARQUE : S’assurer que la protéase III et les réactifs d’incubation dans les processus en aval (mélange de sonde, solutions d’amplification, tampon de blocage et sérums d’anticorps) couvrent entièrement les sections. Une pointe de pipette peut être utilisée pour étaler le réactif sur la section afin de couvrir toute la zone à l’intérieur de la barrière hydrophobe. - Laver les lames deux fois avec 0,1 M PBS, pendant 2 min à chaque fois, dans une grande boîte de Pétri carrée en plastique. Ici, une boîte d’essai biologique carrée de 245 mm x 245 mm a été utilisée (Table des matériaux). Tenez d’un côté du plat et inclinez-le doucement 3 à 5 fois. Après les lavages, retirez l’excédent de 0,1 M de PBS de la lame et ajoutez immédiatement le réactif suivant. Ne laissez pas sécher les sections de tissus.
REMARQUE : Lors de chaque lavage, les lames sont immergées dans une solution à température ambiante. Il s’agit du flux de travail pour toutes les étapes de lavage suivantes. Les sections fixes de 30 μm d’épaisseur se délogent des lames plus facilement que les sections de 14 μm d’épaisseur, soyez doux pendant les lavages.
- Incuber les sections avec la protéase III (tableau des matériaux) à température ambiante pendant 30 minutes.
- Hybridation et amplification
- Après avoir lavé la solution de protéase, placez les lames dans la chambre humidifiée et préchauffée. Incuber les sections avec un mélange de sondes (Table des matériaux) pendant 2 heures à 40 °C à l’intérieur d’un incubateur de paillasse.
REMARQUE : Assurez-vous qu’il y a au moins 2 sections réservées aux sondes de contrôle positives et négatives afin d’évaluer la qualité de l’ARN de l’échantillon et la perméabilisation optimale. Les sondes de contrôle positif ciblent les gènes d’entretien ménager ; ici, il s’agissait d’un cocktail d’ARN ciblant l’ubiquitine C (UBC ; haute abondance), la peptidylpropyl isomérase B (PPIB ; abondance modérée) et l’ARN polymérase 2a (POLR2A ; faible abondance). Les sondes de contrôle négatives ciblent le gène bactérien de la 4-hydroxy-tétrahydrodipicolinate réductase (DapB), qui est normalement absent dans les échantillons de cerveau de souris. Un signal DapB positif indique un signal non spécifique et/ou une contamination bactérienne de l’échantillon. - Après hybridation avec le mélange de sondes, les étapes d’amplification du signal consistent en une incubation avec l’ampli 1-FL (30 minutes), puis avec l’ampli 2-FL (15 minutes), suivie de l’ampli 3-FL (30 minutes) et enfin de l’ampli 4-FL (15 minutes) - chacune à 40 °C. À l’aide des flacons compte-gouttes fournis, recouvrez les sections de tissu avec une solution d’amplification. Procéder au test IHC après la dernière étape d’amplification.
- Rincez les lames avec le tampon de lavage deux fois pendant 2 minutes entre l’hybridation de la sonde et chaque étape d’amplification.
- Après avoir lavé la solution de protéase, placez les lames dans la chambre humidifiée et préchauffée. Incuber les sections avec un mélange de sondes (Table des matériaux) pendant 2 heures à 40 °C à l’intérieur d’un incubateur de paillasse.
4. Dosage IHC
- Étape de blocage IHC
- Pour éviter la liaison non spécifique des anticorps, incuber les coupes pendant 1 h à température ambiante avec une solution bloquante contenant 10 % de sérum de cheval normal, 0,3 % de Tween20 dans 1x TBSm (50 mM de Tris-Cl, pH 7,5, 150 mM de NaCl, 0,05 % de merthiolate) après le test FISH. Préparer les anticorps primaires dans un tampon de dilution contenant 1 TBSm, 5 % de sérum de cheval normal et 0,1 % de Tween20. Les principaux fournisseurs d’anticorps sont répertoriés dans le tableau des matériaux.
- Immunohistochimie
- Retirez l’excès de tampon bloquant en effleurant la lame et incubez les sections avec des anticorps primaires pendant la nuit à 4 °C.
- Laver les lames 3 fois (5 minutes chacune) avec 1x TBSm et incuber avec de l’anticorps secondaire dans un diluant contenant 1x TBSm, 1% de sérum de cheval normal et 0,1% Tween20 pendant 2 heures à température ambiante. Les anticorps secondaires utilisés dans ce protocole sont répertoriés dans le tableau des matériaux.
- Laver les lames 3 fois avec 1x TBSm (5 minutes chacune) avant de les recouvrir avec un support de montage avec ou sans DAPI (Table of Materials).
5. Imagerie
- Examiner l’immunomarquage à l’aide d’un microscope à épifluorescence équipé d’une caméra (voir le tableau des matériaux pour plus de détails). Acquérez des images représentatives à un grossissement de 20x et enregistrez-les sous forme de fichiers TIFF.
- Exportez des images représentatives dans un logiciel de traitement d’image (Table of Materials) pour le réglage de la luminosité et du contraste afin d’augmenter la clarté et de refléter un rendu fidèle.
6. OPTIONNEL : Analyse quantitative des transcriptions cibles
REMARQUE : Il s’agit d’un article sur les méthodes et les résultats quantitatifs ne sont pas fournis. La méthode de quantification présentée ici provient de Dereli et al.10. Le Chapitre 10.
- Acquérir des images des régions d’intérêt comme expliqué dans la section 5.1 et appliquer les mêmes paramètres de microscope et d’appareil photo (tels que le temps d’exposition et l’intensité lumineuse) à toutes les images du même fluorophore.
- Tracez les profils neuronaux à l’aide d’un logiciel d’analyse d’images (Table of Materials).
- Aligner les sections par rapport au niveau de Bregma selon un atlas cérébral stéréotaxique13.
- Appliquez la même luminosité et le même contraste à toutes les images du même fluorophore. Ne considérez que les neurones avec des noyaux colorés par DAPI.
- Comptez manuellement le nombre de cellules co-exprimant l’ARNm, exprimant les protéines, l’ARNm/ARNm, la protéine/protéine et l’ARNm/protéine dans la région d’intérêt.
- Pour réduire les biais dans les résultats expérimentaux, demandez à la personne qui quantifie les résultats expérimentaux de ne pas tenir compte des groupes expérimentaux.
- Appliquez la correction d’Abercrombie14 au nombre total de cellules à l’aide de l’équation d’Abercombie suivante :
Nombre de cellules corrigé = nombre de cellules manuel x épaisseur de section / (épaisseur de section + taille nucléaire)
Par exemple, pour des sections de 14 μm d’épaisseur, la largeur nucléaire moyenne est calculée à 7,7 ± 0,3 μm et l’épaisseur moyenne de la section est de 14 ± 1 μm sur la base de 30 cellules et 10 sections respectivement chez 5 animaux10. Selon l’équation d’Abercrombie, le nombre de cellules corrigé serait le nombre de cellules manuelles x 14/(14+7,7).

Figure 1 : Flux de travail parallèle des étapes de prétraitement des tissus pour les tissus frais congelés et les tissus fixés au paraformaldéhyde. Les étapes de transformation des tissus frais congelés sont indiquées dans les cases encadrées en rouge, tandis que celles des mouchoirs fixes en paraformaldéhyde (PFA) sont affichées dans les cases encadrées en bleu. Veuillez cliquer ici pour voir une version agrandie de cette figure.
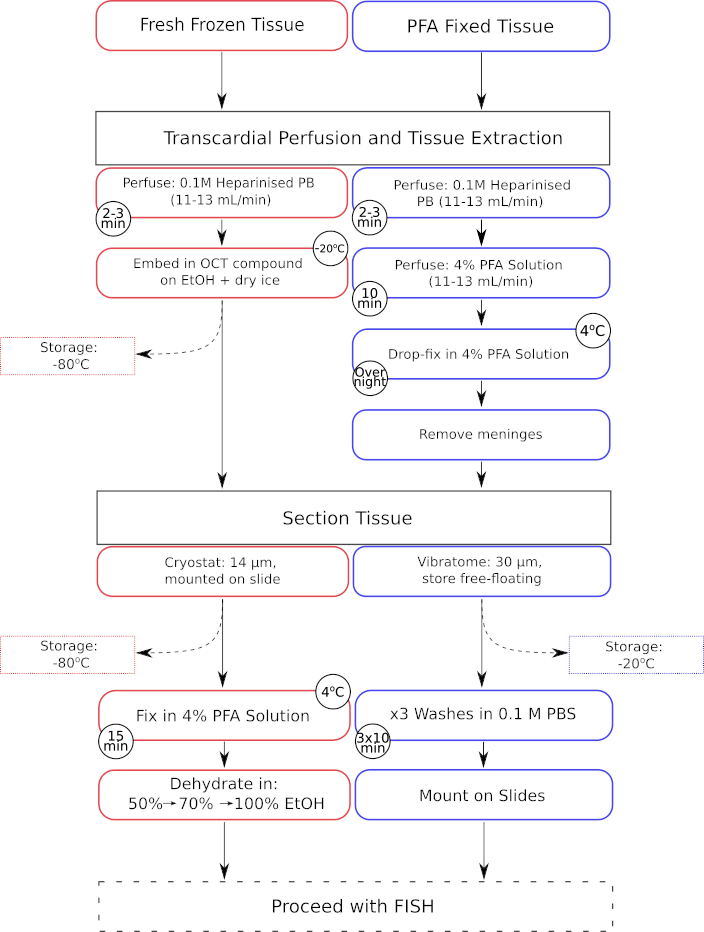{kind=link}

Figure 2 : Résumé de la sonde FISH et de la procédure d’immunohistochimie. Après le prétraitement des tissus, le tissu monté sur lame est encerclé à l’aide d’un stylo barrière hydrophobe, comme on le voit dans la première image, et incubé dans une solution de protéase à température ambiante. Après les lavages, les tissus sont transférés dans un incubateur de paillasse pour hybridation pendant 2 heures avant les étapes d’amplification séquentielle. Le système d’hybridation in situ utilise une conception exclusive de « sonde Z », des préamplificateurs et des amplificateurs, comme on le voit dans les images 3 à6 6. Une fois que le tissu a subi un traitement par sonde FISH, il est lavé avant d’être bloqué avec du sérum de cheval normal. L’incubation primaire de l’anticorps est effectuée pendant la nuit à 4 °C pour maximiser la liaison anticorps-antigène. L’incubation secondaire des anticorps (2 heures) a été réalisée à température ambiante. Veuillez cliquer ici pour voir une version agrandie de cette figure.
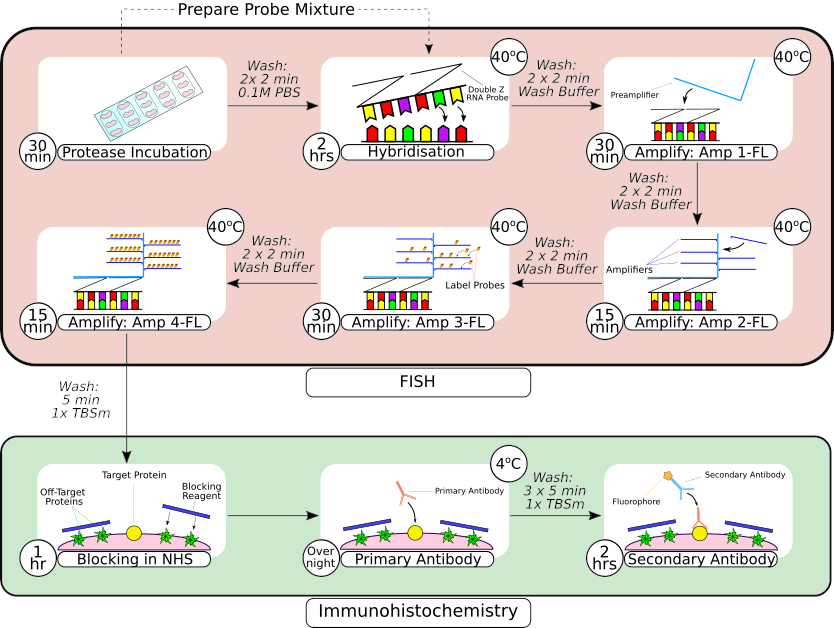{kind=link}
Résultats
Ici, nous décrivons une méthode pour combiner le FISH multiplex avec l’IHC fluorescent pour localiser l’expression de l’ARNm pour GalR1 et GlyT2 en utilisant respectivement des tissus frais congelés et fixés au paraformaldéhyde dans le NTS de souris. Les figures 1 et 2 présentent un pipeline des procédures de traitement des tissus, de FISH et d’IHC décrites dans les méthodes. Le tableau 1 présente un résumé des combinaisons d...
Discussion
Dans le domaine des neurosciences, FISH et IHC sont couramment utilisés pour étudier l’organisation spatiale et la signification fonctionnelle de l’ARNm ou des protéines au sein des sous-populations neuronales. Le protocole décrit dans cette étude améliore la capacité de détection simultanée des ARNm et des protéines dans les sections du cerveau. Notre test multiplex combiné FISH-IHC a permis l’identification phénotypique de sous-populations neuronales distinctes dans le NTS dans les préparations de ce...
Remerciements
Ces travaux ont été financés par la subvention du projet de découverte du Conseil australien de la recherche DP180101890 et la subvention de projet de la Fondation de recherche médicale Rebecca L. Cooper PG2018110
matériels
| Name | Company | Catalog Number | Comments |
| ANIMALS | |||
| C57BL/6 mouse | Australian BioResources, Moss Vale | MGI: 2159769 | |
| Phox2b-eGFP mouse | Australian BioResources, Moss Vale | MGI: 5776545 | |
| REAGENTS | |||
| Cyanoacrylate | Loctite | ||
| Ethylene Glycol | Sigma-Aldrich | 324558 | |
| Heparin-Sodium | Clifford Hallam Healthcare | 1070760 | Consult local veterinary supplier or pharmacy. |
| Lethabarb (Sodium Pentabarbitol) Euthanasia Injection | Virbac (Australia) Pty Ltd | N/A | Consult a veterinarian for local pharmaceutical regulations regarding Sodium Pentabarbitol |
| Molecular grade agarose powder | Sigma Aldrich | 5077 | |
| OCT Compound, 118mL | Scigen Ltd | 4586 | |
| Paraformaldehyde, prilled, 95% | Sigma-Aldrich | 441244-1KG | |
| Polyvinylpyrrolidone, average mol wt 40,000 (PVP-40) | Sigma-Aldrich | PVP40 | |
| ProLong Gold Antifade Mountant | Invitrogen | P36930 | With or without DAPI |
| RNAscope Multiplex Fluorescent Reagent Kit (up to 3-plex capability) | Advanced Cell Diagnostics, Inc. (ACD Bio) | ADV320850 | Includes 50x Wash buffer and Protease III |
| RNase Away | Thermo-Fisher Scientific | 7003 | |
| Tris(hydroxymethyl)aminomethane | Sigma-Aldrich | 252859 | |
| Tween-20, for molecular biology | Sigma-Aldrich | P9416 | |
| EQUIPMENT | |||
| Benchtop incubator | Thermoline scientific micro incubator | Model: TEI-13G | |
| Brain Matrix, Mouse, 30g Adult, Coronal, 1mm | Ted Pella | 15050 | |
| Cryostat | Leica | CM1950 | |
| Drawing-up needle (23 inch gauge) | BD | 0288U07 | |
| Hydrophobic Barrier Pen | Vector labs | H-4000 | |
| Kimtech Science Kimwipes Delicate Task Wipes | Kimberley Clark Professional | 34120 | |
| Olympus BX51 | Olympus | BX-51 | |
| Peristaltic pump | Coleparmer Masterflex | L/S Series | |
| Retiga 2000R Digital Camera | QImaging | RET-2000R-F-CLR | colour camera |
| SuperFrost Plus Glass Slides (White) | Thermo-Fisher Scientific | 4951PLUS4 | |
| Vibrating Microtome (Vibratome) | Leica | VT1200S | |
| Whatman qualitative filter paper, Grade 1, 110 mm diameter | Merck | WHA1001110 | |
| SOFTWARES | |||
| CorelDRAW | Corel Corporation | Version 7 | |
| FIJI (ImageJ Distribution) | Open Source/GNU General Public Licence (GPL) | N/A | ImageJ 2.x: Rueden, C. T.; Schindelin, J. & Hiner, M. C. et al. (2017), "ImageJ2: ImageJ for the next generation of scientific image data", BMC Bioinformatics 18:529, PMID 29187165, doi:10.1186/s12859-017-1934-z and Fiji: Schindelin, J.; Arganda-Carreras, I. & Frise, E. et al. (2012), "Fiji: an open-source platform for biological-image analysis", Nature methods 9(7): 676-682, PMID 22743772, doi:10.1038/nmeth.2019 |
| PRIMARY ANTIBODIES | |||
| Anti-Tyrosine Hydroxylase Antibody | Millipore Sigma | AB1542 | Sheep polyclonal (1:1000 dilution), RRID: AB_90755 |
| Anti-Tyrosine Hydroxylase Antibody, clone LNC1 | Millipore Sigma | MAB318 | Mouse monoclonal (1:1000 dilution), RRID: AB_2201528 |
| Anti-Vesicular Acetylcholine Transporter (VAchT) Antibody | Sigma-Aldrich | ABN100 | Goat polyclonal (1:1000 dilution), RRID: AB_2630394 |
| GFP Antibody | Novus Biologicals | NB600-308 | Rabbit polyclonal (1:1000 dilution), RRID: AB_10003058 |
| Phox2b Antibody (B-11) | Santa Cruz Biotechnology | sc-376997 | Mouse monoclonal (1:1000 dilution), RRID: AB_2813765 |
| SECONDARY ANTIBODIES | |||
| Alexa Fluor 488 AffiniPure Donkey Anti-Rabbit IgG (H+L) (min X Bov, Ck, Gt, GP, Sy Hms, Hrs, Hu, Ms, Rat, Shp Sr Prot) | Jackson ImmunoResearch | 711-545-152 | Donkey anti-Rabbit (1:400 dilution), RRID: AB_2313584 |
| AMCA AffiniPure Donkey Anti-Sheep IgG (H+L) (min X Ck, GP, Sy Hms, Hrs, Hu, Ms, Rb, Rat Sr Prot) | Jackson ImmunoResearch | 713-155-147 | Donkey anti-Sheep (1:400 dilution), RRID: AB_AB_2340725 |
| Cy5 AffiniPure Donkey Anti-Goat IgG (H+L) (min X Ck, GP, Sy Hms, Hrs, Hu, Ms, Rb, Rat Sr Prot) | Jackson ImmunoResearch | 705-175-147 | Donkey anti-Goat (1:400 dilution), RRID: AB_2340415 |
| Cy5 AffiniPure Donkey Anti-Mouse IgG (H+L) (min X Bov, Ck, Gt, GP, Sy Hms, Hrs, Hu, Rb, Rat, Shp Sr Prot) | Jackson ImmunoResearch | 715-175-151 | Donkey anti-Mouse (1:400 dilution), RRID: AB_2619678 |
| Cy5 AffiniPure Donkey Anti-Sheep IgG (H+L) (min X Ck, GP, Sy Hms, Hrs, Hu, Ms, Rb, Rat Sr Prot) | Jackson ImmunoResearch | 713-175-147 | Donkey anti-Sheep (1:400 dilution), RRID: AB_2340730 |
| RNASCOPE PROBES | |||
| Galanin Receptor 1 oligonucleotide probe | ACDBio | 448821-C1 | targets bp 482 - 1669 (Genebank ref: NM_008082.2) |
| Glycine transporter 2 oligonucleotide probe | ACDBio | 409741-C3 | targets bp 925 - 2153 (Genebank ref: NM_148931.3) |
| Phox2b oligonucleotide probe | ACDBio | 407861-C2 | targets bp 1617 - 2790 (Genebank ref: NM_008888.3) |
Références
- Wang, F., et al. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. Journal of Molecular Diagnostics. 14 (1), 22-29 (2012).
- Annese, T., et al. RNAscope dual ISH-IHC technology to study angiogenesis in diffuse large B-cell lymphomas. Histochemistry and Cell Biology. 153 (3), 185-192 (2020).
- Morrison, J. A., McKinney, M. C., Kulesa, P. M. Resolving in vivo gene expression during collective cell migration using an integrated RNAscope, immunohistochemistry and tissue clearing method. Mechanisms of Development. 148, 100-106 (2017).
- Gross-Thebing, T., Paksa, A., Raz, E. Simultaneous high-resolution detection of multiple transcripts combined with localization of proteins in whole-mount embryos. BMC Biology. 12, 55 (2014).
- Stempel, A. J., Morgans, C. W., Stout, J. T., Appukuttan, B. Simultaneous visualization and cell-specific confirmation of RNA and protein in the mouse retina. Molecular Vision. 20, 1366-1373 (2014).
- Kersigo, J., et al. A RNAscope whole mount approach that can be combined with immunofluorescence to quantify differential distribution of mRNA. Cell and Tissue Research. 374 (2), 251-262 (2018).
- Grabinski, T. M., Kneynsberg, A., Manfredsson, F. P., Kanaan, N. M. A method for combining RNAscope in situ hybridization with immunohistochemistry in thick free-floating brain sections and primary neuronal cultures. PLoS One. 10 (3), 0120120 (2015).
- Baleriola, J., Jean, Y., Troy, C., Hengst, U. Detection of axonally localized mRNAs in brain sections using high-resolution in situ hybridization. Journal of Visualized Experiments. (100), e52799 (2015).
- Fe Lanfranco, M., Loane, D. J., Mocchetti, I., Burns, M. P., Villapol, S. Combination of fluorescent in situ hybridization (FISH) and immunofluorescence imaging for detection of cytokine expression in microglia/macrophage cells. Bio-Protocol. 7 (22), (2017).
- Dereli, A. S., Yaseen, Z., Carrive, P., Kumar, N. N. Adaptation of respiratory-related brain regions to long-term hypercapnia: focus on neuropeptides in the RTN. Frontiers in Neuroscience. 13, 1343 (2019).
- Lazarenko, R. M., et al. Acid sensitivity and ultrastructure of the retrotrapezoid nucleus in Phox2b-EGFP transgenic mice. Journal of Comparative Neurology. 517 (1), 69-86 (2009).
- Gage, G. J., Kipke, D. R., Shain, W. Whole animal perfusion fixation for rodents. Journal of Visualized Experiments. (65), e3564 (2012).
- Paxinos, G., Franklin, K. B. . The mouse brain in stereotaxic coordinates. , (2004).
- Abercrombie, M. Estimation of nuclear population from microtome sections. Anatomical Records. 94, 239-247 (1946).
- Kerr, N., et al. The generation of knock-in mice expressing fluorescently tagged galanin receptors 1 and 2. Molecular and Cellular Neurosciences. 68, 258-271 (2015).
- Kachidian, P., Pickel, V. M. Localization of tyrosine hydroxylase in neuronal targets and efferents of the area postrema in the nucleus tractus solitarii of the rat. Journal of Comparative Neurology. 329 (3), 337-353 (1993).
- Stornetta, R. L., et al. Expression of Phox2b by brainstem neurons involved in chemosensory integration in the adult rat. Journal of Neuroscience. 26 (40), 10305-10314 (2006).
- Gilmor, M. L., et al. Expression of the putative vesicular acetylcholine transporter in rat brain and localization in cholinergic synaptic vesicles. Journal of Neuroscience. 16 (7), 2179-2190 (1996).
- Fisher, J. M., Sossin, W., Newcomb, R., Scheller, R. H. Multiple neuropeptides derived from a common precursor are differentially packaged and transported. Cell. 54 (6), 813-822 (1988).
- Towle, A. C., Lauder, J. M., Joh, T. H. Optimization of tyrosine-hydroxylase immunocytochemistry in paraffin sections using pretreatment with proteolytic-enzymes. Journal of Histochemistry and Cytochemistry. 32 (7), 766-770 (1984).
- Biancardi, V., et al. Mapping of the excitatory, inhibitory, and modulatory afferent projections to the anatomically defined active expiratory oscillator in adult male rats. Journal of Comparative Neurology. 529 (4), 853-884 (2021).
- Matthews, D. W., et al. Feedback in the brainstem: an excitatory disynaptic pathway for control of whisking. Journal of Comparative Neurology. 523 (6), 921-942 (2015).
- Ramos-Vara, J. A. Principles and methods of immunohistochemistry. Methods in Molecular Biology. 1641, 115-128 (2017).
- Shi, S. R., Key, M. E., Kalra, K. L. Antigen retrieval in formalin-fixed, paraffin-embedded tissues: an enhancement method for immunohistochemical staining based on microwave oven heating of tissue sections. Journal of Histochemistry and Cytochemistry. 39 (6), 741-748 (1991).
- Yamashita, S., Katsumata, O. Heat-induced antigen retrieval in immunohistochemistry: mechanisms and applications. Methods in Molecular Biology. 1560, 147-161 (2017).
- Yamashita, S., Okada, Y. Mechanisms of heat-induced antigen retrieval: analyses in vitro employing SDS-PAGE and immunohistochemistry. Journal of Histochemistry and Cytochemistry. 53 (1), 13-21 (2005).
- Yamashita, S. Heat-induced antigen retrieval: mechanisms and application to histochemistry. Progress in Histochemistry and Cytochemistry. 41 (3), 141-200 (2007).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.