
Method Article
Generation of Self-assembled Vascularized Human Skin Equivalents
In This Article
Summary
The goal of this protocol is to describe the generation and volumetric analysis of vascularized human skin equivalents using accessible and simple techniques for long term culture. To the extent possible, the rationale for steps is described to allow researchers the ability to customize based on their research needs.
Abstract
Human skin equivalents (HSEs) are tissue engineered constructs that model epidermal and dermal components of human skin. These models have been used to study skin development, wound healing, and grafting techniques. Many HSEs continue to lack vasculature and are additionally analyzed through post-culture histological sectioning which limits volumetric assessment of the structure. Presented here is a straightforward protocol utilizing accessible materials to generate vascularized human skin equivalents (VHSE); further described are volumetric imaging and quantification techniques of these constructs. Briefly, VHSEs are constructed in 12 well culture inserts in which dermal and epidermal cells are seeded into rat tail collagen type I gel. The dermal compartment is made up of fibroblast and endothelial cells dispersed throughout collagen gel. The epidermal compartment is made up of keratinocytes (skin epithelial cells) that differentiate at the air-liquid interface. Importantly, these methods are customizable based on needs of the researcher, with results demonstrating VHSE generation with two different fibroblast cell types: human dermal fibroblasts (hDF) and human lung fibroblasts (IMR90s). VHSEs were developed, imaged through confocal microscopy, and volumetrically analyzed using computational software at 4- and 8-week timepoints. An optimized process to fix, stain, image, and clear VHSEs for volumetric examination is described. This comprehensive model, imaging, and analysis techniques are readily customizable to the specific research needs of individual labs with or without prior HSE experience.
Introduction
Human skin performs many essential biological functions including acting as an immune/mechanical barrier, regulating body temperature, participating in water retention and sensory roles1,2,3,4. Anatomically, skin is the largest organ in the human body and is made up of three main layers (epidermis, dermis, and hypodermis) and possesses a complex system of stromal, vascular, glandular, and immune/nervous system components in addition to epidermal cells. The epidermis itself is composed of four layers of cells that are continuously renewed to maintain barrier function and other structures of native skin (i.e., sweat and sebaceous glands, nails)3. Skin physiology is important in immune function, wound healing, cancer biology, and other fields, leading researchers to use a wide range of models, from in vitro monocultures to in vivo animal models. Animal models offer the ability to study the full complexity of skin physiology, however, commonly used animal models such as mice have significant physiological differences when compared to humans5. These limitations, and the increased cost of animal models, have led many researchers to focus on developing in vitro models that more closely reflect the physiology of human skin1,6. Of these, one of the simpler model types is the human epidermal equivalent (HEE; also referred to as half-thickness skin models) which are composed of only epidermal keratinocytes on an acellular dermal matrix, but capture epidermal differentiation and stratification seen in vivo. Building on this, models containing dermal and epidermal components (keratinocytes and fibroblasts) are often referred to as human skin equivalents (HSE), full-thickness skin models, or organotypic skin constructs (OSC). Briefly, these models are generated by encapsulating dermal cells within gel matrices and seeding epidermal cells on top. Epidermal differentiation and stratification can then be achieved via specialized media and air exposure7. Skin equivalents have most often been generated through self-assembly techniques using dermal gels made of collagen type I (either of rat tail or bovine skin origin)1,8, but similar models have incorporated other matrix components such as fibrin9,10, fibroblast derived11,12, cadaveric de-epidermized membranes13,14,15,16, commercially available gels and others1,12,13,17,18,19. Currently, there are skin equivalents commercially available (as previously reviewed1,2). However, these are primarily developed for therapeutic purposes and cannot be readily customized to specific research questions.
HSEs have been applied in studies of wound healing, grafting, toxicology, and skin disease/developement11,12,13,16,8,20,21,22,23. Although 3D culture more comprehensively models functions of human tissue compared to 2D cultures24, the inclusion of diverse cell types that more accurately reflect the in vivo population enables studies of cell-cell coordination in complex tissues24,25,26. Most HSEs only include dermal fibroblasts and epidermal keratinocytes27, although the in vivo skin environment includes many other cell types. Recent studies have started including more cell populations; these include endothelial cells in vasculature10,28,29,30,31,32,33,34, adipocytes in sub-cutaneous tissue35,36, nerve components19,21, stem cells27,37,38, immune cells10,39,40,41,42, and other disease/cancer specific models16, 40, 43,44,45,46,47. Particularly important among these is vasculature; while some HSEs include vascular cells, overall they still lack comprehensive capillary elements with connectivity across the entire dermis10,29 , extended in vitro stability28, and appropriate vessel density. Further, HSE models are typically assessed through post-culture histological sectioning which limits analysis of the three-dimensional structure of HSEs. Three dimensional analysis allows for volumetric assessment of vascular density48,49 as well as regional variation of epidermal thickness and differentiation.
Although HSEs are one of the most common organotypic models, there are many technical challenges in generating these constructs including identification of appropriate extracellular matrix and cell densities, media recipes, proper air liquid interface procedures, and post-culture analysis. Further, while HEE and HSE models have published protocols, a detailed protocol incorporating dermal vasculature and volumetric imaging rather than histological analysis does not exist. This work presents an accessible protocol for the culture of vascularized human skin equivalents (VHSE) from mainly commercial cell lines. This protocol is written to be readily customizable, allowing for straight-forward adaptation to different cell types and research needs. In the interest of accessibility, availability, and cost, the use of simple products and generation techniques was prioritized over the use of commercially available products. Further, straightforward volumetric imaging and quantification methods are described that allow for assessment of the three-dimensional structure of the VHSE. Translating this procedure into a robust and accessible protocol enables non-specialist researchers to apply these important models in personalized medicine, vascularized tissue engineering, graft development, and drug evaluation.
Protocol
1. Preparation for 3D culture
- Prepare rat tail collagen stock at 8 mg/mL, using established protocols50,51,52. Alternatively, rat tail collagen can be purchased from vendors (see materials list) at appropriate concentrations.
NOTE: Collagen can be prepared or purchased at different concentrations in the range of 3-10 mg/mL, or higher50,51,52. The calculations in the protocol assume an 8 mg/mL concentration but can be adjusted based on the needs of the researcher. - Expand cell lines: Endothelial and fibroblast cells need to be ready for seeding at the start of 3D collagen dermal component generation (step 2). Keratinocytes need to be ready on day 7 of 3D culture. One complete VHSE construct requires 7.5 x 105 endothelial cells; 7.5 x 104 fibroblasts; and 1.7 x 105 keratinocytes for generation (Table 1).
NOTE: These densities are appropriate for 12-well size permeable tissue culture inserts or equivalent. Cell density and format can be scaled up or down based on the needs of the researcher. To clarify, this amount of endothelial and fibroblast cells will seed 1-3 dermal components, while each epidermal component requires 1.7 x 105 keratinocytes. - Perform all cell centrifugation in this protocol for 5 min at 300 x g, but this may be decreased for more fragile cell types.
2. Generation of 3D collagen dermal component
NOTE: Step 2 is a time sensitive procedure and must be completed in one setting. It is advised to complete a quality check of the collagen stock to ensure proper gelation and homogeneity before beginning dermal component seeding, see troubleshooting in discussion.
- Acellular collagen layer preparation and seeding
- Prepare two 1.7 mL capped microcentrifuge tubes, one for the acellular support and one for the cellular dermis. Amounts given in this step will prepare 1 mL of 3 mg/mL collagen (target collagen concentration), sufficient for (3) 12-well size VHSEs. Equations are listed if adjustment is necessary. Both volume and density can be scaled based on the needs of the researcher (common reference numbers are given in Table 2).
- To each tube, add 100 µL of culture grade 10x Phosphate-buffered saline (PBS) (one tube will yield 3 VHSEs) and add 8.6 µL of 1 N NaOH. Place capped tubes on wet ice to chill for at least 10 min.
Cs = Collagen Stock Concentration
Vf = Final Volume of Collagen Needed
Ct = Target Collagen Concentration
Vs = Volume of stock collagen necessary for desired amount (Vf)
Vpbs = Volume of 10X PBS needed for target collagen concentration (Ct)
VNaOH = Volume of 1N NaOH needed for Ct
Vmedia = Volume of media, call suspension, or ddH2O needed for Ct




- Prepare 1000 and 250 µL positive displacement pipettes for use and set aside. As later steps are time sensitive, it is convenient to load pipette tips and set volumes (375 µL and 125 µL, respectively). Additionally, setup a normal 1000 µL pipette for 516 µL.
NOTE: Positive displacement pipettes can be substituted with normal pipettes if necessary, but because of the high viscosity of collagen and the time/temperature sensitivity of this procedure, positive displacement pipettes are recommended to help produce consistent seeding results. If using normal pipettes, use slow movements. - Prepare culture insert well plates: Use sterile forceps to place three 12-well size culture inserts into a sterile 12-well tissue culture plate, place into the center columns.
- Set out cold media appropriate for fibroblast and endothelial cell types.
- After cooling of capped tubes, place one tube (for the acellular support) on a rack with the contents visible. Leave the other tube (for the cellular dermis) on ice.
- Remove 8 mg/mL collagen stock from refrigeration and place on wet ice.
NOTE: Do not use freezer ice or -20 °C benchtop coolers, as this will freeze the collagen. - To the cold capped tube, add 516 µL of media and immediately add 375 µL of cold collagen using the 1000 µL positive displacement pipette. Dispense collagen into the solution (not to side of the tube). Immediately remove the empty pipette tip and switch to the prepared 250 µL positive displacement pipette to mix.
- Mix quickly but gently to prevent bubble formation, do not remove tip from the solution, if possible. Mix until the solution is of homogenous color, which typically takes about 5 pipette cycles or 10 s (if using media with phenol red, the color will become lighter and uniform). When mixing, be sure to draw from different positions of the tube (bottom and top) for uniform mixing.
NOTE: This can be performed with 516 µL of cell culture grade water or other cell culture grade liquid, however, phenol red of most media is a good indicator of the mixing. Use either fibroblast or endothelial media that was used for 2D expansions.
- Mix quickly but gently to prevent bubble formation, do not remove tip from the solution, if possible. Mix until the solution is of homogenous color, which typically takes about 5 pipette cycles or 10 s (if using media with phenol red, the color will become lighter and uniform). When mixing, be sure to draw from different positions of the tube (bottom and top) for uniform mixing.
- Immediately disperse 125 µL of acellular collagen onto the membrane of each of the three 12-well culture inserts. To ensure uniform coverage of the acellular collagen gel, rock the dish; if that does not create uniform membrane coverage then use the pipette tip to essentially paint the membrane by gently spreading collagen around; avoid applying pressure to the membrane. Gelation begins almost immediately; perform this step quickly to ensure even coverage.
NOTE: There will be excess acellular collagen. The volume can be reduced, however, preparing less than 1 mL of collagen suspension can result in difficulties mixing the solution and insufficient gelation. - Immediately move the 12-well plate to a 37 °C cell culture incubator to let it gel for at least 20 min (acellular collagen can gel for longer if needed; during this gelation time, proceed to step 2.2). Remove the collagen suspension from ice and place back into refrigeration (collagen is most stable at 4 °C).
- Cell suspension & seeding preparation
NOTE: For the culture timeline of this protocol, this corresponds to Submersion Day 1 (SD1)- During gelation of the acellular collagen support, trypsinize and count the endothelial and fibroblast cell lines.
- Suspend 7.5 x 105 endothelial cells and 7.5 x 104 fibroblasts in 258 µL of their respective medias and combine cell suspensions to create a 516 µL aliquot. Maintain the cell suspensions on wet ice until use.
- Prepare 1000 and 250 µL positive displacement pipettes for use and set aside. As later steps are time sensitive, it is convenient to load pipette tips and set volumes (375 µL and 250 µL, respectively). Additionally, setup a normal 1000 µL pipette for 516 µL.
- Cell laden collagen seeding of dermal compartment
- After the gelation period, remove the 12 well plate of acellular collagen from the incubator.
NOTE: If this collagen is not gelled after 30 min, do not continue the procedure as there was likely a mistake during seeding or the collagen stock may have a problem (see troubleshooting in discussion). - Remove the 1.7 mL capped tube from wet ice (contains 10x PBS and NaOH). Place the tube in a rack so that the contents are visible. Loosen/open all caps (cell suspension, cold capped tube).
- Remove the stock collagen (8 mg/mL) from 4 °C refrigeration and place it on wet ice. Leave the cap open.
- Add the 516 µL of cooled cell suspension to the cold capped tube. Use the 1000 µL positive displacement pipette to immediately pipette 375 µL of cold collagen solution directly into the solution of the capped tube.
- Expel all collagen from pipette into the tube and discard the positive displacement pipette tip. Immediately switch to the 250 µL positive displacement pipette and mix the collagen solution.
- Mix the collagen solution as completed previously (quickly but gently to prevent bubble formation), do not remove tip from gel if possible. Mix until solution is homogenous (about 5 pipette cycles or 10 s). When mixing be sure to draw from different positions of the tube (bottom and top) for uniform mixing.
- Once mixed, immediately transfer 250 µL of cellular collagen solution onto the acellular collagen supports in each of the three 12-well culture inserts. To ensure uniform coverage of the acellular collagen support, rock the dish and/or use the positive displacement pipette to gently move the freshly seeded cellular collagen around without disturbing the acellular layer.
- Immediately move the 12-well plate to 37 °C cell culture incubator to let it gel for at least 30 min. Place collagen back into 4 °C refrigeration after use.
- After the 30-minute gel time, gently tilt the plate to assess the gelation. Ensure the collagen is solidified.
- Add 500 µL and 1000 µL of blend media (half endothelial and half fibroblast maintenance media) to the upper chamber and lower chamber of the insert, respectively (top first, then bottom to prevent hydrostatic pressure from pushing collagen up). Add media slowly to the side of the well, not directly onto collagen gel, to minimize disruption of the collagen.
- Ensure that the collagen gel is submerged, add more media if necessary. Place the well plate in the cell incubator for overnight incubation. At this stage, media contains 10% FBS; the normal maintenance media for each cell line (timeline and schematic given in Figure 1, A).
NOTE: Media throughout VHSE culture can be adapted for custom cell types; some optimization may be necessary.
- Ensure that the collagen gel is submerged, add more media if necessary. Place the well plate in the cell incubator for overnight incubation. At this stage, media contains 10% FBS; the normal maintenance media for each cell line (timeline and schematic given in Figure 1, A).
- After the gelation period, remove the 12 well plate of acellular collagen from the incubator.
- Submersion Day 2 (SD2) media change
- Change 10% FBS media in VHSE wells to 5% FBS half fibroblast, half endothelial media supplemented with 100 µg/mL L-Ascorbic Acid. Add 500 µL to upper chamber of the culture insert at the side of the well (again, add carefully to the sidewall to minimize disruption of the collagen) and add 1000 µL to lower chamber.
- Renew media every 2 days (SD4 and SD6) until Submersion Day 7 (SD7).
- Use a manual pipette to remove media from the wells. Using an aspirator is possible but can result in damage or destruction of the construct.
NOTE: L-ascorbic acid must be made up fresh every 2-3 days (it oxidizes in solution to produce hydrogen peroxide thus, inducing oxidative stress and eventually cellular damage53). Thus, media must be changed every 2-3 days from SD2 until the end of the VHSE culture since L-ascorbic acid is present. It is easiest to make a stock of media and add a freshly prepared amount of L-ascorbic acid to a media aliquot every feeding day. Use culture grade water or media as a solvent and prepare fresh L-ascorbic acid at 100 mg/mL. L-ascorbic acid stimulates collagen synthesis by fibroblasts at an appropriate rate and promotes collagen stability54,55,56; it also decreases endothelial permeability and maintains vessel wall integrity56,57 and additionally contributes to epidermal barrier formation6,58.
3. Seeding of epidermal component and stratification induction
- Submersion Day 7 (SD7): seed keratinocytes
NOTE: Seed keratinocytes to establish the epidermis on SD7. This time point can be shifted based on the needs of the researcher. Duration of submersion culture without keratinocytes should not exceed 9 days, as a longer submersion often leads to increased dermal contraction. If contraction occurs before SD7, it is recommended to shorten the submersion period to 5 days and seed epidermis on SD5. Optimize the submersion period as required for specific experiments (see troubleshooting in discussion).- Culture keratinocytes (N/TERT-120,59 or other appropriate cells) to their confluency limit before trypsinization and seeding onto VHSEs. For N/TERT-1 cells, confluency should not significantly exceed 30% to prevent un-wanted differentiation of keratinocytes in 2D culture59. For other appropriate cell lines, such as primary human epidermal keratinocytes, a confluency limit of 75-80% is generally used60.
- After trypsinization, count and suspend 510,000 cells in 600 µL of Human Skin Equivalent (HSE) Differentiation media supplemented with 5% FBS (Table 1).
NOTE: 510,000 cells in 600 µL allows 170,000 cells/construct when seeding 200 µL per construct (3 VHSEs). - Using a manual pipette, collect and discard media currently in the bottom and top chamber for each construct well. Be sure to collect as much media as possible. Collect media that may be stuck directly under the permeable membrane by gently placing the pipette tip under the culture insert membrane and knocking the insert out of place temporarily. Media may have been stuck due to surface tension. Be sure that the inserts sit flat in their wells before proceeding. Using an aspirator is possible but can result in damage or destruction of the construct.
- Add 1 mL of HSE media supplemented with 5% FBS to the lower chamber of each well. Then add 200 µL of cell suspension to the top chamber of each well. Seed directly onto the dermal construct surface. Let keratinocytes settle for 2 h in the incubator.
- Two h after seeding the keratinocytes, carefully add 300 µL of HSE media supplemented with 5% FBS to the top chamber of each construct well; slowly pipette media onto side of culture insert. Load media into the top chamber very carefully as to not disturb settled keratinocytes that may not have adhered tightly to the underlying collagen gel yet.
- After loading the media, place construct back into the incubator.
- Submersion Day 8/9 (SD8 or SD9)
- Make up HSE media supplemented with 1% FBS and 100 µg/mL L-ascorbic acid.
- Remove media from both the upper and the lower chambers with a manual pipettor.
- Add 500 µL media into the top chamber first and then 1 mL into the bottom chamber. (This step can be done on SD8 or SD9)
- Submersion Day 9/10 (SD9 or SD10, this should be the day after step 3.2)
- Make up serum free HSE differentiation media with 100 µg/mL L-ascorbic acid.
- Remove media from both the upper and the lower chambers with a manual pipettor.
- Load 500 µL into the upper chamber and 1 mL in the lower chamber.
- Air-Liquid Interface Day 1 (ALI1)
NOTE: ALI is performed the day after step 3.3.- Lift each construct to air-liquid interface (ALI) by removing media waste from the upper chamber only. Use a manual pipette to get as close to the epidermal layer as possible without touching or damaging it.
- Tilt the plate slightly at different angles to collect the media. Remove as much media as possible in this step. Add approximately 2 mL of sterile water to the surrounding wells in the plate to maintain consistent humidity; keep the wells filled with water throughout culture.
- Check the plate a few h later to make sure the keratinocytes are still at the air-liquid interface. If there is media in the upper chamber remove it. Keep track of how much media is removed for each VHSE well, (The initial volume of upper and lower chambers (1500 µL) - media removed = a good starting point for ALI feeding).
NOTE: VHSEs do not necessarily require the same media level for air lift; usually if the VHSEs are seeded together then they need about the same level of media for air lift, but this is not always the case. Adjust the volumes as needed to maintain ALI but ensure that the media levels are not so low that the VHSEs dry out. It is safer to be cautious and remove small media amounts daily until a balance between air lift and hydration has been met.
- ALI Day 2 (ALI2)
- From this point on, only use serum free HSE media supplemented with 100 µg/mL L-ascorbic acid. Change media on ALI Day 2 (ALI2). If there is media in the top chamber, remove it and add it to the amount of removed media recorded previously. Calculate the volume of media needed using the equation in the previous step. For example: If 200 µL of media was removed from the upper chamber then add 1300 µL to establish ALI (as 1500 µL - 200 µL = 1300 µL)
- Use the volume calculated to load into the bottom chamber of each well, then place the plate back into the cell incubator. Keep track of the volume used per day. When using the recommended collagen amounts in 12-well culture inserts, the usual range of ALI values falls between 750 µL and 1300 µL. Typically, the volume decreases over culture maturation and becomes consistent around week 2/3 of ALI. Depending on the culture specifics, this number may change and must be optimized (as described in 3.4.2 - 4.1).
4. Routine maintenance of vascular human skin equivalent
- From ALI Day 3 (ALI3) through culture endpoint: Renew media of the lower chamber every 2-3 days using serum free HSE media with 100 µg/mL L-ascorbic acid. Continue to adjust and track the media level needed in the bottom chamber for ALI as described in Step 3.5.2.
- As the epidermal surface must remain in contact with the air, check and adjust the media level daily until consistent ALI levels are established. The epidermal layer should look hydrated, not dry, but there should not be media pooled on top of the construct. Cultures with 8 weeks of ALI have provided the most consistent morphology and expression; however, depending on the application, cultures of 4 to 12 weeks may be appropriate. Culture duration for different cell and culture conditions may need to be optimized.
NOTE: Changing media Monday, Wednesday, Friday is a good practice. The VHSEs are healthy over the weekend, but media should be changed early on Monday and late on Friday. After entirely completing steps 1-4, the generation of a VHSE is complete. Steps 5-end of the protocol are optional processing and imaging techniques that have been optimized for this type of 3D construct.
5. Fixation and permeabilization of 3D constructs
NOTE: Step 5 has been optimized for imaging techniques specific to this 3D construct that are outlined in the remainder of the protocol. The following steps are not necessary for generating a VHSE.
- Fixation/permeabilization
- Carefully remove all media from upper and lower chambers of each well at the end point of the culture period.
NOTE: The epidermal layer is possibly fragile, handle with care and do not agitate the epidermis with aggressive pipetting. - Add 4% paraformaldehyde (PFA) in PBS (pH 6.9) to the upper chamber wall (not directly on the construct) and then to the lower chamber, to pre-fix each construct. Add 1 mL per chamber and expose for 5 min at room temperature.
CAUTION: PFA is dangerous and should be handled with care and appropriate personal protection equipment (PPE), including eye protection. - Remove 4% PFA solution after 5 min and add the 0.5% Triton X 100 in 4% PFA solution to the upper and lower chambers as described in the previous step. Expose for 1 hour at room temperature; VHSE construct does not require a sterile environment from now on.
- After 1 hour, carefully remove the permeabilization/fixation solution from both the chambers and wash the sample 3 times with 1x PBS.
- Store samples in PBS in 4 °C refrigeration or immediately stain. To store the samples, wrap the dish in a plastic wrap and then foil to minimize evaporation and light exposure
NOTE: Pause point - After fixation and permeabilization, this procedure can be paused since the samples are stable for several weeks if prepared as outlined in step 5.1.5. Alternatively, staining (as described in step 6) can be completed immediately following step 5.
- Carefully remove all media from upper and lower chambers of each well at the end point of the culture period.
6. Immunofluorescent staining of 3D constructs
- Construct Preparation
NOTE: VHSEs stain well when separated from porous membrane of the culture insert; separation from the membrane is also necessary for un-obstructed imaging and enable reduced volumes for staining.- To prepare the construct for immunofluorescent staining, turn an insert upside down and place it over its well on the well plate (if the VHSE falls, it will fall into the well with PBS) (Supplemental Figure 1A).
- Stabilize the insert with one hand over the well while using fine tip forceps and/or an precision knife to cut about half of the circumference of the insert membrane. Cut as close to the plastic housing as possible with a gentle hand to prevent damage of the VHSE construct.
- Using the fine tip forceps, grab the edge of the cut membrane flap and gently peel the porous membrane off the insert as well as the VHSE construct. Do this very carefully and slowly to prevent damage to the VHSE construct structure. If the VHSE construct separates easily then it should fall into the well below, if it gets stuck on the side of the chamber then use the fine tip forceps or a small scoopula to move it to the well. Be very mindful of the epidermal layer as it is usually fragile (Supplemental Figure 1A).
NOTE: Sometimes the membrane does not come off easily or comes off in pieces, if this happens use the tools to carefully pull the membrane and VHSE construct apart. Ensure the VHSEs do not dry out during this process by dipping in PBS, if necessary. - Once the VHSE is in the well, discard any remaining pieces of the insert membrane and keep the culture insert housing in each well to hold the VHSEs in a submerged position during staining.
- Staining
NOTE: Staining and associated handling/manipulation and washes should be performed as gently as possible since VHSEs can be fragile. If portions of the epidermis lift off, the pieces can be stained separately; upper layers of the epidermis are fragile and go through natural desquamation4, but for analysis it is important to maintain integrity as much as possible.- Prepare the chosen primary antibody stains in 700 µL of blocking buffer per construct well (typically, all primary antibodies can be in the same staining solution, but this should be confirmed for new antibodies). 700 µL works for 12-well size, but may be adjusted for other culture formats. Recommended concentrations of primary and secondary antibodies with blocking buffer recipe are given in Table 3 (optimization may be required).
- Remove any PBS from the well using a manual pipette, be careful to pipette away from VHSEs (as VHSEs are floating, vacuum aspiration is not recommended).
- Add the primary stain solution to each well and place the culture insert housing into the well to keep the VHSE submerged in fluid (Supplemental Figure 1B). Wrap the well plate with plastic wrap. Foil and stain for 48 h in 4 °C refrigeration without agitation or rocking (rocking may damage the VHSE construct).
- After 48 h, prepare secondary antibodies and chemical stains in 700 µL of blocking buffer (per well).
- Remove the culture insert housing and the primary stain solution and wash with 1x PBS, 3x for 5 min before adding the secondary stain solution. Place the culture insert housing back into the well to keep VHSE construct submerged (Supplemental Figure 1B). Expose for 48 h in 4 °C refrigeration without agitation or rocking.
- After 48 h exposure, remove the stain solution with a manual pipettor and gently wash 3x with PBS; do not pipette fluid straight onto the VHSEs as they may be fragile. Rehydrate with excess PBS and place the culture insert housing back into the well to keep the VHSE submerged and hydrated during storage (store by wrapping in plastic wrap and foil to minimize evaporation and light exposure)
- Clearing (optional & terminal)
NOTE: Clearing is optional for imaging. If completed, it should be done after staining/imaging the sample completely since clearing prevents further staining, may alter fluorophore performance, and may damage VHSE structure. Multiple tissue clearing methods exist49,61,62 and can be optimized for specific projects. Methyl salicylate clearing, described below, is both simple and effective for VHSE. The following clearing technique must be completed in glass containers and pipette tips must be glass or polypropylene (polystyrene will dissolve in contact with methyl salicylate). Complete all clearing procedure in a well-ventilated area or fume hood.- Add 100% methanol to a small shallow glass container (glass Petri dishes work well). Use the smallest possible container that will fit the construct (to minimize reagent waste).
- Remove the construct from the well plate using forceps/scoopula (Supplemental Figure 1C) and place in the methanol filled container. Add more methanol if construct is not submerged.
- Dehydrate the VHSE construct in methanol for 3 x 10 min immersions; fully replace methanol after each immersion and promptly remove methanol after the last bath. Over the course of this procedure, the construct may become more opaque and shrink slightly.
NOTE: These durations and repetitions have been optimized but methanol and the following methyl salicylate procedures may need to be customized, depending on the specific culture format and stains. - Immediately after removing methanol, add methyl salicylate and submerge the VHSE in 5 x 5 min immersions. Fully replace the reagent after each immersion and leave the VHSE in the 5th immersion solution for storage. Over the course of this procedure, the construct becomes transparent.
- Image the construct or store at 4 °C. After clearing, complete all imaging as soon as possible, as the fluorophores may degrade in methyl salicylate within days. Clearing causes the constructs to become brittle and the extended storage, while not recommended, needs a regular check to ensure that there is a sufficient amount of methyl salicylate.
7. Confocal Imaging of 3D constructs
NOTE: Imaging through tissue culture plastic will not yield the same quality of image as imaging through coverslip glass, this method describes fabrication of a custom glass-bottom well to prevent drying during confocal imaging. Typically, this is sufficient for at least 3 h of imaging.
- Two days before imaging: prepare polydimethylsiloxane (PDMS)
- Prepare PDMS48,63,64 at a suggested concentration of [9:1], base: crosslinker. Prepare 30 g of PDMS total: 27 g of base component and 3 g of crosslinker. Place any clean mixing vessel on a weighing balance and tare the scale. Add the base (27 g) and then add the crosslinker (3 g) to achieve a total of 30 g. Always add base prior to crosslinker.
- Stir the solution vigorously for at least 4 min; this will create small bubbles. After sufficient mixing, pour the PDMS into a 100-mm Petri dish, or similar flat bottom heat resistant container.
- De-gas the PDMS in a vacuum chamber until all bubbles from mixing disappear and PDMS is clear. Release the vacuum slowly and remove the PDMS (slowly). Place the dish into an oven to cure overnight (50-60 °C); ensure the dish is sitting flat for PDMS to cure evenly.
NOTE: After curing, PDMS should be clear and the surface should be smooth and not sticky (stickiness may indicate inadequate mixing).
- One day before imaging: PDMS well preparation
- Using a steel punch or handheld precision knife, punch or cut out a circular well from the PDMS sheet prepared in 7.1. The well should be around the same size as the VHSE construct. Cut a square patch around the circular well to create a single PDMS well. The 30 g PDMS amount prepared should yield at least four custom wells.
NOTE: The PDMS well must be close to the size of the VHSE construct. It must constrict sample motion during imaging. Multiple wells can be fabricated at once and stored indefinitely in a clean container. - Using a glass coverslip of a similar size to the PDMS well, add cyanoacrylate glue (e.g., super glue) to the bottom surface of the PDMS (the smooth surface that was in contact with the Petri dish) and smear evenly with a disposable pipette tip. Center, and press the PDMS well onto the glass while leaving a clear glass window within the punched circle (ensure the glue is not smeared over the viewing window).
NOTE: If available, plasma bonding of the PDMS to the coverslip is an alternative65,66,67. - Let the glue dry for several hours, or overnight, before using. These are reusable until they break from normal wear and tear.
NOTE: It is not recommended to stain the samples in the glued PDMS well used for imaging. These wells hold fluid for several hours but can leak during longer staining.
- Using a steel punch or handheld precision knife, punch or cut out a circular well from the PDMS sheet prepared in 7.1. The well should be around the same size as the VHSE construct. Cut a square patch around the circular well to create a single PDMS well. The 30 g PDMS amount prepared should yield at least four custom wells.
- VHSE imaging
NOTE: If imaging uncleared samples, use PBS as imaging solution. If imaging with cleared samples, use methyl salicylate (or the chosen clearing solution) as the imaging solution.- Add a few drops of imaging solution into the PDMS well and check for leaks (if there is a leak, repair it with a dot/smear of cyanoacrylate super glue or use another well).
- Keep the imaging solution in the PDMS well when adding the VHSE. Using scoopula or fine tip forceps (Supplemental Figure 1C), remove construct from 12-well plate and place into the PDMS well onto the glass coverslip. Place construct with the orientation of interest facing toward the objective. For example, to image the epidermis using an inverted microscope, make sure the epidermis is facing down, toward the glass.
- Alternatively, for an upright microscope, face the epidermis up. The below imaging procedures are described for an inverted microscope, but could be readily adapted for an upright.
NOTE: Be cautious when manipulating the VHSE to avoid damage. Transfer over the well plate in case the VHSE falls. A bent, flat tip scoopula is the easiest way to transfer the construct (Supplemental Figure 1C).
- Alternatively, for an upright microscope, face the epidermis up. The below imaging procedures are described for an inverted microscope, but could be readily adapted for an upright.
- Make sure the sample is sitting flat in the well and that no portions of the epidermis or dermis are folded under the sample. If folding occurs, gently manipulate the sample with forceps or a scoopula; adding extra imaging solution temporarily to float the VHSE may help it straighten out. Folding or wrinkling of the sample can be seen by eye or using the microscope.
- Fill the well with imaging solution, using just enough fluid to keep sample hydrated; too much fluid will float the sample, resulting in motion during imaging. The construct should be sitting on the glass viewing window; test for movement by tilting the PDMS well. If there is movement, remove some fluid; add and remove fluid drop wise until the movement stops.
- Place a glass slide over the well to minimize evaporation during imaging (Supplemental Figure 1D). For longer imaging sessions, check sample frequently to ensure proper fluid levels. If accessible, a humidified chamber during imaging can be used (although it is typically not necessary).
- Place sample on the microscope stage and image through the glass coverslip window (Supplemental Figure 1D). This technique allows for at least 3 h of continuous confocal imaging, but the hydration of the sample should be checked regularly, with imaging solution added when needed.
NOTE: If sample is cleared, methyl salicylate will degrade the glue over time. The glue bonding the PDMS can be re-applied between imaging runs; or the sample can be transferred to new wells periodically. In wells with plasma bonding, this will not be an issue. - After imaging, float the sample with imaging fluid as much as possible in the well. Use a scoopula or fine tip forceps to transfer the sample into its storage well. Perform the transfer over a well plate in case the sample falls.
- Each PDMS well and top glass coverslip can be re-used until they break. Clean bottom glass before imaging, both inside and outside the well. Before re-using, always check for leaks and repair with glue, as necessary.
- Store samples as described in step 6.3.6 and add PBS every few months to maintain; if samples are cleared, store in glass using methyl salicylate and check the levels regularly. Cleared samples may degrade rapidly (within days) and should be imaged as soon as possible.
Results
Here is presented a protocol for generation of in vitro vascularized human skin equivalents (VHSE) using telomerase reverse transcriptase (TERT) immortalized keratinocytes (N/TERT-120,59), adult human dermal fibroblasts (hDF), and human microvascular endothelial cells (HMEC-1) (Figure 1). Additionally, the customizable nature of this protocol is highlighted by also demonstrating VHSE generation and stability when using commonly available lung fibroblasts (IMR90) instead of hDF. Generation of the VHSE is completed in steps 1-4, while steps 5-7 are optional end point processing and imaging techniques that were optimized for these VHSEs. It is important to note that the VHSEs can be processed according to specific research questions and steps 5-7 are not required to generate the construct. Volumetric imaging, analysis, and 3D renderings were completed to demonstrate a volumetric analysis method. These volumetric construct preparation and imaging protocols preserve VHSE structure at both the microscopic and macroscopic levels, allowing for comprehensive 3D analysis.
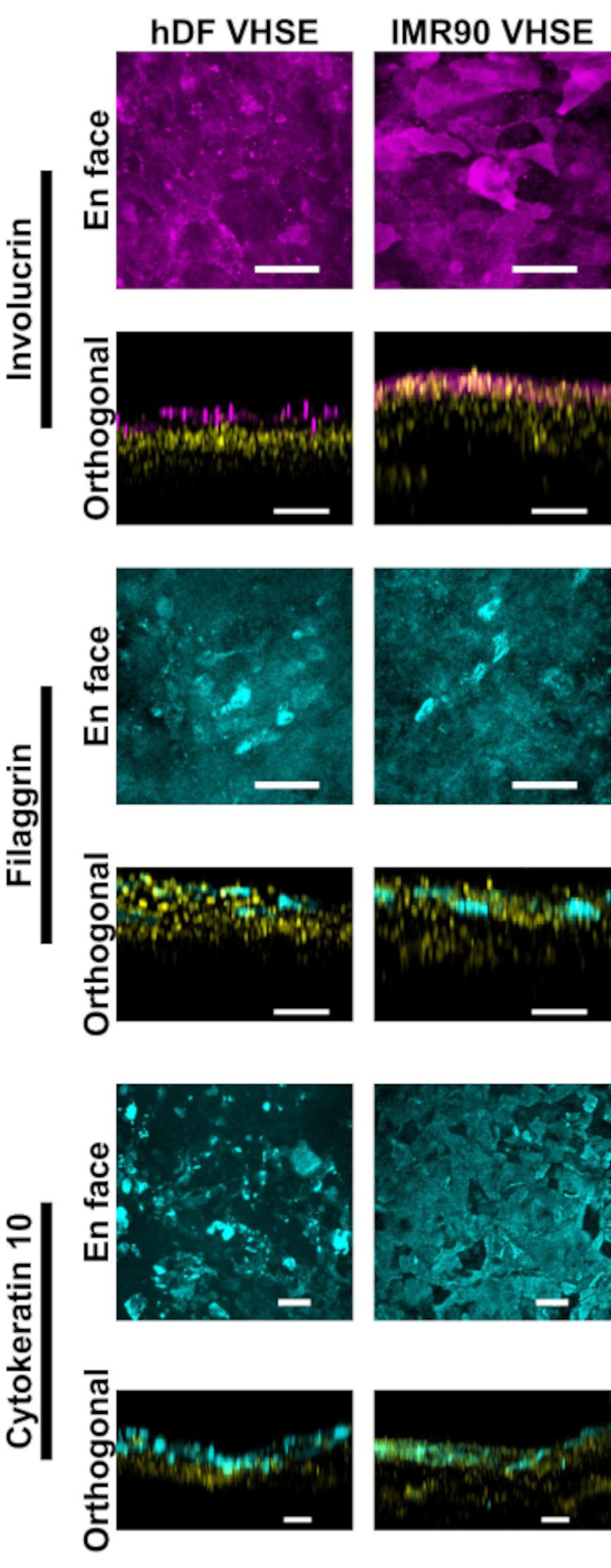
Characterization of the epidermis and dermis show appropriate immunofluorescent markers for human skin in the VHSE constructs (Figure 2, 3). Cytokeratin 10 (CK10) is an early differentiation keratinocyte marker which usually marks all suprabasal layers in skin equivalents18,30,68 (Figure 2). Involucrin and filaggrin are late differentiation markers in keratinocytes and mark the uppermost suprabasal layers in skin equivalents12,30,68,69 (Figure 2). A far-red fluorescent nuclear dye (see materials list) was used to mark nuclei in both the epidermis and dermis, with Col IV marking the vasculature of the dermis (Figure 2, Figure 3, Figure 4). Epidermal basement membrane (BM) components are not always properly expressed in HSE cultures15,16; and Col IV staining of the BM is not consistently observed using this protocol. Research focused BM components and structure would benefit from additional media, cell, and imaging optimization14.
Though confocal imaging through the bulk of the VHSE cultures often yields high resolution images that are sufficient for computational analysis of the dermis and epidermis, the clearing method described allows for deeper tissue imaging. Clearing improves confocal laser penetration depth, and effective imaging in VHSEs can be achieved to over 1 mm for cleared samples (compared to ~250 µm for uncleared). The described clearing technique (methanol dehydration and methyl salicylate) sufficiently matches refractive index throughout VHSE sample tissue61. Clearing the VHSE allowed for straightforward imaging through the entire construct without manipulation (e.g., reorienting the construct to image the dermis and epidermis separately), (Figure 3).
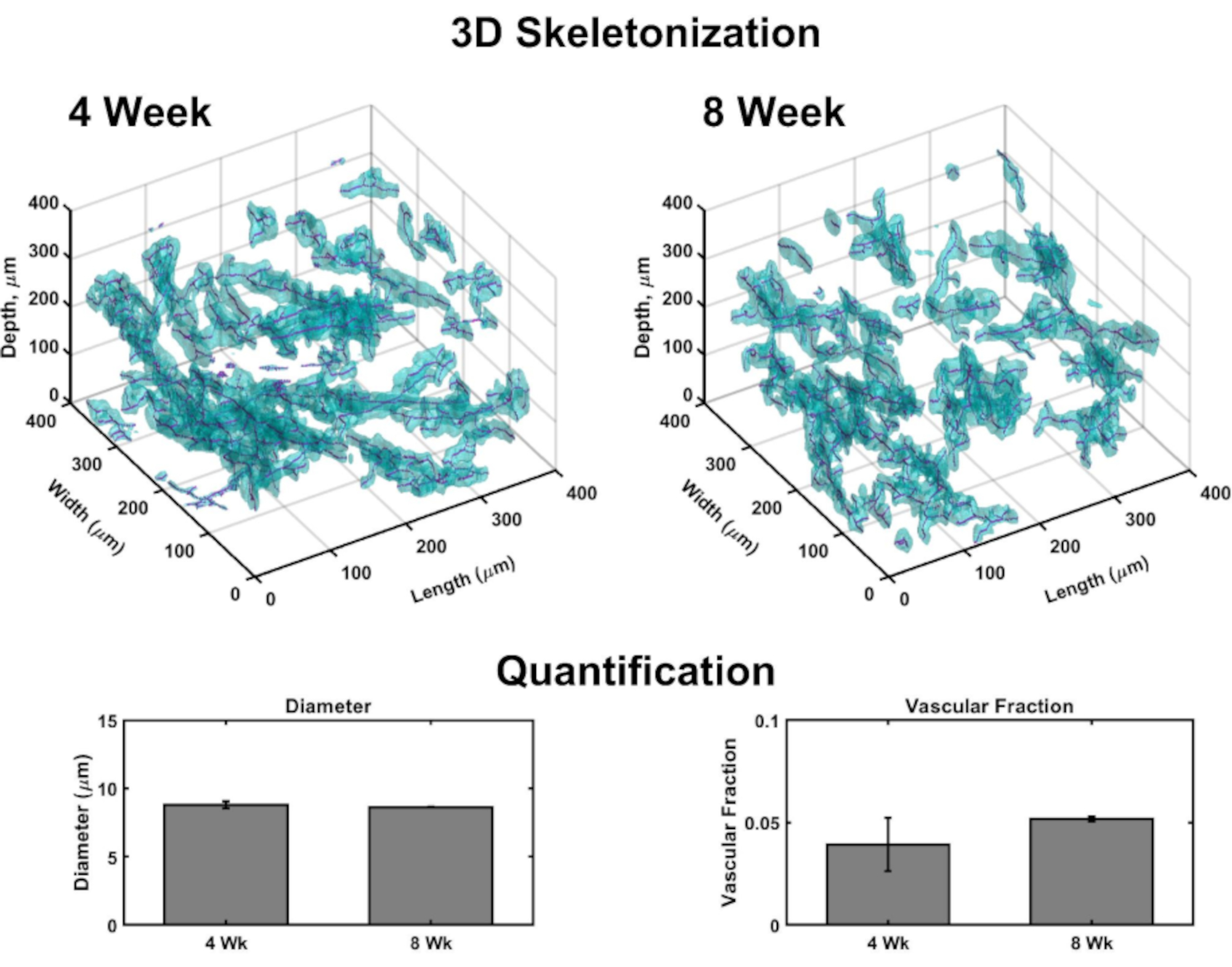
Volumetric images allow for generation of 3D rendering to map vasculature throughout each construct (Figure 4). Briefly, confocal image sets were taken in dermal to epidermal orientation of several sub-volumes of VHSEs to detect Collagen IV stain (marking vessel walls) and nuclei (marked by a far-red fluorescent nuclear dye). Image stacks are loaded into computational software (see materials list)and a custom algorithm (based on these sources 48,70,71,72,73,74,75) is used for 3D rendering and quantification as described previously48. This algorithm automatically segments the vascular component based on the Col IV stain. The volumetric segmentation is passed to a skeletonization algorithm based on fast marching75,76,77. Skeletonization finds the definitive center of each Col IV marked vessel and the resulting data can be used to calculate vessel diameter as well as vascular fraction (Figure 4). Widefield fluorescent microscopy is an accessible option if laser scanning microscopy is not available; the vascular network and epidermis can be imaged with widefield fluorescent microscopy (Supplemental Figure 2). Three-dimensional quantification is possible using widefield imaging of VHSEs rather than laser scanning microscopy, although it may require more filtering and deconvolution of images due to out-of-plane light.

Figure 1: Schematic timeline of vascularized human skin equivalent generation. A) Shows progression of the VHSE model from 1) dermal component seeding, 2) keratinocyte seeding onto the dermal component, 3) epithelial stratification via air liquid interface and culture maintenance. Post-culture processing and volumetric imaging can be performed at culture endpoint. B) Camera images of hDF VHSE macrostructure in the culture inserts at their culture endpoint, 8 weeks. Various levels of contraction are normal for VHSEs; contraction can be reduced as protocol describes. (1 & 2) Less contracted samples. (3 & 4) More contracted samples still yield proper skin elements. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Epidermal characterization via immunofluorescent markers. All images were taken of VHSEs at 8wk culture timepoint via confocal microscopy. Corresponding staining methods are described in protocol step 6. Proper epithelial markers are present in both hDF VHSEs (left column) and IMR90 VHSEs (right column). Involucrin and Filaggrin are late differentiation markers of keratinocytes and demonstrate that the epidermis is fully stratified in both VHSE types. Cytokeratin 10 is an early differentiation marker which is identifying suprabasal layers in the VHSEs. Nuclei are shown in orthogonal views in yellow. En face and orthogonal max projection images were rendered via computational software; Images are individually scaled with background subtraction and median filtering for clarity. Scale bars are 100 µm. (Primary and secondary antibodies with in-house blocking buffer recipe are given in Table 3). Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Comparison of uncleared vs. cleared VHSE. This VHSE was generated with IMR90s and images were taken at 4wk culture timepoint via confocal microscopy. Collagen IV is shown in cyan; Nuclei are shown in magenta; magenta in the cleared 3D rendering represents consolidation of nuclei in the epidermal layer of the VHSE. The uncleared VHSE image is an example of laser attenuation in thicker VHSE constructs, through clearing (methanol and methyl salicylate) the whole construct can be imaged with little/no laser attenuation from the dermal side of the construct. Imaging settings including laser line, gain, and pinhole were lowered for cleared VHSE to reduce oversaturation. Clearing and imaging were completed as described in steps 6 & 7 in the protocol. Orthogonal max projection images and 3D rendering were completed with computational software, 3D rendering was generated from cleared construct images. Images are individually scaled with background subtraction and median filtering for clarity. Scale bars are 100 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Three-Dimensional analysis of vasculature within VHSEs. Volumetric images taken via confocal microscopy enable vascular parameter quantification at the culture endpoints through computational image analysis. From VHSE sub-volumes, detection of Collagen IV stain (cyan) marks endothelial walls of vasculature and allows for segmentation of vascular based on collagen IV location; segmentation data is then skeletonized, and the center of each vessel is found (magenta). Examples of 3D skeletonization are shown for 4 week and 8 week IMR90 VHSE samples, un-cleared. Resulting data of an IMR90 VHSE experiment set was used to calculate the vessel diameters and the vascular fractions for four sub-volumes (each 250 µm in the z-direction) within each construct, data was averaged per VHSE and further averaged per culture timepoint. These data show the vascular network homeostasis spanning 4 and 8 week culture durations with diameters relevant to in vivo human skin78, and the vascular fraction within the same order as in vivo human skin79 (vascular fraction in collagen constructs has been shown to be customizable48 and could be further optimized for increased values). Data is represented as means ± standard error mean (S.E.M); n = 3 for each timepoint. Please click here to view a larger version of this figure.
{kind=link}
| Media | Components |
| Human Dermal Fibroblast cell line (hDF) | DMEM HG |
| 5% Fetal Bovine Serum (FBS) | |
| 1% Penicillin/Streptomycin (P/S) | |
| IMR90 Fibroblast cell line | DMEM/HAM’S F12 50:50 |
| 10% FBS | |
| 1% P/S | |
| HMEC-1 Endothelial cell line | MCDB131 Base medium |
| 10% FBS | |
| 1% P/S | |
| L-Glutamine [10 mM] | |
| Epidermal Growth Factor (EGF) [10 ng/mL] | |
| Hydrocortisone [10 µg/mL] | |
| N/TERT-1 Keratinocyte cell line | K-SFM media base |
| 1% P/S | |
| Bovine Pituitary Extract (BPE) [25 µg/mL], from K-SFM supplement kit | |
| Epidermal Growth Factor (EGF) [0.2 ng/mL], from K-SFM supplement kit | |
| CaCl2 [0.3 mM] | |
| Human Skin Equivalent (HSE) Differentiation | 3:1 DMEM: Ham’s F12 |
| 1% P/S | |
| 0.5 µM Hydrocortisone | |
| 0.5 µM Isoproterenol | |
| 0.5 µg/mL Insulin |
Table 1: Media recipes. Media recipes for 2D culture of the human dermal fibroblasts, IMR90 fibroblasts, HMEC-1, and N/TERT-1 keratinocytes are given. These recipes were used to expand cell lines before generating VHSEs. Human skin equivalent (HSE) differentiation media is used to generate VHSEs; a base recipe is given, during portions of submersion culture and stratification induction, tapering amounts of FBS should be added as described in protocol step 3. HSE recipe based on these sources11,80.
| Collagen stock concentration (Cs) : | 8 | mg/mL | ||
| Desired Volume (Vf): | 1 | mL | ||
| Normalizing NaOH Adjustment*: | 1 | X | ||
| *Each lot of collagen needs to be tested to determine the amount of NaOH needed to set pH 7 - 7.4 | ||||
| Desired Collagen Concentration (mg/mL) | ||||
| 2 | 3 | 4 | 5 | |
| 10X PBS (Vpbs) | 0.1 | 0.1 | 0.1 | 0.1 |
| Collagen stock (Vs) | 0.25 | 0.375 | 0.5 | 0.625 |
| 1N NaOH (VNaOH) | 0.00575 | 0.008625 | 0.0115 | 0.014375 |
| Media (Vmedia) | 0.64425 | 0.516375 | 0.3885 | 0.260625 |
Table 2: Collagen calculation reference table. Reference table gives commonly desired collagen concentrations calculated assuming an 8 mg/mL collagen stock concentration and a desired final volume of 1 mL; all values are in mL. The equations used to calculate these amounts are given in protocol step 2.2. It is important to check pH for each collagen stock; if necessary, NaOH amounts should be added to achieve pH 7 - 7.4 (after PBS, NaOH, collagen stock, media are added). The protocol has been optimized for VHSEs using a 3 mg/mL collagen concentration; changes in collagen concentration may be necessary for different cell lines/desired end results48.
| Primary Antibody | Source | Concentration | Use |
| Filaggrin (AKH1) mouse monoclonal IgG | Santa Cruz; sc-66192 (200 µg/mL) | [1:250] | Late differentiation marker15 |
| Involucrin rabbit polyclonal IgG | Proteintech; 55328-1-AP (30 µg/150 µL) | [1:250] | Late terminal differentiation marker15 |
| Cytokeratin 10 (DE-K10) mouse IgG, supernatant | Santa Cruz; sc-52318 | [1:350] | Suprabasal epidermal marker14,36,59 |
| Collagen IV rabbit polyclonal | Proteintech; 55131-1-AP | [1:500] | Endothelial vascular wall67 |
| DRAQ 7 | Cell Signaling; 7406 (0.3 mM) | [1:250] | Nuclear marker |
| Secondary Antibody | Source | Concentration | Use |
| Goat Anti-Rabbit IgG DyLight™ 488 Conjugated | Invitrogen; 35552 (1 mg/mL) | [1:500] | Collagen IV secondary |
| Anti-Rabbit IgG (H&L) (GOAT) Antibody, DyLight™ 549 Conjugated | Rockland Immunochemicals; 611-142-002 | [1:500] | Involucrin secondary |
| Goat Anti-Mouse IgG (H&L), DyLight™ 488 | Thermo Scientific; 35502 (1 mg/mL) | [1:500] | Filaggrin or Cytokeratin 10 secondary |
| BLOCKING BUFFER (500 mL) | |||
| Reagent | Amount | ||
| ddH2O | 450 mL | ||
| 10 x PBS | 50 mL | ||
| Bovine Serum Albumin (BSA) | 5 g | ||
| Tween 20 | 0.5 mL | ||
| Cold water Fish Gelatin | 1 g | ||
| Sodium Azide (10% Sodium Azide in diH2O) | 5 mL (0.1 % final concentration) | ||
Table 3: Primary and secondary antibodies with blocking buffer recipe. The listed antibodies and chemical stains were used for staining shown in Figure 2, Figure 3, Figure 4. The staining was completed as given in protocol step 6 using the blocking buffer recipe listed here. Some optimizations of the staining concentrations and the duration may be required depending on the chosen culture techniques and the cell lines.
Supplemental Table 1: Abbreviations List. Abbreviations list included for the reader's convenience. Please click here to download this table.
Supplemental Figure 1: VHSE technical aid for handling. Handling of VHSEs is challenging especially during fixation, processing, and staining. A-D corresponds to instructions in steps 5-7. A shows the technical handling of removing the porous membrane from a culture insert to ensure proper staining. B shows how to keep each VHSE submerged during staining and storage. C shows the safest and easiest way to move constructs to PDMS imaging wells. D shows a VHSE sitting in a PDMS imaging well: PDMS well is glued to a glass slide on the bottom, creating a window for imaging, a glass slide is placed on top to maintain the humidity through long imaging runs. Please click here to download this file.
Supplemental Figure 2: Standard widefield fluorescence microscopy can be used to assess VHSEs. Widefield imaging can be used for volumetric imaging for routine assessment when laser scanning microscopy is not available. As an example, imaging of VHSEs from both the apical and basolateral aspects are shown as en face and orthogonal (Ortho.) maximum projections. (Top) The epidermis was imaged using involucrin and nuclei as markers. (Bottom) Dermal vasculature was imaged using collagen IV as a marker. Images are background subtracted for clarity. Out-of-plane light leads to the "streaking" or "flare" artifacts evident in the orthogonal views. Widefield imaging can be used for quantification but may require more image processing. Please click here to download this file.
Discussion
This protocol has demonstrated a simple and repeatable method for the generation of VHSEs and their three-dimensional analysis. Importantly, this method relies on few specialized techniques or equipment pieces, making it accessible for a range of labs. Further, cell types can be replaced with limited changes in the protocol, allowing researchers to adapt this protocol to their specific needs.
Proper collagen gelation is a challenging step in establishing skin culture. Especially when using crude preparations without purification, trace contaminants could influence the gelation process. To help ensure consistency, groups of experiments should be performed with the same collagen stock that will be used for VHSE generation. Further, the gelation should ideally occur at a pH of 7-7.4, and trace contaminants may shift the pH. Before using any collagen stock, a practice acellular gel should be made at the desired concentration and the pH should be measured prior to gelation. Completing this collagen quality check before beginning dermal component seeding will identify the problems with proper gelation and collagen homogeneity prior to setting up a complete experiment. Instead of seeding acellular collagen directly onto a culture insert, seed some collagen onto a pH strip that evaluates the whole pH scale and verify a pH of 7-7.4. Gelation can be evaluated by applying a droplet of the collagen gel solution onto a coverslip or tissue culture plastic well plate (a well plate is recommended to simulate the confined sides of a culture insert). After gelation time, the collagen should be solid, i.e., it should not flow when the plate is tilted. Under phase contrast microscopy, the collagen should look homogeneous and clear. Occasional bubbles from collagen seeding are normal but large amorphous blobs of opaque collagen within the clear gel indicates a problem-likely due to insufficient mixing, wrong pH, and/or failure to keep the collagen chilled during mixing.
The cell seeding amounts and media may be adjusted. In the protocol above, the encapsulated cell amounts have been optimized for a 12-well insert at 7.5 x 104 fibroblasts and 7.5 x 105 endothelial cells per mL of collagen with 1.7 x 105 keratinocytes seeded on top of the dermal construct. Cell densities have been optimized for this VHSE protocol based on the preliminary studies and the previous research investigating 3D vascular network generation in various collagen concentrations48 and HSE generation22,80,81. In similar systems, the published endothelial cell densities are 1.0 x 106 cells/mL collagen48; the fibroblast concentrations often range from 0.4 x 105 cells/mL of collagen22,28,82 to 1 x 105 cells/mL of collagen8,58,83,84,85; and the keratinocyte concentrations range from 0.5 x 105 [cells/cm2]80 to 1 x 105 [cells/cm2]8. Cell densities can be optimized for specific cells and research question. Three-dimensional cultures with contractile cells, such as fibroblasts, can contract leading to viability reduction and culture loss86,87. Preliminary experiments should be completed to test contraction of the dermal compartment (which can occur with more dermal cells, more contractile dermal cells, longer submersion cultures, or softer matrices) and to test epidermal surface coverage. Additionally, the number of days in submersion and the rate of tapering the serum content can also be customized if excessive dermal contraction is occurring or a different rate of keratinocyte coverage is required. For example, if contraction is noticed during the period of dermal submersion or while keratinocytes are establishing a surface monolayer, moving more quickly through the serum tapering process and raising VHSEs to ALI can aid in preventing additional contraction. Similarly, if keratinocyte coverage is not ideal, changing the number of days that the VHSE is submerged without serum may help increase the epidermal monolayer coverage and mitigate the contraction since serum is left out. Changes in cell densities or other suggestions above must be optimized for the specific cultures and research goals.
To establish a proper stratification of the epidermis during the air liquid interface (ALI) period, it is critical to regularly check and maintain fluid levels in each well so that ALI and appropriate hydration of each construct is kept throughout the culture length. Media levels should be checked and tracked daily until consistent ALI levels are established. The epidermal layer should look hydrated, not dry, but there should not be pools of media on the construct. During ALI, the construct will develop an opaque white/yellow color which is normal. The epidermal layer will likely develop unevenly. Commonly, the VHSEs are tilted due to the collagen seeding or dermal contraction. It is also normal to observe a higher epidermal portion in the middle of the construct in smaller constructs (24 well size) and a ridge formation around the perimeter of the VHSE in 12 well size. Contraction of the constructs13 may change these topographical formations, and/or may not be observed at all.
Staining and imaging of VHSEs introduces mechanical manipulation to the VHSEs. It is very important to plan and limit manipulation of each culture. When manipulation is necessary, maintain gentle movements when removing VHSEs from the insert membranes, when adding staining or wash solutions to the construct surface, and when removing and replacing VHSEs in their storage/imaging wells during imaging preparation. Specifically, the apical layers of the epidermal component may be fragile and are at risk of sloughing off the basal epidermal layers. Apical layers of the epidermis are fragile and go through desquamation even in native tissue4, but for accurate analysis of epidermal structure it is important to minimize damage or loss. If epidermal layers lift off the construct, they can be imaged separately. The basal layers of the epidermis are most likely still attached to the dermis while portions of the apical layers may detach. For visualization of the epidermis, a nuclear stain is helpful in observing this since dense nuclei is a characteristic of lower and mid layers of the epidermis.
Confocal imaging of the VHSE post-fixation has been discussed in the protocol, but it is also possible to image the VHSEs throughout the culture via upright based optical coherence tomography (OCT)88,89,90,91,92,93. VHSE are stable enough to withstand imaging without incubation or humidification for at least two hours without noticeable effects. As OCT is label free and noninvasive, it is possible to track the epidermal thickness during maturation. Other noninvasive imaging modalities can likely be employed as well.
Volumetric imaging of the combined dermal and epidermal structures can be challenging due to laser attenuation deeper in the VHSE. This can be mitigated by imaging the construct in two orientations, from the epidermal side (Figure 1) and from the dermal side (Figure 2), allowing for good resolution of dermal vascular structures and the epidermis. Additionally, the sample can be cleared, allowing for volumetric images of the entire structure with minimal attenuation. Several clearing methods were attempted, however, the methanol/methyl salicylate method described yielded the best results. Researchers interested in optimizing other clearing methods are directed towards these reviews49,61,62. If clearing, it is suggested to fully image the sample prior to clearing, as the method can damage the fluorophores and/or the structure. Further, the imaging should be completed as soon as possible after clearing, as the fluorescence may fade within days.
For simplicity and accessibility, this protocol utilized the simplest media blends found in previous literature11,80. Although there are many advantages to using simple media blends, the limitations of this choice are also recognized. Other groups have studied the effects of specific media components on epidermal and dermal health and found that other media additives94, such as external free fatty acids/lipids, enhance the stratum corneum of the epidermis and improve the skin barrier function. Although our immunofluorescent markers show appropriate differentiation and stratification in the epidermis, depending on the studies being conducted, additional media optimization may be needed. Further, an extensive analysis of the epidermal BM was not conducted when evaluating the VHSEs presented here. The integrity of the BM is an important indication of skin equivalents; various groups have done research on the culture duration and its effect on BM markings95 as well as analysis of fibroblast presence and added growth factor effects on BM expression14. It is important to note that analysis of the BM component should be evaluated and optimized when using this protocol.
In this protocol is described a procedure for VHSE generation, demonstrating results after 8 weeks at ALI. VHSE cultures have been cultured up to 12 weeks at ALI without noticeable change or loss of viability, and it is possible that they may be viable longer. Importantly, this protocol is readily adaptable to commonly available cell types, as demonstrated by the replacement of dermal fibroblasts with IMR90 lung fibroblasts. Depending on the researcher's need and available resources, the cell types and media blends on the culture can be adjusted, although more dissimilar cell types may require media optimization. In summary, these procedures are meant to provide clarity on the culture of VHSEs for the study of skin biology and disease. To maximize accessibility, the protocol was developed this simple and robust using common equipment, cell lines, and reagents as a minimal effective approach that can be further customized to the specific needs of research studies.
Disclosures
The authors have nothing to disclose.
Acknowledgements
The authors thank Dr. Jim Rheinwald59 and Dr. Ellen H. van den Bogaard20 for their generous gift of N/TERT cell lines. This work was supported by the American Heart Association (19IPLOI34760636).
Materials
| Name | Company | Catalog Number | Comments |
| 1 N NaOH | Fisher Chemical | S318-100 | (Dilute from Lab stock) |
| 4% Paraformaldehyde | ACROS Organics | #41678-5000, Lot # B0143461 | Made up using solid Paraformaldehyde in PBS, pH adjusted to 6.9 |
| Autoclaved forceps | Fine Science Tools | #11295-00 | Dumont #5 forceps |
| CaCl2 | Fisher bioreagents | Cat # BP510-250, Lot # 190231 | Rnase, Dnase, Protease-Free |
| Cell line, Endothelial: Microvascular Endothelial Cell (HMEC1) | ATCC | CRL-3243 | SV40 Immortalized microvascular endothelial cell. Note that 750,000 cells/mL of collagen were used. |
| Cell line, Fibroblasts: dermal Human fibroblast, adult | ATCC | PCS-201-012 | Primary dermal cells. Note that 75,000 cells/mL of collagen were used. |
| Cell line, Fibroblasts: human lung firbroblast (IMR90) | ATCC | CCL-186 | Primary embryonic cells. Note that 75,000 cells/mL of collagen were used. |
| Cell line, Keratinocyte: N/TERT-1 | Immortalized via hTERT expression. N/TERT-1 was made using a retroviral vector conferring hygromycin resistance. Cell line established by Dickson et al. 2000. Can be replaced with ATCC PCS-200-010 or PCS-200-011. Note that 170,000 cells were used per construct; N/TERT1 cells must be used from plates that are 30% confluent- two 30% confluent 90 mm tissue culture dishes give more than enough cells.The authors thank Dr. Jim Rheinwald and Dr. Ellen H. van den Bogaard for their generous gift of N/TERT cell lines. | ||
| Centrifuge | Thermo Scientific; Sorvall Legend X1R | (standard lab equipment) | |
| Computational Software | MATLAB | MATLAB 2020a | MathWorks, Natick, MA. |
| Confocal Microscope | Leica TCS SPEII confocal | Laser scanning confocal. Can be replaced with other confocals or deconvolution microscopy. | |
| Cover Glass (22 x 22) | Fisher Scientific | 12-545F | 0.13-0.17 mm No.1 Thickness |
| Cyanoacrylate super glue or silicone grease | Glue Masters | #THI0102 | Glue Masters, THICK, Instant Glue, Cyanoacrylate; super glue is preferred |
| DMEM media base | Corning; Mediatech, Inc | REF # 10-013-CM; Lot # 26119007 | DMEM, 1X (Dulbecco's Modification of Eagle's Medium) with 4.5 g/L glucose, L-glutamine & sodium pyruvate |
| DMEM/F-12 50/50 | Corning; Mediatech, Inc | REF # 10-090-CV; Lot # 21119006 | DMEM/F-12 50/50, 1X (Dulbecco's Mod. Of Eagle's Medium/Ham's F12 50/50 Mix) with L-glutamine |
| Ethanol | Decon Labs | #V1101 | (standard lab reagent) |
| Fetal Bovine Serum | Fisher Scientific | Cat # FB12999102, Lot # AE29451050 | Research Grade Fetal Bovine Serum, Triple 0.1 um sterile filtered |
| Fine tip forceps | Fine Science Tools | #11295-00 | Dumont #5 forceps |
| Human Epidermal Growth Factor (EGF) | Peprotech | Cat # AF-100-15-1MG, Lot # 0318AFC05 D0218 | Made up in 0.1% BSA in PBS |
| Hydrocortisone | Alpha Easar | Lot # 5002F2A | made up in DMSO |
| Insulin (human) | Peprotech | Lot # 9352621 | |
| Inverted Light/Phase Contrast Microscope | VWR | 76317-470 | (standard lab equipment) |
| Isoproterenol | Alfa Aesar | #AAJ6178806 | DL-Isoproterenol hydrochloride, 98% |
| Keratinocyte-SFM (1x) media base | Gibco; Life Technologies Corporation | REF #: 10724-011; Lot # 2085518 | Keratinocyte-SFM (1X); serum free medium |
| L-Ascorbic Acid | Fisher Chemical | Cat # A61-100, Lot # 181977, CAS # 50-81-7 | Crystalline. L-Ascorbic acid can also be purchased as a salt |
| L-glutamine (solid) | Fisher Bioreagents | CAT # BP379-100, LOT # 172183, CAS # 56-85-9 | L-Glutamine, white crystals or Crystalline powder |
| MCDB 131 media base | Gen Depot | CM034-050, Lot # 03062021 | MCDB 131 Medium Base, No L-Glutamine, sterile filtered |
| Metal punches | Sona Enterprises (SE) | 791LP, 12PC | Hollow Leather Punch Set, High Carbon Steel, Hardness: 48HRC; (various sizes including): 1/8", 5/32", 3/16", 7/32", 1/4", 9/32", 5/16", 3/4", 7/16", 1/2", 5/8:, 3/4". This punch set is helpful, but x-acto knife can work as well. Size of metal punch that works well for 12 well transwell VHSE is 3/8" or 1/2". |
| Methanol | Fisher Chemical | CAS # 67-56-1 | (optional). For clearing dehydration step. |
| Methyl Salicylate | Fisher Chemical | O3695-500; Lot # 164535; CAS # 119-36-8 | (optional). For clearing. |
| Microtubes, 1.7 mL | Genesee Scientific Corporation; Olympus Plastics | Cat # 24-282; Lot # 19467 | 1.7 ml Microtubes, Clear; Boilproof, Polypropulene, Certified Rnase, Dnase, DNA, PCR inhibitor and endotoxin-Free |
| PBS, 10x Culture grade or autoclaved | APEX Bioresearch Products | Cat # 20-134, Lot # 202237 | PBS Buffer, 10x Dry Pack; add contents of pack into container and add water to 1 liter to produce 10x concentrated. |
| PBS, 1x Culture grade, (-) Calcium, (-) magnesium | Genesee Scientific Corporation | Ref # 25-508; Lot # 07171015 | |
| PBS, 1x non-Culture grade | APEX Bioresearch Products | Cat # 20-134, Lot # 202237 | PBS Buffer, 10x Dry Pack; add contents of pack into container and add water to 1 liter to produce 10x concentrated. Dilute to 1x with water. |
| Penicillin/Streptomycin | Gibco; Life Technologies Corporation | Ref # 15140-122, Lot # 2199839 | Pen Strep (10,000 Units/mL Penicillin; 10,000 ug/mL Streptomycin) |
| Petri Dish, glass, small | Corning | PYREX 316060 | (optional). To be used as a clearing container |
| Petri Dishes, 100 mm | Fisher Scientific | FB0875713 | Use for making up PDMS. |
| Polydimethylsiloxane (PDMS) | Dow Corning | GMID 02065622, Batch # H04719H035 | Sylgard 184 Silicone Elastomer Base, Dow Corning, Midland, MI) |
| Dow Corning | GMID 02065622, Batch # H047JC4003 | DOWSIL 184, Silicone Elastomer Curing Agent | |
| note: PDMS is usually sold as a kit that includes both the base and curing agent components. | |||
| Positive Displacement Pipettes (1000 & 250 uL) | Gilson | 1000 uL: HM05136, M1000. 250 uL: T12269L | M1000 pipette capacity (100-1000 uL); M250 pipette capacity to 250 uL |
| Positive Displacement Tips/Pistons (1000 & 250 uL) | Gilson | 1000 uL: CAT # F148180, BATCH # B01292902S; 250 uL: CAT # F148114, BATCH # B05549718S | Sterilized capillaries and pistons |
| Round tipped scoopula | (optional; standard lab equipment) For manipulation of VHSEs prior to imaging | ||
| Supplements for Keratinocyte-SFM media | Gibco; Life Technologies Corporation | Ref # 37000-015, Lot # 2154180 | Contains EGF Human Recombinant (Cat # 10450-013), Bovine Pituitary Extract (Cat # 13028-014) |
| Tissue Culture Plate Inserts, 12 well size, 3 µm pore size | Corning; Costar | REF # 3462 - Clear; Lot # 14919057 | Transwell; 12 mm Diameter Inserts, 3.0 um pore size, tissue culture treated, polyester membrane, polystyrene plates, |
| Tissue Culture Plates, 12 well size | Greiner Bio-one; CellStar | Cat # 665 180; Lot # E18103QT | 12 well cell culture plate; sterile, with lid; products are sterile, free of detectable Dnase, Rnase, human DNA and pyrogens. Contents non-cytotoxic |
| Tissue Culture Plates, 60.8cm^2 growth area | Genesee Scientific Corporation | Cat # 25-202; Lot No: 191218-177B | Tissue culture dishes; treated, growth area 60.8 cm^2; sterile, Dnase, Rnase, Pyrogen Free; virgin polystyrene |
| Triton x 100 | Ricca Chemical Company | Cat # 8698.5-16, Lot # 4708R34 | Wetting agent |
| Trypsin 0.25% | Corning; Mediatech, Inc | Ref # 25-053-CI | 0.25% Trypsin, 2.21 mM EDTA, 1x [-] sodium bicarbonate |
| Type 1 Collagen isolated from Rat tail | Pel-Freez Biologicals | 56054-1 | Sprauge-Dawley rat tails can be purchased frozen from Pel-Freez or other suppliers. Collagen can be isolated from the tail tendons. Isolation Protocol references [Cross et al.,2010; Rajan et al.,2007; Bornstein, 1958] .Alternatively, high concentration rat-tail collagen can be purchased from suppliers including Corning (Catalog Number: 354249) |
| Vaccum chamber, benchtop | Bel-Art | F42010-0000 | (standard lab equipment) |
| Handheld Precision knife | X-Acto | X3311 | (X-Acto knife optional if purchased steel punches) |
References
- Stojic, M., et al. Skin tissue engineering 3. Biomaterials for Skin Repair and Regeneration. , 59 (2019).
- Shevchenko, R. V., James, S. L., James, S. E. A review of tissue-engineered skin bioconstructs available for skin reconstruction. Journal of The Royal Society Interface. 7 (43), 229-258 (2010).
- Kolarsick, P. A. J., Kolarsick, M. A., Goodwin, C. Anatomy and Physiology of the Skin. Journal of the Dermatology Nurses' Association. 3 (4), (2011).
- McGrath, J. A., Eady, R. A. J., Pope, F. M. Anatomy and organization of human skin. Rook's textbook of dermatology. 10, 9781444317633 (2004).
- Zomer, H. D., Trentin, A. G. Skin wound healing in humans and mice: Challenges in translational research. Journal of Dermatological Science. 90 (1), 3-12 (2018).
- Zhang, Z., Michniak-Kohn, B. B. Tissue Engineered Human Skin Equivalents. Pharmaceutics. 4 (1), (2012).
- Oh, J. W., Hsi, T. -. C., Guerrero-Juarez, C. F., Ramos, R., Plikus, M. V. Organotypic skin culture. The Journal of investigative dermatology. 133 (11), 1-4 (2013).
- El-Ghalbzouri, A., Gibbs, S., Lamme, E., Van Blitterswijk, C. A., Ponec, M. Effect of fibroblasts on epidermal regeneration. British Journal of Dermatology. 147 (2), 230-243 (2002).
- Sun, T., Haycock, J., MacNeil, S. In situ image analysis of interactions between normal human keratinocytes and fibroblasts cultured in three-dimensional fibrin gels. Biomaterials. 27 (18), 3459-3465 (2006).
- Kreimendahl, F., et al. Macrophages significantly enhance wound healing in a vascularized skin model. Journal of Biomedical Materials Research Part A. 107, 1340-1350 (2019).
- El Ghalbzouri, A., Commandeur, S., Rietveld, M. H., Mulder, A. A., Willemze, R. Replacement of animal-derived collagen matrix by human fibroblast-derived dermal matrix for human skin equivalent products. Biomaterials. 30 (1), 71-78 (2009).
- Roger, M., et al. Bioengineering the microanatomy of human skin. Journal of Anatomy. 234, 438-455 (2019).
- Carlson, M. W., Alt-Holland, A., Egles, C., Garlick, J. A. Three-Dimensional Tissue Models of Normal and Diseased Skin. Current Protocols in Cell Biology. 41 (1), 1-17 (2008).
- El Ghalbzouri, A., Jonkman, M. F., Dijkman, R., Ponec, M. Basement Membrane Reconstruction in Human Skin Equivalents Is Regulated by Fibroblasts and/or Exogenously Activated Keratinocytes. Journal of Investigative Dermatology. 124 (1), 79-86 (2005).
- Pruniéras, M., Régnier, M., Woodley, D. Methods for cultivation of keratinocytes with an air-liquid interface. The Journal of Investigative Dermatology. 81, 28-33 (1983).
- Ali, N., Hosseini, M., Vainio, S., Taieb, A., Cario-André, M., Rezvani, H. R. Skin equivalents: skin from reconstructions as models to study skin development and diseases. British Journal of Dermatology. 173 (2), 391-403 (2015).
- Mathes, S. H., Ruffner, H., Graf-Hausner, U. The use of skin models in drug development. Innovative tissue models for drug discovery and development. 69-70, 81-102 (2014).
- Mieremet, A., Rietveld, M., Absalah, S., van Smeden, J., Bouwstra, J. A., El Ghalbzouri, A. Improved epidermal barrier formation in human skin models by chitosan modulated dermal matrices. PLOS ONE. 12 (3), 0174478 (2017).
- Vidal, S. E. L., Tamamoto, K. A., Nguyen, H., Abbott, R. D., Cairns, D. M., Kaplan, D. L. 3D biomaterial matrix to support long term, full thickness, immuno-competent human skin equivalents with nervous system components. Organoids and Ex Vivo Tissue On-Chip Technologies. 198, 194-203 (2019).
- Smits, J. P. H., et al. Immortalized N/TERT keratinocytes as an alternative cell source in 3D human epidermal models. Scientific Reports. 7 (1), 11838 (2017).
- Lebonvallet, N., et al. Effects of the re-innervation of organotypic skin explants on the epidermis. Experimental Dermatology. 21 (2), 156-158 (2011).
- Van Drongelen, V., et al. Barrier Properties of an N/TERT-Based Human Skin Equivalent. Tissue Engineering Part A. 20 (21-22), 3041-3049 (2014).
- . Establishment and initial characterization of a simple 3D organotypic wound healing model Available from: https://opus.hs-furtwangen.de/frontdoor/index/index/docld/4852 (2018)
- Breslin, S., O'Driscoll, L. Three-dimensional cell culture: the missing link in drug discovery. Drug Discovery Today. 18 (5), 240-249 (2013).
- Shamir, E. R., Ewald, A. J. Three-dimensional organotypic culture: experimental models of mammalian biology and disease. Nature Reviews Molecular Cell Biology. 15 (10), 647-664 (2014).
- Amelian, A., Wasilewska, K., Megias, D., Winnicka, K. Application of standard cell cultures and 3D in vitro tissue models as an effective tool in drug design and development. Pharmacological Reports. 69 (5), 861-870 (2017).
- Lu, W., et al. Mixture of Fibroblasts and Adipose Tissue-Derived Stem Cells Can Improve Epidermal Morphogenesis of Tissue-Engineered Skin. Cells Tissues Organs. 195 (3), 197-206 (2012).
- Marino, D., Luginbühl, J., Scola, S., Meuli, M., Reichmann, E. Bioengineering Dermo-Epidermal Skin Grafts with Blood and Lymphatic Capillaries. Science Translational Medicine. 6 (221), 221 (2014).
- Martins-Green, M., Li, Q. -. J., Yao, M. A new generation organ culture arising from cross-talk between multiple primary human cell types. The FASEB Journal. 19 (2), 222-224 (2004).
- Kim, B. S., Gao, G., Kim, J. Y., Cho, D. -. W. 3D Cell Printing of Perfusable Vascularized Human Skin Equivalent Composed of Epidermis, Dermis, and Hypodermis for Better Structural Recapitulation of Native Skin. Advanced Healthcare Materials. 8 (7), 1801019 (2019).
- Baltazar, T., et al. 3D bioprinting of a vascularized and perfusable skin graft using human keratinocytes. Tissue Engineering Part A. 26 (5-6), 227-238 (2019).
- Klar, A. S., et al. Tissue-engineered dermo-epidermal skin grafts prevascularized with adipose-derived cells. Biomaterials. 35 (19), 5065-5078 (2014).
- Grebenyuk, S., Ranga, A. Engineering Organoid Vascularization. Frontiers in Bioengineering and Biotechnology. 7, (2019).
- Black, A. F., Berthod, F., L'heureux, N., Germain, L., Auger, F. A. In vitro reconstruction of a human capillary-like network in a tissue-engineered skin equivalent. The FASEB Journal. 12 (13), 1331-1340 (1998).
- Huber, B., Link, A., Linke, K., Gehrke, S. A., Winnefeld, M., Kluger, P. J. Integration of Mature Adipocytes to Build-Up a Functional Three-Layered Full-Skin Equivalent. Tissue Engineering Part C: Methods. 22 (8), 756-764 (2016).
- Monfort, A., Soriano-Navarro, M., García-Verdugo, J. M., Izeta, A. Production of human tissue-engineered skin trilayer on a plasma-based hypodermis. Journal of Tissue Engineering and Regenerative Medicine. 7 (6), 479-490 (2013).
- Shamis, Y., et al. Fibroblasts derived from human embryonic stem cells direct development and repair of 3D human skin equivalents. Stem Cell Research & Therapy. 2 (1), 10 (2011).
- Kim, Y., et al. Establishment of a complex skin structure via layered co-culture of keratinocytes and fibroblasts derived from induced pluripotent stem cells. Stem Cell Research & Therapy. 9 (1), 217 (2018).
- Chau, D. Y. S., Johnson, C., MacNeil, S., Haycock, J. W., Ghaemmaghami, A. M. The development of a 3D immunocompetent model of human skin. Biofabrication. 5 (3), 035011 (2013).
- Vanden Bogaard, E. H., et al. Crosstalk between Keratinocytes and T Cells in a 3D Microenvironment: A Model to Study Inflammatory Skin Diseases. Journal of Investigative Dermatology. 134 (3), 719-727 (2014).
- Linde, N., Gutschalk, C. M., Hoffmann, C., Yilmaz, D., Mueller, M. M. Integrating Macrophages into Organotypic Co-Cultures: A 3D In Vitro Model to Study Tumor-Associated Macrophages. PLOS ONE. 7 (7), 40058 (2012).
- Ouwehand, K., Spiekstra, S. W., Waaijman, T., Scheper, R. J., de Gruijl, T. D., Gibbs, S. Technical Advance: Langerhans cells derived from a human cell line in a full-thickness skin equivalent undergo allergen-induced maturation and migration. Journal of Leukocyte Biology. 90 (5), 1027-1033 (2011).
- Weinmüllner, R., et al. Organotypic human skin culture models constructed with senescent fibroblasts show hallmarks of skin aging. npj Aging and Mechanisms of Disease. 6 (1), 4 (2020).
- Barker, C. L., et al. The Development and Characterization of an In Vitro Model of Psoriasis. Journal of Investigative Dermatology. 123 (5), 892-901 (2004).
- Larcher, F., Espada, J., Díaz-Ley, B., Jaén, P., Juarranz, A., Quintanilla, M. New Experimental Models of Skin Homeostasis and Diseases. Actas Dermo-Sifiliográficas (English Edition). 106 (1), 17-28 (2015).
- Varkey, M., Ding, J., Tredget, E. E. Fibrotic Remodeling of Tissue-Engineered Skin with Deep Dermal Fibroblasts Is Reduced by Keratinocytes. Tissue Engineering Part A. 20 (3-4), 716-727 (2013).
- Moulin, V. J. Reconstitution of skin fibrosis development using a tissue engineering approach. Methods in Molecular Biology. 961, 287-303 (2013).
- Morgan, J. T., Shirazi, J., Comber, E. M., Eschenburg, C., Gleghorn, J. P. Fabrication of centimeter-scale and geometrically arbitrary vascular networks using in vitro self-assembly. Biomaterials. 189, 37-47 (2019).
- Silvestri, L., Costantini, I., Sacconi, L., Pavone, F. S. Clearing of fixed tissue: a review from a microscopist's perspective. Journal of Biomedical Optics. 21 (8), 1-8 (2016).
- Cross, V. L., et al. Dense type I collagen matrices that support cellular remodeling and microfabrication for studies of tumor angiogenesis and vasculogenesis in vitro. Biomaterials. 31 (33), 8596-8607 (2010).
- Rajan, N., Habermehl, J., Coté, M. -. F., Doillon, C. J., Mantovani, D. Preparation of ready-to-use, storable and reconstituted type I collagen from rat tail tendon for tissue engineering applications. Nature Protocols. 1, 2753 (2007).
- Bornstein, M. B. Reconstituted rat-tail collagen used as substrate for tissue cultures on coverslips in Maximow slides and roller tubes. Laboratory Investigation. 7 (2), 134-137 (1958).
- Clément, M. -. V., Ramalingam, J., Long, L. H., Halliwell, B. The In Vitro Cytotoxicity of Ascorbate Depends on the Culture Medium Used to Perform the Assay and Involves Hydrogen Peroxide. Antioxidants & Redox Signaling. 3 (1), 157-163 (2001).
- Tajima, S., Pinnell, S. R. Ascorbic acid preferentially enhances type I and III collagen gene transcription in human skin fibroblasts. Journal of Dermatological Science. 11 (3), 250-253 (1996).
- Murad, S., Tajima, S., Johnson, G. R., Sivarajah, A., Pinnell, S. R. Collagen Synthesis in Cultured Human Skin Fibroblasts: Effect of Ascorbic Acid and Its Analogs. Journal of Investigative Dermatology. 81 (2), 158-162 (1983).
- Villacorta, L., Azzi, A., Zingg, J. -. M. Regulatory role of vitamins E and C on extracellular matrix components of the vascular system. Vitamin E: An Overview of Major Research Directions. 28 (5), 507-537 (2007).
- Ashino, H., et al. Novel Function of Ascorbic Acid as an Angiostatic Factor. Angiogenesis. 6 (4), 259-269 (2003).
- Ponec, M., et al. The Formation of Competent Barrier Lipids in Reconstructed Human Epidermis Requires the Presence of Vitamin C. Journal of Investigative Dermatology. 109 (3), 348-355 (1997).
- Dickson, M. A., et al. Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Molecular and Cellular Biology. 20 (4), 1436-1447 (2000).
- Johansen, C. Generation and Culturing of Primary Human Keratinocytes from Adult Skin. Journal of visualized experiments: JoVE. (130), e56863 (2017).
- Richardson, D. S., Lichtman, J. W. Clarifying Tissue Clearing. Cell. 162 (2), 246-257 (2015).
- Ariel, P. A beginner's guide to tissue clearing. The International Journal of Biochemistry & Cell Biology. 84, 35-39 (2017).
- Friend, J., Yeo, L. Fabrication of microfluidic devices using polydimethylsiloxane. Biomicrofluidics. 4 (2), 026502 (2010).
- Ng, J. M. K., Gitlin, I., Stroock, A. D., Whitesides, G. M. Components for integrated poly(dimethylsiloxane) microfluidic systems. ELECTROPHORESIS. 23 (20), 3461-3473 (2002).
- Eddings, M. A., Johnson, M. A., Gale, B. K. Determining the optimal PDMS-PDMS bonding technique for microfluidic devices. Journal of Micromechanics and Microengineering. 18 (6), 067001 (2008).
- Markov, D. A., Lillie, E. M., Garbett, S. P., McCawley, L. J. Variation in diffusion of gases through PDMS due to plasma surface treatment and storage conditions. Biomedical microdevices. 16 (1), 91-96 (2014).
- Katzenberg, F. Plasma-bonding of poly(dimethylsiloxane) to glass. e-Polymers. 5 (1), (2005).
- El Ghalbzouri, A., Lamme, E., Ponec, M. Crucial role of fibroblasts in regulating epidermal morphogenesis. Cell and Tissue Research. 310 (2), 189-199 (2002).
- Kanitakis, J. Anatomy, histology and immunohistochemistry of normal human skin. European journal of dermatology: EJD. 12 (4), 390-399 (2002).
- Hessian based Frangi Vesselness filter. MATLAB Central File Exchange Available from: https://www.mathworks.com/matlabcentral/fileexchange/24409-hessian-based-frangi-vesselness-filter (2010)
- Jerman, T., Pernuš, F., Likar, B. Špiclin Enhancement of Vascular Structures in 3D and 2D Angiographic Images. IEEE Transactions on Medical Imaging. 35 (9), 2107-2118 (2016).
- Kovesi, P. Phase Preserving Denoising of Images. signal. 4, 6 (1999).
- Vincent, L. Morphological grayscale reconstruction in image analysis: applications and efficient algorithms. IEEE Transactions on Image Processing. 2 (2), 176-201 (1993).
- Xie, L., et al. Quantitative susceptibility mapping of kidney inflammation and fibrosis in type 1 angiotensin receptor-deficient mice. NMR in Biomedicine. 26 (12), 1853-1863 (2013).
- Van Uitert, R., Bitter, I. Subvoxel precise skeletons of volumetric data based on fast marching methods. Medical Physics. 34 (2), 627-638 (2007).
- Sethian, J. A. A fast marching level set method for monotonically advancing fronts. Proceedings of the National Academy of Sciences of the United States of America. 93 (4), 1591-1595 (1996).
- Sethian, J. A. Fast Marching Methods. SIAM Review. 41 (2), 199-235 (1999).
- Braverman, I. M. The Cutaneous Microcirculation. Journal of Investigative Dermatology Symposium Proceedings. 5 (1), 3-9 (2000).
- Men, S. J., Chen, C. -. L., Wei, W., Lai, T. -. Y., Song, S. Z., Wang, R. K. Repeatability of vessel density measurement in human skin by OCT-based microangiography. Skin research and technology: official journal of International Society for Bioengineering and the Skin (ISBS) [and] International Society for Digital Imaging of Skin (ISDIS) [and] International Society for Skin Imaging (ISSI). 23 (4), 607-612 (2017).
- Commandeur, S., Ho, S. H., de Gruijl, F. R., Willemze, R., Tensen, C. P., El Ghalbzouri, A. Functional characterization of cancer-associated fibroblasts of human cutaneous squamous cell carcinoma. Experimental Dermatology. 20 (9), 737-742 (2011).
- Thakoersing, V. S., Danso, M. O., Mulder, A., Gooris, G., Ghalbzouri, A. E., Bouwstra, J. A. Nature versus nurture: does human skin maintain its stratum corneum lipid properties in vitro. Experimental Dermatology. 21 (11), 865-870 (2012).
- Thakoersing, V. S., Gooris, G. S., Mulder, A., Rietveld, M., El Ghalbzouri, A., Bouwstra, J. A. Unraveling barrier properties of three different in-house human skin equivalents. Tissue Engineering. Part C, Methods. 18 (1), 1-11 (2012).
- Bouwstra, J. A., Groenink, H. W. W., Kempenaar, J. A., Romeijn, S. G., Ponec, M. Water distribution and natural moisturizer factor content in human skin equivalents are regulated by environmental relative humidity. The Journal of Investigative Dermatology. 128 (2), 378-388 (2008).
- Thakoersing, V. S., van Smeden, J., Mulder, A. A., Vreeken, R. J., El Ghalbzouri, A., Bouwstra, J. A. Increased Presence of Monounsaturated Fatty Acids in the Stratum Corneum of Human Skin Equivalents. Journal of Investigative Dermatology. 133 (1), 59-67 (2013).
- Smola, H., Thiekötter, G., Fusenig, N. Mutual induction of growth factor gene expression by epidermal-dermal cell interaction. The Journal of Cell Biology. 122 (2), 417 (1993).
- Fluck, J., Querfeld, C., Cremer, A., Niland, S., Krieg, T., Sollberg, S. Normal Human Primary Fibroblasts Undergo Apoptosis in Three-Dimensional Contractile Collagen Gels. Journal of Investigative Dermatology. 110 (2), 153-157 (1998).
- Nakagawa, S., Pawelek, P., Grinnell, F. Long-Term Culture of Fibroblasts in Contracted Collagen Gels: Effects on Cell Growth and Biosynthetic Activity. Journal of Investigative Dermatology. 93 (6), 792-798 (1989).
- Smith, L. E., Bonesi, M., Smallwood, R., Matcher, S. J., MacNeil, S. Using swept-source optical coherence tomography to monitor the formation of neo-epidermis in tissue-engineered skin. Journal of Tissue Engineering and Regenerative Medicine. 4 (8), 652-658 (2010).
- Pierce, M. C., Strasswimmer, J., Park, H., Cense, B., De Boer, J. F. Birefringence measurements in human skin using polarization-sensitive optical coherence tomography. Biomed Opt. 2 (2), 287-291 (2004).
- Pierce, M. C., Strasswimmer, J., Hyle Park, B., Cense, B., de Boer, J. F. Advances in Optical Coherence Tomography Imaging for Dermatology. Journal of Investigative Dermatology. 123 (3), 458-463 (2004).
- Yeh, A. T., Kao, B., Jung, W. G., Chen, Z., Nelson, J. S., Tromberg, B. J. Imaging wound healing using optical coherence tomography and multiphoton microscopy in an in vitro skin-equivalent tissue model. Journal of Biomedical Optics. 9 (2), 9 (2004).
- Derr, K., et al. Fully Three-Dimensional Bioprinted Skin Equivalent Contructs with Validated Morphology and Barrier Funtion. Tissue Engineering Part C: Methods. 25 (6), 334-343 (2019).
- Park, B. H., de Boer, J. F. Polarization Sensitive Optical Coherence Tomography. Optical Coherence Tomography: Technology and Applications. , 1055-1101 (2015).
- Batheja, P., Song, Y., Wertz, P., Michniak-Kohn, B. Effects of Growth Conditions on the Barrier Properties of a Human Skin Equivalent. Pharmaceutical Research. 26 (7), 1689-1700 (2009).
- Dos Santos, M., Metral, E., Boher, A., Rousselle, P., Thepot, A., Damour, O. In vitro 3-D model based on extending time of culture for studying chronological epidermis aging. Matrix Biology. 47, 85-97 (2015).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved