
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Utilisation du système GAL4-UAS pour la génétique fonctionnelle chez Anopheles gambiae
Dans cet article
Résumé
Le système bipartite GAL4-UAS est un outil polyvalent pour la modification de l’expression des gènes de manière spatio-temporelle contrôlée qui permet une analyse génétique fonctionnelle chez Anopheles gambiae. Les procédures décrites pour l’utilisation de ce système sont une stratégie de clonage semi-standardisée, le sexage et le dépistage des pupes pour les marqueurs de protéines fluorescentes et la fixation des embryons.
Résumé
Le système bipartite GAL4-UAS est un outil polyvalent et puissant pour l’analyse génétique fonctionnelle. L’essence du système est de croiser des lignées transgéniques « pilotes » qui expriment le facteur de transcription de levure GAL4 d’une manière spécifique au tissu, avec des lignées transgéniques « répondeurs » portant une construction d’interférence gène / ARN candidate dont l’expression est contrôlée par des séquences d’activation en amont (UAS) qui se lient à GAL4. Dans la descendance qui s’ensuit, le gène ou la construction de silençage est donc exprimé d’une manière spatio-temporelle prescrite, ce qui permet de tester les phénotypes résultants et d’en déduire la fonction du gène. Le système binaire permet une flexibilité dans les approches expérimentales pour dépister les phénotypes générés par l’expression du transgène dans de multiples modèles spécifiques aux tissus, même si des coûts de condition physique élevés sont induits. Nous avons adapté ce système à Anopheles gambiae, le principal vecteur du paludisme en Afrique.
Dans cet article, nous fournissons certaines des procédures courantes utilisées lors de l’analyse GAL4-UAS. Nous décrivons les lignes An. gambiae GAL4-UAS déjà générées, ainsi que le clonage de nouvelles constructions de répondeurs pour la régulation à la hausse et l’ARNi. Nous spécifions un guide étape par étape pour le sexage des pupes de moustiques afin d’établir des croisements génétiques, qui comprend également le dépistage de la progéniture pour suivre l’héritage des marqueurs génétiques fluorescents qui marquent les insertions du conducteur et du répondeur. Nous présentons également un protocole pour le nettoyage des embryons d’An. gambiae afin d’étudier le développement embryonnaire. Enfin, nous introduisons des adaptations potentielles de la méthode pour générer des lignes pilotes par insertion CRISPR/Cas9 de GAL4 en aval des gènes cibles.
Introduction
Le système bipartite GAL4-UAS est le cheval de bataille de la caractérisation fonctionnelle des gènes de l’organisme modèle d’insecte Drosophila melanogaster1,2,3. Pour utiliser le système GAL4-UAS, des lignées pilotes transgéniques, exprimant le facteur de transcription de levure GAL4 sous contrôle d’une séquence régulatrice, sont croisées avec des lignes de répondeur portant un gène d’intérêt ou une construction d’interférence ARN (ARNi) contrôlée par une séquence d’activation en amont (UAS) reconnue par GAL4. La descendance de ce croisement exprime le transgène d’intérêt pour un schéma spatio-temporel dicté par le promoteur contrôlant l’expression GAL4 (Figure 1). Les phénotypes affichés par la descendance des croisements conducteur-répondeur peuvent être évalués pour élucider la fonction des gènes candidats. Bien que D. melanogaster ait été utilisé pour examiner les gènes d’autres organismes4,5,6,7, le système GAL4-UAS a maintenant été adapté pour être utilisé chez les insectes d’importance médicale et agricole afin de fournir une analyse directe des espèces d’intérêt 8,9,10,11,12,13,14.
Chez le moustique africain du paludisme, Anopheles gambiae, le système GAL4-UAS a d’abord été testé par co-transfection de lignée cellulaire9. Plusieurs constructions ont été testées pour l’efficacité dans différentes combinaisons par paires et ont constaté que 14 UAS répétés en tandem complétés par un petit intron artificiel (UAS-14i) présentaient la plus large gamme de potentiel d’activation lorsqu’ils étaient utilisés avec un panel de pilotes GAL4. Pour démontrer la fonctionnalité in vivo, ces constructions ont ensuite été utilisées pour créer deux lignées transgéniques distinctes d’An. gambiae par transformation PiggyBac8 : une ligne pilote portant GAL4 entraînée par un promoteur spécifique de l’intestin moyen, et une ligne répondeur contenant à la fois les gènes de la luciférase et de la protéine fluorescente jaune améliorée (eYFP) sous régulation des séquences UAS. L’activité de la luciférase spécifique à l’intestin et la fluorescence dans la progéniture ont indiqué que le système était efficace chez les anophèles. Depuis lors, des lignées conductrices ont été créées exprimant des transgènes dans d’autres tissus importants pour la capacité vectorielle et la résistance aux insecticides, y compris les œnocytes15 et les hémocytes16, et dans un schéma proche de l’omniprésence10. De nombreuses lignées d’UAS ont également été générées pour tester les gènes soupçonnés d’être impliqués dans le métabolisme et la résistance aux insecticides médiés par la séquestration, la synthèse des hydrocarbures cuticulaires et pour marquer par fluorescence différents types de cellules et de tissus (tableau 1). Pour les lignes répondeuses, l’intégration dirigée par site du transgène est maintenant réalisée par échange de cassettes de recombinaison catalysée ΦC31111111111 pour fixer le contexte génomique des gènes régulés par UAS. De cette façon, l’expression du transgène est normalisée en ce qui concerne l’emplacement d’insertion génomique, ce qui permet une comparaison plus précise des effets phénotypiques de différents gènes candidats.
Les lignes de répondeur créées à ce jour sont conçues pour exprimer le transgène à des niveaux élevés ou pour réduire l’expression des gènes par interférence ARN (ARNi). Habituellement, les clones d’ADNc sont fusionnés à la séquence UAS pour générer des plasmides d’expression appropriés, mais des séquences génomiques complètes sont également réalisables en supposant qu’elles ne sont pas trop grandes pour le clonage. Pour générer des constructions de silencieux, nous avons utilisé trois méthodes différentes pour obtenir des séquences inversées en tandem appropriées qui forment un ARNd en épingle à cheveux qui stimule l’ARNi. Ceux-ci ont inclus la PCR par fusion, la PCR asymétrique et la synthèse commerciale de constructions en épingle à cheveux. Chaque méthode a en commun l’inclusion d’une séquence d’introns entre les séquences inversées pour assurer la stabilité du clonage. Des plasmides répondeurs dans lesquels un gène d’intérêt/construction d’ARNi peut être inséré ont été développés15. Ces plasmides portent également les sites ΦC31 attB requis pour le RMCE (décrits dans l’article d’Adolfi accompagnant JoVE qui décrit en détail la technique RCME). Des protocoles couvrant les étapes importantes requises lors de la sélection de la séquence à insérer dans l’un de ces plasmides pour la surexpression sont inclus dans ce manuscrit. De plus, deux protocoles pour la création de construction en épingle à cheveux ARNi sont décrits et illustrés.
Lors de la création de nouvelles lignées, l’identification d’individus transgéniques rares est cruciale pour se reproduire afin d’établir et de maintenir des colonies transgéniques. Plus important encore, pour le système GAL4-UAS, il est nécessaire de distinguer les lignes d’intervention et de conducteur afin d’établir des croisements et d’identifier les descendants individuels porteurs des deux transgènes. Ceci est réalisé en utilisant différents gènes marqueurs sélectionnables dominants liés aux cassettes du conducteur et du répondeur. Le plus souvent, il s’agit de gènes marqueurs fluorescents qui peuvent être clairement distingués à l’aide de filtres optiques (par exemple, eYFP, eCFP, dsRed). Il est important que les marqueurs soient exprimés selon un schéma spatio-temporel connu et fiable, car cela facilite l’identification des anomalies et de la contamination. L’expression du gène marqueur fluorescent est régulièrement régulée par le promoteur synthétique 3xP3, qui provoque une expression spécifique des ganglions oculaires et ventraux à tous les stades du développement d’An. gambiae19. Les marqueurs fluorescents contrôlés par 3xP3 sont inclus dans tous les plasmides de transformation décrits dans cet article. Un protocole détaillant les méthodes courantes utilisées pour filtrer les lignes fluorescentes An. gambiae pupae GAL4-UAS est inclus ici.
L’un des éléments clés du système GAL4-UAS est la nécessité de traverser les lignes de conduite et d’intervention marquées différentiellement. Pour ce faire, les mâles et les femelles de chaque lignée doivent être séparés avant l’accouplement. Les adultes se distinguent facilement par la vue, cependant, pour établir des croisements génétiques, il est judicieux de séparer les sexes avant l’émergence de l’adulte pour s’assurer que l’accouplement n’a pas eu lieu. La différence de taille générale entre les pupes an. gambiae mâles et femelles est trop variable pour être une méthode efficace et fiable de détermination du sexe20. Au lieu de cela, des différences morphologiques claires dans les organes génitaux externes fournissent une base fiable pour le sexage chez An. gambiae. Dans cet article, nous décrivons une méthode fiable pour sexer les pupes d’An. gambiae afin de mettre en place des croisements appropriés.

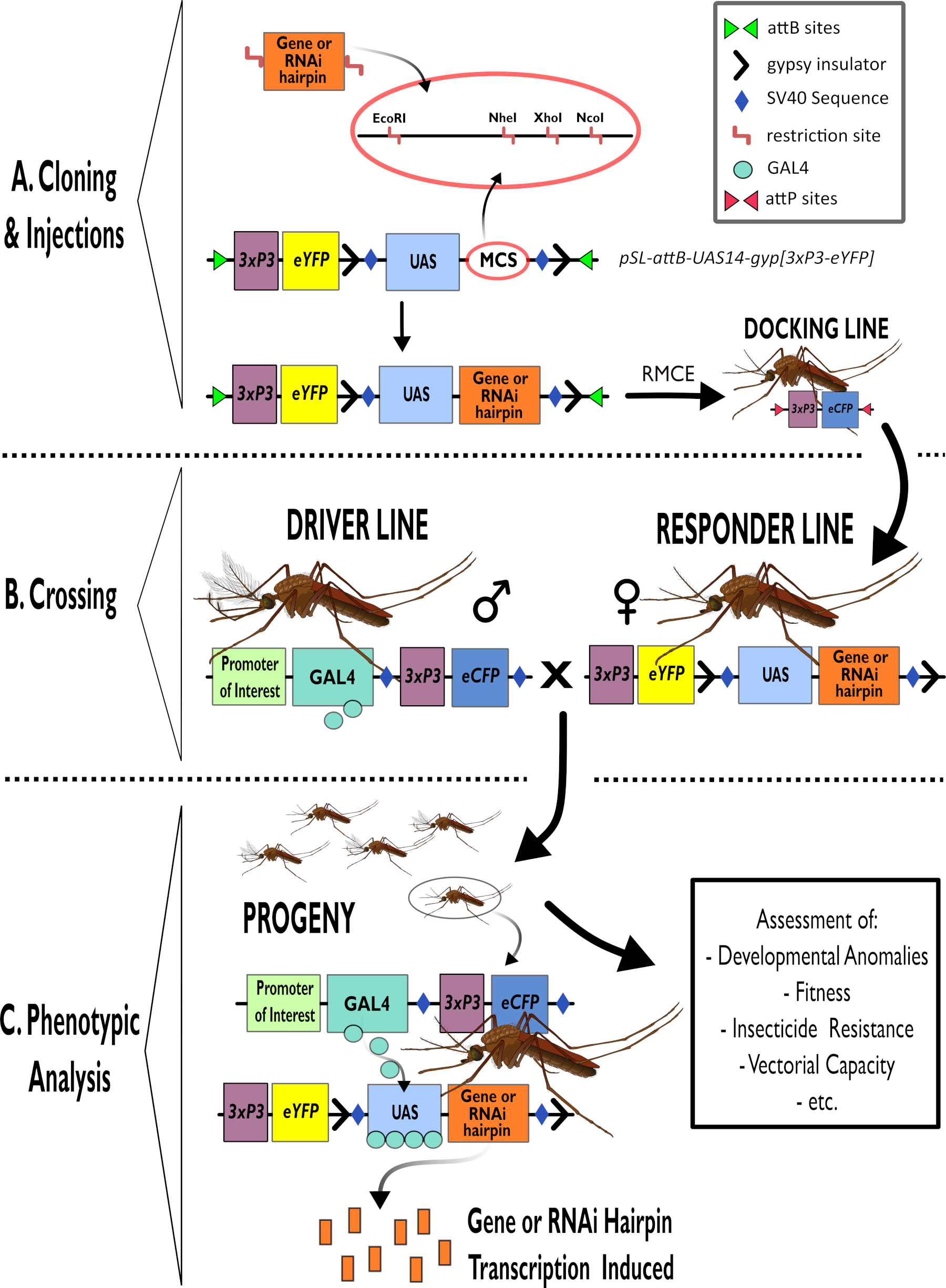
Figure 1 - Représentation schématique du processus d’utilisation du système bipartite GAL4-UAS dans Anopheles gambiae. (A) Les principaux composants d’un exemple de vecteur (pSL-attB-UAS14-gyp[3xp3-eYFP]) sont représentés, détaillant les sites de restriction disponibles (EcoRI, NheI, XhoI et NcoI) dans les multiples sites de clonage qui peuvent être utilisés pour insérer la construction en épingle à cheveux ou la séquence codante pour le gène d’intérêt. La structure de la ligne d’amarrage est également représentée. (B) L’étape de croisement est illustrée en indiquant l’utilisation d’hommes de la ligne de conduite (transportant le conducteur GAL4 par un promoteur d’intérêt et eCFP piloté par le promoteur 3xP3) et de femelles de la ligne de réponse (portant le gène d’intérêt ou la construction en épingle à cheveux contrôlée par un promoteur UAS et un marqueur eYFP contrôlé par le promoteur 3xP3). (C) Une représentation schématique de GAL4 conduisant l’expression du gène d’intérêt dans la descendance du croisement dans B et une liste de certains des phénotypes typiques qui sont évalués. Abréviations : Site de clonage multiple (MCS), échange de cassettes médié par la recombinase (RMCE), séquence d’activateur en amont (UAS), protéine fluorescente jaune améliorée (eYFP), protéine fluorescente cyan améliorée (eCFP). Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
C’est l’utilisation de croisements qui fournit la nature bipartite du système GAL4-UAS, qui présente des avantages distincts par rapport aux approches plus linéaires. Par exemple, beaucoup plus de combinaisons de lignées conductrices et répondeuses peuvent être évaluées que ce qui serait faisable si une nouvelle lignée transgénique devait être générée et maintenue pour chaque combinaison promoteur/gène. Plus important encore, il permet l’analyse de gènes qui produisent des phénotypes mortels ou stériles lorsque leur expression est perturbée et qui sont difficiles à créer / maintenir dans un système linéaire. De tels phénotypes mortels peuvent se manifester à tous les stades de développement, en fonction de la fonction du gène et de l’expression spatio-temporelle, mais sont le plus souvent observés au cours du développement embryonnaire. La visualisation du développement embryonnaire de moustique nécessite l’élimination du chorion opaque qui recouvre les œufs. En suivant les méthodes décrites dans Trpiš (1970)21 et Kaiser et al. (2014)22, nous décrivons les protocoles que nous utilisons pour fixer les embryons, tout en maintenant l’intégrité structurelle, et le blanchiment pour éliminer l’endochorion qui permet la visualisation et l’imagerie microscopiques.
Protocole
1. Conception et construction de constructions UAS
- Conception et assemblage de vecteurs pour l’expression de gènes candidats
- Déterminer la séquence à utiliser pour la régulation à la hausse des gènes candidats.
- Séquencer l’ADNc/ADNg de la souche d’intérêt et le comparer à la séquence publiée pour vérifier son identité et identifier les SNP potentiels et les sites de restriction pour le digest diagnostique.
- Assurez-vous que l’amorce avant utilisée pour l’amplification des gènes couvre la séquence de Kozak native et commencez le codon, le cas échéant. Une amorce avec une liaison d’environ 10 pb en amont du codon de départ englobera la séquence de Kozak.
- Inclure le codon d’arrêt dans le fragment amplifié à partir de l’amorce inverse dans la plupart des cas. Utiliser les séquences de terminaison 3' fournies dans les vecteurs plasmidiques décrits, ou amplifier à partir de séquences génomiques de gènes candidats.
- Commandez des séquences commerciales avec un biais de codon spécifique si vous le souhaitez.
- Utilisez des procédures de sous-clonage standard pour insérer des cassettes de gènes dans les vecteurs plasmidiques des UAS, par exemple, pSL-attB-UAS14-gyp[3xP3-eYFP]15 (Figure 1) pour les constructions de régulation à la hausse et d’ARNi.
- Produire des moustiques transgéniques créés à l’aide d’un échange de cassettes médié par recombinaison ΦC31111111111111111111111111111111111111111111111110,17,18,23.
- Déterminer la séquence à utiliser pour la régulation à la hausse des gènes candidats.
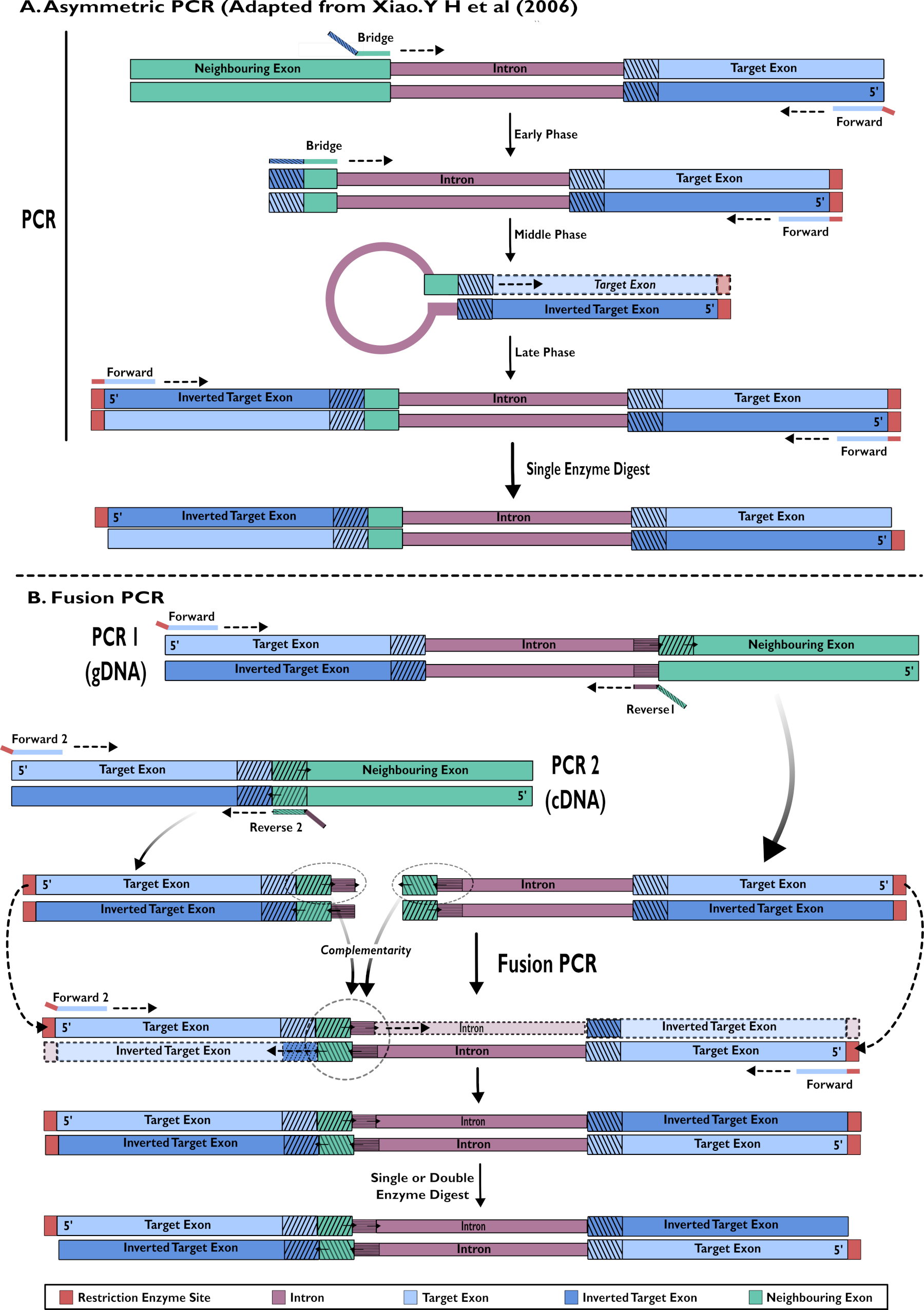
- Création de constructions en épingle à cheveux ARNi : amplification en une seule étape à l’aide de la PCR asymétrique15,24
- Extraire l’ADN génomique (ADNg) d’une femelle adulte An. gambiae portant le gène candidat souhaité en utilisant la méthode Livak25.
- Concevez l’amorce avant pour qu’elle se lie à l’exon cible au 5' du fragment désiré dirigé vers l’intron voisin. Concevez l’extrémité 3' d’un amorce de pont pour se lier à l’extrémité de l’exon précédent afin d’amplifier l’intron. L’extrémité 5' est complémentaire à un petit fragment de l’exon cible immédiatement après l’intron.
- Exécutez une réaction de PCR asymétrique comme décrit dans Xiao (2006)24 (Figure 2).
- Clonez le produit PCR purifié dans un vecteur approprié portant le promoteur UAS (par exemple, pSL-attB-UAS14-gyp[3xP3-eYFP]15).
REMARQUE : Les enzymes du site de clonage multiple qui conviennent au clonage pSL-attB-UAS14-gyp[3xP3-eYFP]15 et les étapes suivantes requises sont indiquées à la figure 1. La digestion enzymatique unique est essentielle car un seul site de restriction est ajouté. La déphosphorylation du plasmide améliorera l’efficacité du clonage.
- Extraire l’ADN génomique (ADNg) d’une femelle adulte An. gambiae portant le gène candidat souhaité en utilisant la méthode Livak25.
- Construction de constructions en épingle à cheveux ARNi : Fusion PCR de l’ADNc et de l’ADNg15
- Extraire l’ADN génomique (ADNg) d’une femelle adulte An. gambiae portant le gène candidat souhaité en utilisant la méthode Livak25.
- Inclure l’ADNg dans une réaction de PCR pour amplifier la zone cible des séquences d’exons et d’introns ensemble (Figure 2).
- Concevez l’extrémité 3' de l’amorce avant pour se lier à la séquence d’exons cible inverse pour amplifier vers la séquence d’intron cible et l’extrémité 5' pour porter un site de restriction pour faciliter le clonage.
- Concevoir l’amorce inverse (1) pour se lier à l’extrémité 5' de l’intron et le porte-à-faux de l’extrémité 5' porte les premières bases de la séquence avant de l’exon voisin. Ce porte-à-faux est utilisé dans la PCR de fusion.
- Purifier le produit de réaction souhaité.
- Extraire l’ARN, éliminer l’ADN à l’aide de DNase et préparer l’ADNc d’une femelle adulte An. gambiae portant le gène candidat souhaité en suivant les protocoles du fabricant.
- Utilisez l’ADNc dans une réaction de PCR pour amplifier uniquement la zone cible de l’exon (Figure 2).
- Concevez l’amorce avant (2) de sorte que l’extrémité 3' se lie à l’extrémité 3' de la séquence d’exons cible complémentaire et que l’extrémité 5' de l’amorce porte un site de restriction pour une utilisation dans le clonage.
REMARQUE: L’amorce avant de 1.3.1.2 peut être utilisée à nouveau dans cette deuxième réaction. Cependant, cela signifie qu’une seule digestion enzymatique est essentielle. L’utilisation d’un deuxième amorce avant avec un site de restriction différent permettra une double digestion qui peut augmenter l’efficacité du clonage. - Amorce inverse de conception (2) - l’extrémité 3' se lie à l’extrémité 5' de l’exon voisin amplifiant l’exon cible. L’extrémité 5' se lie à l’extrémité 3' du brin avant des introns. Ce porte-à-faux est utilisé dans la PCR de fusion.
- Purifier le produit de réaction souhaité.
- Concevez l’amorce avant (2) de sorte que l’extrémité 3' se lie à l’extrémité 3' de la séquence d’exons cible complémentaire et que l’extrémité 5' de l’amorce porte un site de restriction pour une utilisation dans le clonage.
- Inclure les produits des étapes 1.3.1 et 1.3.2 comme modèles pour une réaction de PCR de fusion utilisant des concentrations standard avec des amorces avant 1 et 2. Purifier le produit désiré.
- Digérez le produit purifié pour générer les surplombs pour le clonage. Cloner dans un vecteur approprié en aval du promoteur UAS. Les enzymes appropriées pour le clonage pSL-attB-UAS14-gyp[3xP3-eYFP]15 et les étapes suivantes requises sont indiquées à la figure 1.
- Extraire l’ADN génomique (ADNg) d’une femelle adulte An. gambiae portant le gène candidat souhaité en utilisant la méthode Livak25.

Figure 2 - Représentation schématique de la création de constructions ARNi à insérer dans pSL-attB-UAS14-gyp[3xP3-eYFP] par deux méthodes : (A) PCR asymétrique en une seule étape (adaptée de Xiao. Y H et al (2006) et (B) PCR par fusion en plusieurs étapes. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
2. Dépistage des pupes an. gambiennes
- Collection de pupes pour la caractérisation microscopique
REMARQUE: Tout au long de ces protocoles, l’eau fait référence à l’eau distillée complétée par 0,01% de sel d’étang.- Moustiques an. gambiens arrière utilisant des protocoles standard (par exemple, MR426) au stade nymphal.
ATTENTION : Veillez à ne pas blesser les nymphes tout au long de ce processus. - Collectez les nymphes sur un plat transparent adapté à une utilisation avec un stéréomicroscope (par exemple, une boîte de Petri en plastique de 100 x 15 mm, en évitant les bords).
REMARQUE: Pour recueillir les pupes, nous utilisons une pipette Pasteur en plastique de 3 mL avec environ 10 mm coupés de l’extrémité pour élargir l’extrémité et éviter de blesser les moustiques. Le dépistage et le sexage peuvent être effectués sur les individus, cependant, c’est très lent. Il est recommandé d’effectuer un dépistage et un sexage sur des groupes de 50 à 200 pupes (la taille du groupe possible est limitée par la taille du plat utilisé et est soumise à une préférence personnelle). Si un grand nombre d’entre eux sont examinés, l’efficacité peut être augmentée en alignant d’abord les nymphes à environ 4 à 5 profondeurs dans les lignes et en déplaçant les nymphes cibles hors de cette ligne. - À l’aide d’une pipette Pasteur, retirez soigneusement presque toute l’eau autour des nymphes. Laissez juste assez d’eau autour des nymphes pour qu’elles soient effectivement immobiles mais puissent être déplacées facilement avec une brosse fine. S’ils deviennent difficiles à déplacer, ajoutez plus d’eau.
REMARQUE: Lorsque suffisamment d’eau est retirée, les nymphes se trouvent sur le côté, ce qui permet de visualiser les yeux pour la détection par fluorescence et l’identification des organes génitaux dimorphes (Figure 4DE).
ATTENTION : Assurez-vous que les nymphes ne se dessèchent pas. S’il ne reste qu’un très petit volume d’eau, il peut réduire davantage avec la chaleur de la lampe du microscope et lorsqu’il est divisé entre des bassins de pupes. De l’eau supplémentaire doit parfois être ajoutée au cours du processus à l’aide d’une pipette Pasteur de 3 mL au(x) groupe(s) souhaité(s).
- Moustiques an. gambiens arrière utilisant des protocoles standard (par exemple, MR426) au stade nymphal.
- Identification des marqueurs fluorescents chez les pupes
REMARQUE: L’utilisation d’un stéréoscope à faible grossissement permet un criblage à grand champ, le tri peut être effectué sur un microscope composé inversé, mais doit être effectué individuellement.- Lors du dépistage d’un marqueur fluorescent, il est d’abord crucial de connaître les modèles d’expression et d’héritage attendus. Tenez compte des éléments suivants :
- Couleur(s) : déterminez le(s) filtre(s) pour visualiser l’expression.
- Modèle d’expression spatio-temporelle: Comprenez où et à quel stade de la vie vous vous attendez à voir l’expression.
- Ratio de différents phénotypes: établir quel pourcentage de la population devrait porter les marqueurs d’intérêt.
- Effectuez un dépistage fluorescent dans l’obscurité, car même une faible lumière peut interférer avec la résolution de la fluorescence. Cependant, utilisez une lampe à côté du stéréoscope lorsque la lumière est nécessaire pour d’autres manipulations.
ATTENTION : Assurez-vous que l’espace de travail autour du stéréoscope fluorescent est dégagé avant d’éteindre les lumières. - Allumez l’ampoule fluorescente et laissez chauffer pendant la période recommandée par le fabricant (normalement 10-15 min). Sélectionnez le filtre requis sur le stéréoscope fluorescent et vérifiez qu’il y a un faisceau de lumière coloré visible qui est dirigé vers le centre de la plaque de scène. Si cela n’est pas visible ou est très faible, l’ampoule fluorescente peut ne pas s’être complètement réchauffée, l’obturateur est fermé ou l’optique du microscope n’est pas bien alignée.
- À l’aide de la lumière blanche, centrez les nymphes dans le champ de vision et mettez-les au point. Ce grossissement peut devoir être modifié lors de la commutation entre différents filtres en fonction de l’intensité de fluorescence.
- L’utilisation d’un pinceau à détails fins permet de s’assurer que les nymphes examinées ne se chevauchent pas.
- Éteignez la lumière blanche du stéréoscope et utilisez la mise au point fine pour mettre au point la zone des pupes portant le phénotype d’intérêt. Le motif fluorescent doit être visible. Des exemples de fluorescence contrôlée par le promoteur 3xP3 sont fournis à la figure 3.
- Utilisez le grossissement le plus bas auquel le phénotype fluorescent attendu peut être distingué de manière fiable des individus sans fluorescence.
- Pour les souches à fluorescence vive, utilisez également une lumière de champ lumineux de faible intensité lors du criblage, si le signal fluorescent est toujours clairement identifiable.
- Une fois le dépistage primaire terminé, scannez rapidement les populations sous d’autres filtres pour détecter une contamination potentielle.
ATTENTION : Assurez-vous qu’il y a une distance claire entre les groupes de nymphes triées pour éviter toute contamination par le mouvement des nymphes. Sachez que la taille des groupes changera à mesure que les nymphes seront sexuées et que les distances peuvent sembler plus grandes lorsque vous regardez sous grossissement. Faites particulièrement attention lorsque les piscines ne sont pas dans le champ de vision.
- Lors du dépistage d’un marqueur fluorescent, il est d’abord crucial de connaître les modèles d’expression et d’héritage attendus. Tenez compte des éléments suivants :

Figure 3 - Pupes anophèles gambiennes exprimant des marqueurs fluorescents entraînés par le promoteur 3xP3 (A) eYFP, (B) dsRed et (C) eCFP. Grossissement: A = 16X, B, C = 20X.
-
Sexage des pupes
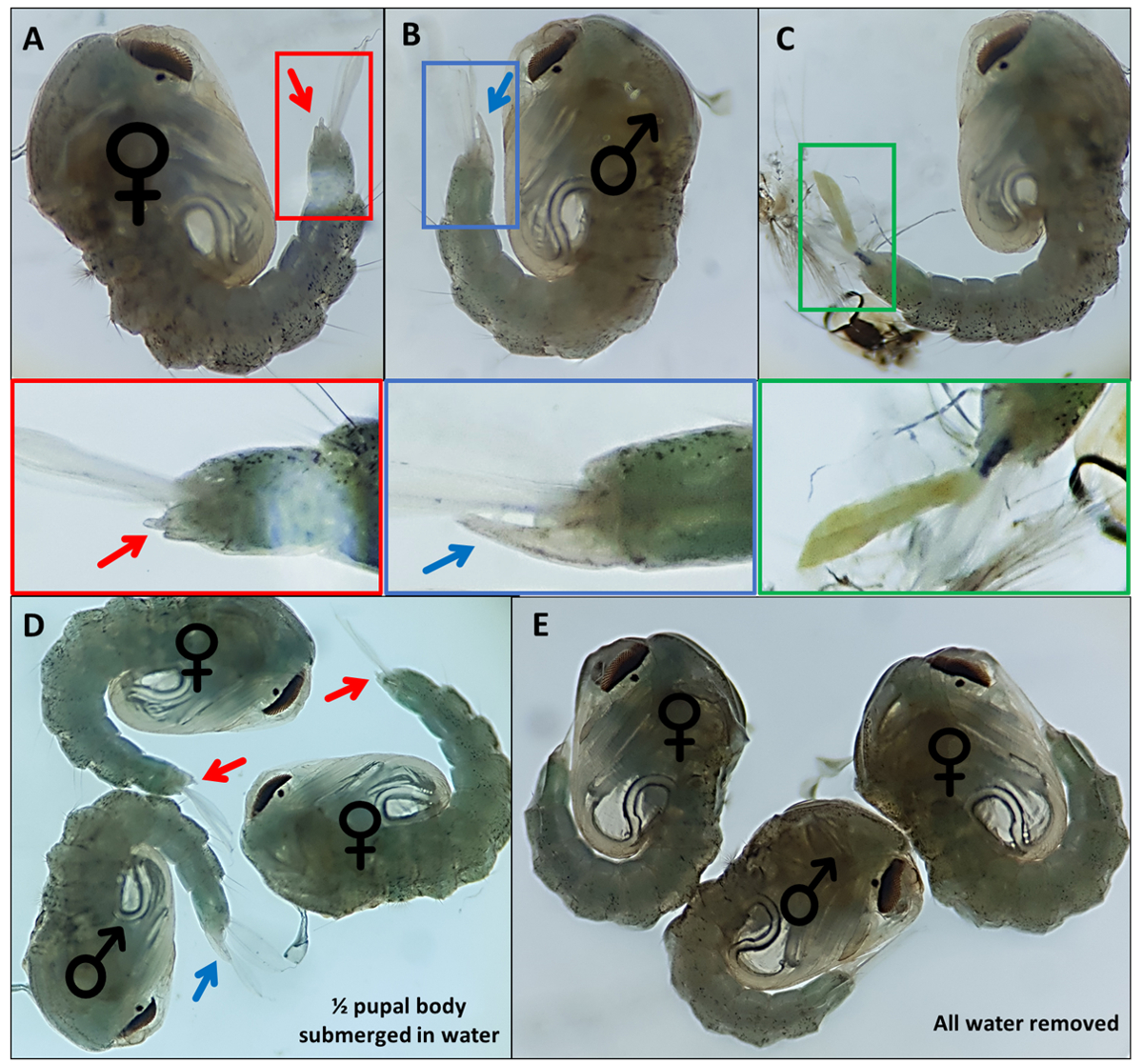
- Collectez les nymphes. Enlevez l’excès d’eau, mais fournissez suffisamment pour que les pagaies anales se séparent légèrement des organes génitaux pour faciliter la visualisation et la caractérisation morphologique (Figure 4D, E).
- Si une nymphe / e n’est pas sur le côté, utilisez un pinceau à détails fins pour tourner doucement la nymphe et déplacer les pagaies anales afin que les organes génitaux externes puissent être identifiés.
- Nymphes séparées en fonction d’organes génitaux externes distinctifs; les mâles ont un long tube qui s’extrude du segment dorsal final d’environ la moitié de la longueur des pagaies anales (Figure 4B). Les organes génitaux externes des nymphes femelles sont considérablement plus courts et bifurqués (figure 4A).
REMARQUE: À l’occasion, si l’exosquelette larvaire du 4e stade reste attaché ou si les organes génitaux externes sont endommagés (Figure 4C), l’identification confiante du sexe est plus difficile. Lorsque le sexe d’une nymphe n’est pas clair, il est préférable de le jeter. Si l’individu doit être gardé, la nymphe doit être autorisée à émerger de manière isolée et son sexe déterminé à l’aide de caractéristiques morphologiques adultes. Il est probable que si ses organes génitaux sont endommagés, l’individu peut ne pas s’accoupler avec succès. - Faites une piscine pour chaque sexe à l’extrémité opposée du plat à la piscine non sexuée, en déplaçant les nymphes identifiées à travers le plat à l’aide d’un pinceau aux détails fins. Étiquetez le dessous du plat où les deux piscines seront rassemblées pour les identifier plus tard.
- Lorsque le sexage et le dépistage fluorescent sont nécessaires, effectuez d’abord un dépistage fluorescent, car c’est le processus le plus rapide des deux.

Figure 4 - Sexing Anopheles gambiae pupee. Pupes individuelles indiquant les organes génitaux externes de (A) une femelle (B) un mâle et (C) un individu qui ne peut pas être facilement identifié en raison du détachement incomplet de l’exosquelette larvaire. Images agrandies ci-dessous mettant en évidence les organes génitaux externes. Pupes avec ♀ (femelle) et ♂ (mâle) indiquant les organes génitaux externes des nymphes avec (D) ~ 50% de la nymphe immergée dans l’eau et avec (E) toute l’eau enlevée soulignant la différence de facilité de visualisation des organes génitaux externes. Grossissement : A,B,C=40x, D,E=30x. Veuillez cliquer ici pour agrandir cette figure.
{kind=link}
- Confirmation du sexe à l’âge adulte
- Jusqu’à ce qu’un taux d’erreur très faible ait été démontré, confirmez le sexage des pupes par morphologie adulte après l’émergence. Séparer les nymphes sexées en groupes de 10 ou moins dans un tube clair de 20 mL avec quelques mL d’eau, en scellant avec une boule de coton, étiqueté avec le sexe attendu et en laissant émerger pendant la nuit.
REMARQUE: Comme les adultes sont transférés le lendemain matin, il n’est pas nécessaire de fournir de la nourriture aux adultes émergents. - Confirmez le sexe des adultes émergés en utilisant des caractéristiques morphologiques le lendemain.
- Si des mâles sont présents dans les collections femelles, jetez les femelles, au cas où l’accouplement aurait déjà eu lieu.
- Si des femelles sont présentes dans la collection masculine, retirez la ou les femelle(s) et gardez les mâles pour le croisement.
- Jusqu’à ce qu’un taux d’erreur très faible ait été démontré, confirmez le sexage des pupes par morphologie adulte après l’émergence. Séparer les nymphes sexées en groupes de 10 ou moins dans un tube clair de 20 mL avec quelques mL d’eau, en scellant avec une boule de coton, étiqueté avec le sexe attendu et en laissant émerger pendant la nuit.
- Configuration des croisements du système GAL4-UAS
- Aspirer le nombre souhaité d’adultes mâles et femelles des tubes à l’étape 2.4 dans une cage ou un petit seau mis en place de la manière standard pour l’élevage d’An. gambiae.
REMARQUE: Veillez à ne pas endommager les adultes pendant ce transfert. - Utilisez environ 50 femelles avec un nombre égal de mâles, quand ~ 2000 adultes sont nécessaires de la progéniture.
REMARQUE: Lorsqu’une croix doit être nourrie plusieurs fois pour générer plusieurs lots, jusqu’à 200 de chaque sexe peuvent être installés dans des cages de 30 cm x 30 cm x 30 cm. Lorsque seul un petit nombre de femelles (<20) sont disponibles pour la croix, nous ajoutons ~ 4 fois le nombre de mâles pour augmenter la probabilité d’accouplement réussi. - Le sang nourrit les femelles croisées et la progéniture arrière au stade approprié, conformément aux protocoles standard26, pour effectuer une évaluation phénotypique (p. ex., résistance aux insecticides, capacité vectorielle et tests de coût de la condition physique).
- Lorsque l’effet maternel de l’expression transgénique est probable, mettre en place des croisements réciproques des lignes du conducteur et du répondeur et analyser le phénotype attendu.
REMARQUE: Les croisements utilisant des populations « hétérozygotes » ou mixtes de lignées conductrices et répondeuses produisent une progéniture avec chacun des 4 génotypes possibles. Cela fournit des contrôles de type sauvage, UAS uniquement et GAL4 uniquement, ainsi que les transhétérozygotes GAL4-UAS avec lesquels analyser le phénotype. Si des populations homozygotes sont croisées, mettez en place des croisements supplémentaires pour fournir des contrôles appropriés pour comparer les phénotypes. La descendance doit être dépistée comme ci-dessus en séparant la progéniture portant les deux ou seulement l’un ou l’autre marqueur, ainsi que des négatifs, pour une évaluation phénotypique.
- Aspirer le nombre souhaité d’adultes mâles et femelles des tubes à l’étape 2.4 dans une cage ou un petit seau mis en place de la manière standard pour l’élevage d’An. gambiae.
- Établir des populations homozygotes à partir de lignées générées par RCME portant des marqueurs fluorescents alternatifs
REMARQUE: Il est essentiel que le marqueur fluorescent des deux lignées soit présent au même endroit génomique et qu’ils soient complètement distinguables.- Mettre en place un croisement parental d’environ 200 adultes avec un nombre égal d’hommes marqués différemment d’une ligne et de femmes de l’autre ligne après le dépistage pour sélectionner des individus présentant une fluorescence et un sexe corrects, comme décrit ci-dessus. Environ une semaine plus tard, le sang alimente le croisement en utilisant des protocoles établis26.
- Élevez la progéniture F1 aux nymphes en utilisant des protocoles standard et collectez les nymphes comme décrit précédemment.
- Dépistage de la fluorescence en sélectionnant ceux qui portent les deux marqueurs parentaux (transhétérozygotes). Mettez en place un intercroiseur F1 avec ces pupes.
- Une semaine plus tard, le sang nourrit les femelles F1 et la progéniture arrière jusqu’au stade nymphal en suivant les protocoles standard.
- Filtrez les nymphes F2 en sélectionnant celles qui n’affichent QU’un seul des marqueurs. Ceux-ci seront homozygotes pour l’insertion. Seulement 25% de la progéniture sera homozygote pour chaque insertion, alors assurez-vous que suffisamment de progéniture est élevée pour fournir une cage de stock (400-500).
REMARQUE: La sélection de la progéniture transhétérozygote doit être entièrement rigoureuse, sinon le processus devient contaminé et une homozygotie complète peut ne pas être atteinte. Vérifiez toutes les descendants sélectionnés pour l’intercross F1.
- Mettre en place un croisement parental d’environ 200 adultes avec un nombre égal d’hommes marqués différemment d’une ligne et de femmes de l’autre ligne après le dépistage pour sélectionner des individus présentant une fluorescence et un sexe corrects, comme décrit ci-dessus. Environ une semaine plus tard, le sang alimente le croisement en utilisant des protocoles établis26.
3. Protocole d’élimination des embryons d’An. gambie
-
Alimentation sanguine et entretien
- Moustiques an. gambiaes à l’âge adulte suivant les protocoles standard (par exemple, MR4).
- Le sang nourrit des femelles adultes âgées de 5 à 7 jours, ce qui garantit que la plupart sont complètement engorgées.
ATTENTION: Tout au long de ce protocole, il est essentiel de travailler rapidement pour s’assurer que les œufs ne sont pas autorisés à se dessécher.
-
Ponte induite
- 3 jours après l’alimentation sanguine, prélever les œufs par ponte induite.
- Assemblez la chambre de ponte.
- Remplissez le pot de ponte avec de l’eau jusqu’à une profondeur d’environ 5 mm. Fixez le pot à une extrémité d’un tube en polypropylène de 50 mL, préalablement coupé avec une scie à métaux afin que les deux extrémités soient ouvertes. (Nous utilisons un disque en plastique pour un pot (Figure 5); cependant, le couvercle d’origine du tube peut être utilisé à la place).
- Couvrir l’autre extrémité du tube en polypropylène coupé avec un matériau (tuyau / collants) ou des sections de gant en latex fixées par une bande élastique, afin que les adultes puissent être introduits mais ne puissent pas s’échapper (Figure 5). D’autres modèles de chambre de ponte alternatifs existent et peuvent être utilisés26.
- Introduisez soigneusement 10 à 15 femelles (nourries au sang à l’étape 3.1.2) dans la chambre de ponte. Couvrir la chambre de ponte pour produire l’obscurité et laisser reposer pendant 20 minutes.
ATTENTION : Évitez de déplacer le pot de ponte une fois que les œufs ont été pondus pour éviter l’échouage et la dessiccation des œufs. - Détachez soigneusement le tube en polypropylène de 50 mL du pot de ponte, tout en veillant à ne pas relâcher les moustiques. Les œufs blancs doivent être visibles. Vérifier qu’il y en a eu suffisamment pour des raisons proscrites. Répétez l’opération si nécessaire.
- Couvrir le pot (pour protéger la poussière) et laisser les œufs mûrir jusqu’au stade de développement d’intérêt.
- Utilisez un pinceau à détails fins pour ramasser les œufs du pot et placez-les sur l’eau dans un bloc de verre excavé de 40 mm2.

Figure 5 - Exemple d’une chambre de ponte (A) démontée pour mettre en évidence les composants et (B) assemblée. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
-
Fixation d’embryons
ATTENTION : Effectuer toutes les étapes de fixation (étape 3.3) dans une hotte en raison de l’utilisation de formaldéhyde.- Préparer la solution FAA comme décrit dans Kaiser et al. (2014)22. La FAA comprend 3,6 M de formaldéhyde, 0,87 M d’acide acétique et 8,5 M d’éthanol absolu composé en volume avec de l’eau distillée (dH2O).
- Pour 10 mL de FAA, combiner 2,68 mL de formaldéhyde 13,42 M, 4,96 mL d’éthanol 17,14 M et 0,5 mL d’acide acétique 17,4 M avec 1,86 mL de H2O distillé. Le fixateur peut être conservé pendant au moins 3 mois dans un récipient en verre hermétiquement fermé, conservé dans un placard à produits chimiques désigné.
- Retirez soigneusement l’eau du bloc de verre avec une micropipette et couvrez les œufs dans 500 μL de FAA et oscillez doucement (~ 25 TR / min) sur un agitateur orbital à température ambiante pendant 30 minutes. Aucun changement de couleur n’est visible à ce stade.
- Rincer abondamment les œufs à l’eau distillée. Effectuez un rinçage 15 fois pour éliminer toute trace de formaldéhyde. À l’aide d’une micropipette de 1000 μL, ajoutez puis retirez 1 mL de dH2O à la fois en veillant à ne pas endommager les œufs en le faisant.
- Entreposer les eaux usées provenant des rinçages dans un contenant de formaldéhyde désigné pour les éliminer conformément aux directives de sécurité.
- À ce stade, les œufs fixes peuvent être conservés à 4 °C pendant la nuit dans de l’eau pour les garder hydratés.
- Préparer la solution FAA comme décrit dans Kaiser et al. (2014)22. La FAA comprend 3,6 M de formaldéhyde, 0,87 M d’acide acétique et 8,5 M d’éthanol absolu composé en volume avec de l’eau distillée (dH2O).
-
Blanchiment d’embryons
ATTENTION : Effectuer toutes les étapes de blanchiment (étape 4) dans une hotte en raison de la libération potentielle de chlore gazeux lorsque l’hypochlorite de sodium et l’acide acétique sont combinés.- Préparer une solution de blanchiment (solution de Trpiš - décrite dans Trpiš (1970)21 et modifiée selon Kaiser et al. (2014)22). La solution de Trpiš est constituée d’hypochlorite de sodium 0,59 M et d’acide acétique 0,35 M dissous dans du H2O distillé.
- Pour un volume de 10 mL de solution de Trpiš, combiner 2,68 mL d’hypochlorite de sodium de 2,2 M et 0,2 mL d’acide acétique de 17,4 M avec 7,12 mL de H2O distillé.
REMARQUE: La solution de Trpiš peut être stockée pendant au moins 3 mois dans un récipient en verre hermétiquement fermé et conservée dans un placard à produits chimiques sécurisé. La solution peut devoir être vortexée après le stockage et doit toujours être ouverte dans une hotte en cas de rejet de chlore gazeux.
- Pour un volume de 10 mL de solution de Trpiš, combiner 2,68 mL d’hypochlorite de sodium de 2,2 M et 0,2 mL d’acide acétique de 17,4 M avec 7,12 mL de H2O distillé.
- Couvrir les œufs fixes avec 1 mL de solution de Trpiš et incuber à température ambiante pendant 30 minutes. Les œufs commenceront à développer des taches pâles après environ 5 minutes d’incubation, atteignant finalement une couleur blanche laiteuse une fois éliminés.
- Rincer les œufs comme à l’étape 3.3.3 pour enlever la solution de Trpiš.
- Entreposez les eaux usées dans un conteneur à déchets désigné et éliminez-les avec l’excès d’eau dans le drain.
- Préparer une solution de blanchiment (solution de Trpiš - décrite dans Trpiš (1970)21 et modifiée selon Kaiser et al. (2014)22). La solution de Trpiš est constituée d’hypochlorite de sodium 0,59 M et d’acide acétique 0,35 M dissous dans du H2O distillé.
-
Stockage
- Conserver dans 500 μL de dH2O et conserver entre 2 et 8 °C pendant quelques jours. Retirez soigneusement la majeure partie de l’eau avant de regarder et d’imager la masse, mais évitez la dessiccation des œufs en laissant un petit volume d’eau dans le verre de la montre. Cela ne perturbera pas la photographie des œufs. Les œufs individuels peuvent être placés sur une lame de microscope pour une imagerie à grossissement plus élevé.
Résultats
L’expression 3xP3 d’eYFP, dsRed et eCFP fournit une identification fiable et facilement reconnaissable des individus possédant les gènes marqueurs produisant l’expression dans les yeux et les ganglions ventraux des nymphes d’An. gambiae (Figure 3). La morphologie différentielle observée dans les organes génitaux externes mâles et femelles utilisés pour le sexage et un exemple de pupe non identifiable sont mis en évidence à la figure 4
Discussion
Comprendre la fonction des gènes des moustiques est essentiel pour développer de nouvelles approches pour contrôler les anophèles et avoir un impact sur la transmission du paludisme. Le système GAL4-UAS décrit est un système polyvalent et puissant pour l’analyse fonctionnelle des gènes candidats et, à ce jour, nous avons utilisé le système pour examiner la base génétique de la résistance aux insecticides17 et de la production d’hydrocarbures cuticulaire...
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Nous remercions le financement de LSTM et IVCC (Adriana Adolfi), BBSRC (New Investigator Award (AL), MRC (phD studentship to BCP:MR/P016197/1), Wellcome (Sir Henry Wellcome Postdoctoral fellowship to LG: 215894/Z/19/Z) qui ont intégré l’analyse Gal4UAS dans les propositions.
matériels
| Name | Company | Catalog Number | Comments |
| 100 x 15 mm plastic Petri dish | SLS | 2175546 | Pack of 10 |
| 1000 µL Gilson Pipette | Gilson | F144059P | |
| 20/25 mL Universal Tubes | Starlab | E1412-3020 | Pack of 400 |
| 3 mL Pasteur Pipettes | SLS | G612398 | Greiner Pasteur pipette 3 mL sterile individually wrapped |
| 50 mL Falcon Tubes | Fisher Scientific | 11512303 | |
| Absolute Ethanol | Fisher Scientific | BP2818-500 | 500 mL |
| Acetic Acid | SLS | 45726-1L-F | 1 L |
| Cages | SLS | E6099 | 30x30x30 with screen port |
| Fine Paint Brushes | Amazon | UKDPB66 | KOLAMOON 9 Pieces Detail Painting Brush Set Miniture Brushes for Watercolor, Acrylic Painting, Oil Painting (Wine Red) |
| Fish food | Amazon | Tetra Min Fish Food, Complete Food for All Tropical Fish for Health, Colour and Vitality, 10 L | |
| Formaldehyde Solution | Sigma Aldrich | F8775 | |
| Mouth Aspirator | John Hock | 612 | |
| Pond Salt | Amazon | Blagdon Guardian Pond Tonic Salt, for Fish Health, Water Quality, General Tonic, pH Buffer, 9.08 kg, treats 9,092 L | |
| Pupae Pots | Cater4you | SP8OZ | 250 pots with lids |
| Small Plastic Buckets | Amazon | 2.5 L White Plastic Pail Complete with White Lid (Pack of 10) | |
| Sodium Hypochlorite | Fisher Scientific | S25552 |
Références
- Brand, A. H., Perimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 118 (2), 401-415 (1993).
- Duffy, J. B. GAL4 system in drosophila: A fly geneticist's swiss army knife. Journal of Genetics and Development. 34 (1-2), 1-15 (2002).
- Dow, J. A. . ELS. , (2012).
- Edi, C. V., et al. CYP6 P450 Enzymes and ACE-1 Duplication Produce Extreme and Multiple Insecticide Resistance in the Malaria Mosquito Anopheles gambiae. PLoS Genetics. 10 (3), 1004236 (2014).
- Daborn, P. J., et al. Using Drosophila melanogaster to validate metabolism-based insecticide resistance from insect pests. Insect Biochemistry and Molecular Biology. 42 (12), 918-924 (2012).
- Riveron, J. M., et al. Genome-wide transcription and functional analyses reveal heterogeneous molecular mechanisms driving pyrethroids resistance in the major malaria vector Anopheles funestus across Africa. Genes Genomes Genetics. 7 (6), 1819-1832 (2017).
- Riveron, J. M., et al. A single mutation in the GSTe2 gene allows tracking of metabolically based insecticide resistance in a major malaria vector. Genome Biology. 15 (2), (2014).
- Lynd, A., Lycett, G. J. Development of the Bi-Partite Gal4-UAS System in the African Malaria Mosquito, Anopheles gambiae. PLoS ONE. 7 (2), 31552 (2012).
- Lynd, A., Lycett, G. J. Optimization of the Gal4-UAS system in an Anopheles gambiae cell line. Insect Molecular Biology. 20 (5), 599-608 (2011).
- Adolfi, A., Pondeville, E., Lynd, A., Bourgouin, C., Lycett, G. J. Multi-tissue GAL4-mediated gene expression in all Anopheles gambiae life stages using an endogenous polyubiquitin promoter. Insect Biochemistry and Molecular Biology. 96, 1-9 (2018).
- Kokoza, V. A., Raikhel, A. A. Targeted gene expression in the transgenic Aedes aegypti using the binary Gal4-UAS system. Insect Biochemistry and Molecular Biology. 41, 637-644 (2011).
- O'Brochta, D. A., Pilitt, K. L., Harrell, R. A., Aluvihare, C., Alford, R. T. Gal4-based Enhancer-Trapping in the Malaria Mosquito Anopheles stephensi. Genes Genomes Genetics. 2, 21305-21315 (2012).
- Zhao, B., et al. Regulation of the Gut-Specific Carboxypeptidase: A Study Using the Binary Gal4/UAS System in the Mosquito Aedes Aegypti. Insect Biochemistry and Molecular Biology. 54, 1-10 (2014).
- Imamura, M., et al. Targeted Gene Expression Using the GAL4/UAS System in the Silkworm Bombyx mori. Genetics. 165 (3), 1329-1340 (2003).
- Lynd, A., et al. Development of a functional genetic tool for Anopheles gambiae oenocyte characterisation: application to cuticular hydrocarbon synthesis. bioRxiv. , (2019).
- Pondeville, E., et al. Hemocyte-targeted gene expression in the female malaria mosquito using the hemolectin promoter from Drosophila. Insect Biochemistry and Molecular Biology. 120, 103339 (2020).
- Adolfi, A., et al. Functional genetic validation of key genes conferring insecticide resistance in the major African malaria vector, Anopheles gambiae. Proceedings of the National Academy of Sciences of the United States of America. 116 (51), 25764-25772 (2019).
- Pondeville, E., et al. Efficient integrase-mediated site-specific germline transformation of Anopheles gambiae. Nature Protocols. 9 (7), 1698-1712 (2014).
- Horn, C., Schmid, B. G. M., Pogoda, F. S., Wimmer, E. A. Fluorescent transformation markers for insect transgenesis. Insect Biochemistry and Molecular Biology. 32, 1221-1235 (2002).
- Clements, A. . A. Biology of Mosquitoes, Volume 1: Development, Nutrition and Reproduction. 1, (1992).
- Trpiš, M. A new bleaching and decalcifying method for general use in zoology. Canadian Journal of Zoology. 48, 892-893 (1970).
- Kaiser, M. L., Duncan, F. D., Brooke, B. D. Embryonic Development and Rates of Metabolic Activity in Early and Late Hatching Eggs of the Major Malaria Vector Anopheles gambiae. PLoS ONE. 9 (12), 114381 (2014).
- Grigoraki, L., Grau-Bové, X., Yates, H. C., Lycett, G. J., Ranson, H. Isolation and transcriptomic analysis of Anopheles gambiae oenocytes enables the delineation of hydrocarbon biosynthesis. eLife. 9, 58019 (2020).
- Xiao, Y. -. H., Yin, M. -. H., Hou, L., Pei, Y. Direct amplification of intron-containing hairpin RNA construct from genomic DNA. BioTechniques. 41 (5), 548-552 (2006).
- Livak, K. J. Organization and Mapping of a Sequence on the Drosophila melanogaster X and Y Chromosomes That Is Transcribed during Spermatogenesis. Genetics. 107 (4), 611-634 (1984).
- MR4, CDC, NEI & beiResources. . The MR4 Methods in Anopheles Research Laboratory Manual. 5th Edition. , (2015).
- Sik Lee, Y., Carthew, R. W. Making a better RNAi vector for Drosophila: use of intron spacers. Methods. 30 (4), 322-329 (2003).
- Cha-aim, K., Hoshida, H., Fukunaga, T., Akada, R., Peccoud, J. . Gene Synthesis: Methods and Protocols. , 97-110 (2012).
- Cavener, D. R. Comparison of the consensus sequence flanking translational start sites in Drosophila and vertebrates. Nucleic Acids Research. 15 (4), 1353-1361 (1987).
- Wang, Y., Wang, F., Wang, R., Zhao, P., Xia, Q. 2A self-cleaving peptide-based multi-gene expression system in the silkworm Bombyx mori. Scientific Reports. 5, (2015).
- Galizi, R., et al. A synthetic sex ratio distortion system for the control of the human malaria mosquito. Nature Communications. 5, 3977 (2014).
- Kondo, S., et al. Neurochemical organisation of the Drosophila Brain Visualised by Endogenously Tagged Neurotransmitter Receptors. Cell Reports. 30 (1), 284-297 (2020).
- Lee, P. -. T., et al. A gene-specific T2A-GAL4 library for Drosophila. eLife. 7, 35574 (2018).
- Marois, E., et al. High-throughput sorting of mosquito larvae for laboratory studies and for future vector control interventions. Malaria Journal. 11, 302 (2012).
- Crawford, J. E., et al. Efficient production of male Wolbachia-infected Aedes aegypti mosquitoes enables large-scale suppression of wild populations. Nature Biotechnology. 38 (4), 482-492 (2020).
- Goltsev, Y., et al. Developmental and evolutionary basis for drought tolerance of the Anopheles gambiae embryo. Developmental Biology. 330 (2), 462-470 (2009).
- Rezende, G. L., et al. Embryonic desiccation resistance in Aedes aegypti: presumptive role of the chitinized Serosal Cuticle. BMC Developmental Biology. 8 (1), 82 (2008).
- Vargas, H. C. M., Farnesi, L. C., Martins, A. J., Valle, D., Rezende, G. L. Serosal cuticle formation and distinct degrees of desiccation resistance in embryos of the mosquito vectors Aedes aegypti, Anopheles aquasalis and Culex quinquefasciatus. Journal of Insect Physiology. 62, 54-60 (2014).
- Chang, C. -. H., et al. The non-canonical Notch signaling is essential for the control of fertility in Aedes aegypti. PLOS Neglected Tropical Diseases. 12 (3), 0006307 (2018).
- Clemons, A., Flannery, E., Kast, K., Severson, D., Duman-Scheel, M. Immunohistochemical Analysis of Protein Expression during Aedes aegypti Development. Spring Harbor Protocols. 10, 1-4 (2010).
- Juhn, J., James, A. A. Hybridization in situ of Salivary Glands, Ovaries and Embryos of Vector Mosquitoes. Journal of Visualized Experiments. , e3709 (2012).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.