
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Utilizzo del sistema GAL4-UAS per la genetica funzionale in Anopheles gambiae
In questo articolo
Riepilogo
Il sistema bipartito GAL4-UAS è uno strumento versatile per la modifica dell'espressione genica in modo spaziotemporale controllato che consente l'analisi genetica funzionale in Anopheles gambiae. Le procedure descritte per l'utilizzo di questo sistema sono una strategia di clonazione semi-standardizzata, sexing e screening delle pupe per marcatori proteici fluorescenti e fissazione dell'embrione.
Abstract
Il sistema bipartito GAL4-UAS è uno strumento versatile e potente per l'analisi genetica funzionale. L'essenza del sistema è quella di incrociare linee transgeniche "driver" che esprimono il fattore di trascrizione del lievito GAL4 in modo specifico per il tessuto, con linee transgeniche "responder" portatrici di un costrutto di interferenza gene/RNA candidato la cui espressione è controllata da Upstream Activation Sequences (UAS) che legano GAL4. Nella progenie che ne consegue, il gene o il costrutto di silenziamento è quindi espresso in modo spaziotemporale prescritto, consentendo di dosare i fenotipi risultanti e di dedurre la funzione genica. Il sistema binario consente flessibilità negli approcci sperimentali per lo screening dei fenotipi generati dall'espressione transgenica in più modelli tessuto-specifici, anche se sono indotti gravi costi di fitness. Abbiamo adattato questo sistema per Anopheles gambiae, il principale vettore di malaria in Africa.
In questo articolo vengono fornite alcune delle procedure comuni utilizzate durante l'analisi GAL4-UAS. Descriviamo le linee An. gambiae GAL4-UAS già generate, così come la clonazione di nuovi costrutti di responder per l'upregulation e il knockdown RNAi. Specifichiamo una guida passo passo per il sexing delle pupe di zanzara per stabilire incroci genetici, che include anche lo screening della progenie per seguire l'ereditarietà di marcatori genetici fluorescenti che etichettano le inserzioni del conducente e del soccorritore. Presentiamo anche un protocollo per la rimozione degli embrioni di An. gambiae per studiare lo sviluppo embrionale. Infine, introduciamo potenziali adattamenti del metodo per generare linee driver attraverso l'inserimento CRISPR/Cas9 di GAL4 a valle dei geni bersaglio.
Introduzione
Il sistema bipartito GAL4-UAS è il cavallo di battaglia della caratterizzazione funzionale dei geni nell'organismo modello di insetto Drosophila melanogaster1,2,3. Per utilizzare il sistema GAL4-UAS, le linee driver transgeniche, che esprimono il fattore di trascrizione del lievito GAL4 sotto il controllo di una sequenza regolatoria, vengono incrociate con linee di responder portatrici di un costrutto di genico di interesse o di interferenza dell'RNA (RNAi) controllato da una sequenza di attivazione a monte (UAS) riconosciuta da GAL4. La progenie di questa croce esprime il transgene di interesse per un pattern spaziotemporale dettato dal promotore che controlla l'espressione GAL4 (Figura 1). I fenotipi visualizzati dalla progenie di incroci driver-responder possono essere valutati per chiarire la funzione dei geni candidati. Sebbene D. melanogaster sia stato utilizzato per esaminare geni di altri organismi4,5,6,7, il sistema GAL4-UAS è stato ora adattato per l'uso in insetti di importanza medica e agricola per fornire analisi dirette nelle specie di interesse 8,9,10,11,12,13,14.
Nella zanzara africana della malaria, Anopheles gambiae, il sistema GAL4-UAS è stato testato per la prima volta dalla co-trasfezione della linea cellulare9. Sono stati analizzati più costrutti per l'efficienza in diverse combinazioni a coppie e hanno scoperto che 14 UAS ripetuti in tandem integrati con un piccolo introne artificiale (UAS-14i) mostravano la più ampia gamma di potenziale di attivazione quando utilizzati con un pannello di driver GAL4. Per dimostrare la funzionalità in vivo, questi costrutti sono stati poi utilizzati per creare due linee transgeniche Separate an. gambiae mediante la trasformazione piggyBac8: una linea driver che trasporta GAL4 guidata da un promotore specifico del midgut e una linea di risposta contenente sia la luciferasi che i geni della proteina fluorescente gialla potenziata (eYFP) sotto la regolazione delle sequenze UAS. L'attività della luciferasi specifica dell'intestino e la fluorescenza nella progenie indicavano che il sistema era efficiente in Anopheles. Da allora, sono state create linee driver che esprimono transgeni in altri tessuti importanti per la capacità vettoriale e la resistenza agli insetticidi, compresi gli enociti15 e gli emociti16, e in un modello quasi onnipresente10. Sono state inoltre generate numerose linee UAS per analizzare geni ritenuti coinvolti nel metabolismo e nel sequestro mediato dalla resistenza agli insetticidi, dalla sintesi di idrocarburi cuticolari e per etichettare fluorescentemente diversi tipi di cellule e tessuti (Tabella 1). Per le linee di risposta, l'integrazione site-directed del transgene viene ora eseguita mediante scambio di cassette di ricombinazione catalizzate ΦC31111117,18 per fissare il contesto genomico dei geni regolati UAS. In questo modo, l'espressione transgenica viene normalizzata per quanto riguarda la posizione di inserimento genomico, consentendo un confronto più accurato degli effetti fenotipici di diversi geni candidati.
Le linee di risposta create fino ad oggi sono progettate per esprimere il transgene a livelli elevati o per ridurre l'espressione genica attraverso l'interferenza dell'RNA (RNAi). Di solito i cloni di cDNA sono fusi alla sequenza UAS per generare plasmidi di espressione adatti, tuttavia anche le sequenze genomiche complete sono fattibili supponendo che non siano troppo grandi per la clonazione. Per generare costrutti di silenziamento, abbiamo usato tre diversi metodi per ottenere sequenze invertite in tandem adatte che formano dsRNA a forcina che stimola l'RNAi. Questi hanno incluso la PCR di fusione, la PCR asimmetrica e la sintesi commerciale di costrutti a forcina. Comune a ciascun metodo è l'inclusione di una sequenza di introni tra le sequenze invertite per fornire stabilità di clonazione. Sono stati sviluppati plasmidi responder in cui può essere inserito un costrutto gene di interesse/RNAi15. Questi plasmidi portano anche i siti ΦC31 attB richiesti per RMCE (descritti in Adolfi che accompagna l'articolo JoVE che descrive la tecnica RCME in dettaglio). I protocolli che coprono i passaggi importanti richiesti quando si seleziona la sequenza per l'inserimento in uno di questi plasmidi per la sovraespressione sono inclusi in questo manoscritto. Inoltre, vengono descritti e illustrati due protocolli per la creazione di costrutti a forcina RNAi.
Quando si creano nuove linee, l'identificazione di individui transgenici rari è fondamentale per riprodursi per stabilire e mantenere colonie transgeniche. Ancora più importante per il sistema GAL4-UAS c'è la necessità di distinguere le linee di risposta e driver per stabilire incroci e identificare la progenie individuale che trasporta entrambi i transgeni. Ciò si ottiene utilizzando diversi geni marcatori selezionabili dominanti collegati alle cassette del conducente e del risponditore. Più comunemente si tratta di geni marcatori fluorescenti che sono chiaramente distinguibili utilizzando filtri ottici (ad esempio, eYFP, eCFP, dsRed). È importante che i marcatori siano espressi in un modello spaziotemporale noto e affidabile in quanto ciò rende più facile l'identificazione delle anomalie e della contaminazione. L'espressione genica dei marcatori fluorescenti è regolata di routine dal promotore sintetico 3xP3, che causa l'espressione specifica dei gangli oculari e ventrali in tutte le fasi dello sviluppo di An. gambiae19. I marcatori fluorescenti controllati da 3xP3 sono inclusi in tutti i plasmidi di trasformazione descritti in questo articolo. Un protocollo che descrive in dettaglio i metodi comuni utilizzati per schermare le linee fluorescenti An. gambiae pupae GAL4-UAS è incluso qui.
Uno degli elementi chiave del sistema GAL4-UAS è la necessità di attraversare le linee di guida e di risposta con marcatura differenziale. Per fare questo maschio e femmine da ogni linea devono essere separati prima dell'accoppiamento. Gli adulti sono facilmente distinguibili dalla vista, tuttavia, per stabilire incroci genetici è sensato separare i sessi prima dell'emergere degli adulti per garantire che l'accoppiamento non si sia verificato. La differenza generale di dimensioni tra maschio e femmina An. gambiae pupae è troppo variabile per essere un metodo efficiente e affidabile di determinazione del sesso20. Invece chiare differenze morfologiche nei genitali esterni forniscono una base affidabile per il sexing in An. gambiae. In questo articolo, descriviamo un metodo affidabile per il sexing An. gambiae pupae per impostare croci appropriate.

Figura 1 - Rappresentazione diagrammatica del processo per l'utilizzo del sistema bipartito GAL4-UAS in Anopheles gambiae. (A) Sono rappresentati i componenti principali di un vettore di esempio (pSL-attB-UAS14-gyp[3xp3-eYFP]), che descrivono in dettaglio i siti di restrizione disponibili (EcoRI, NheI, XhoI e NcoI) all'interno dei siti di clonazione multipli che sono adatti all'uso per inserire il costrutto a forcina o la sequenza codificante per il gene di interesse. Anche la struttura della linea di attracco è raffigurata. (B) La fase di attraversamento è illustrata indicando l'uso di maschi dalla linea di guida (che trasportano il conducente GAL4 da un promotore di interesse e eCFP guidato dal promotore 3xP3) e femmine dalla linea di risposta (portatori del gene di interesse o del costrutto a forcina controllato da un promotore UAS e da un marcatore eYFP controllato dal promotore 3xP3). (C) Una rappresentazione diagrammatica dell'espressione trainante GAL4 del gene di interesse nella progenie della croce in B e un elenco di alcuni dei fenotipi tipici che vengono valutati. Abbreviazioni: Multiple Cloning Site (MCS), Recombinase mediated cassette exchange (RMCE), Upstream Activator Sequence (UAS), enhanced yellow fluorescent protein (eYFP), enhanced cyan fluorescent protein (eCFP). Fare clic qui per visualizzare una versione più grande di questa figura.
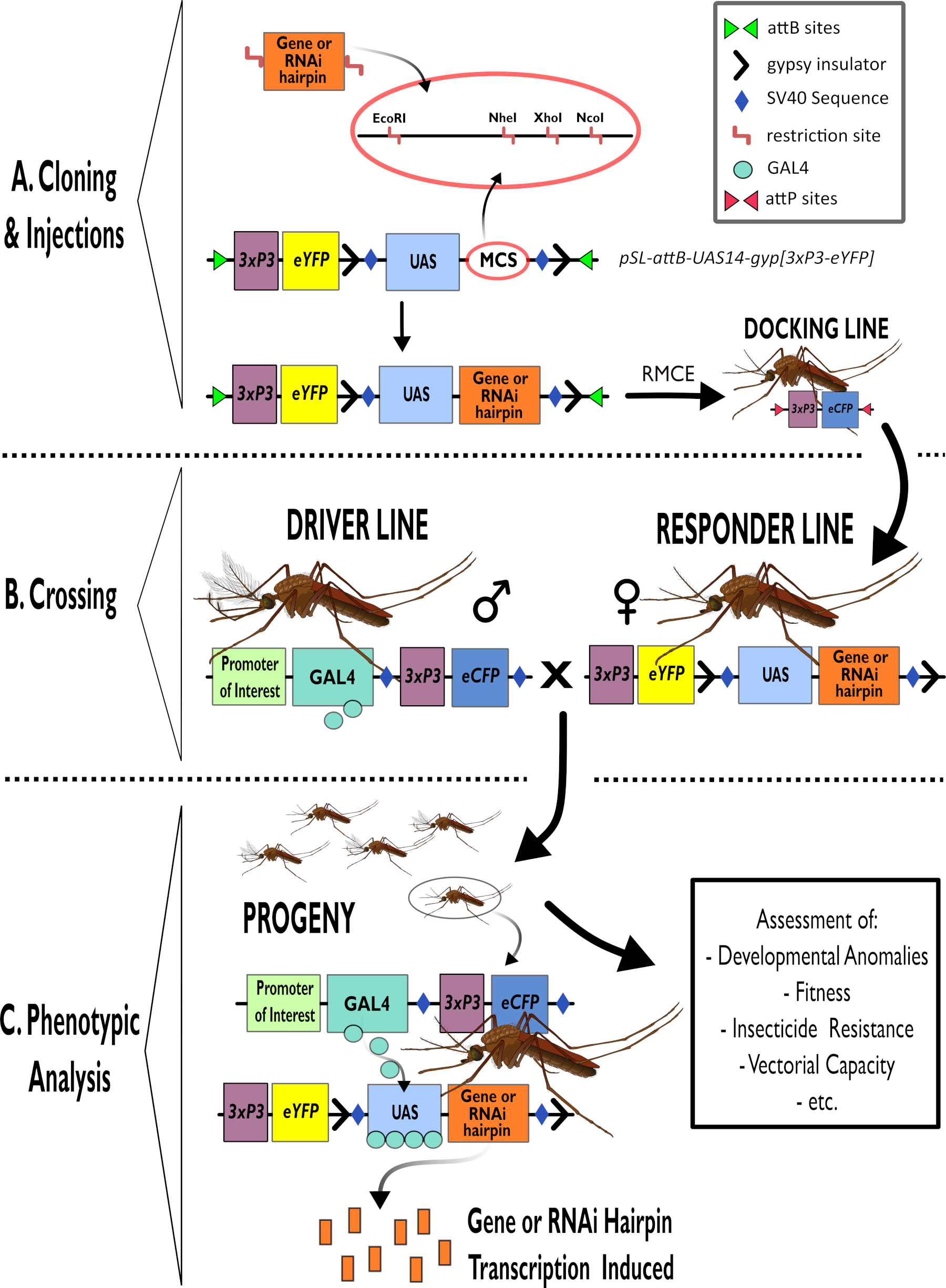{kind=link}
È l'uso delle croci che fornisce la natura bipartita del sistema GAL4-UAS, che presenta vantaggi distinti rispetto agli approcci più lineari. Ad esempio, è possibile valutare molte più combinazioni di linee driver e responder di quanto sarebbe fattibile se una nuova linea transgenica dovesse essere generata e mantenuta per ciascuna combinazione promotore/gene. Ancora più importante, consente l'analisi di geni che producono fenotipi letali o sterili quando la loro espressione è perturbata che sono difficili da creare / mantenere in un sistema lineare. Tali fenotipi letali possono manifestarsi in tutte le fasi dello sviluppo, a seconda della funzione genica e dell'espressione spaziotemporale, ma sono più spesso osservati durante lo sviluppo embrionale. Visualizzare lo sviluppo dell'embrione di zanzara richiede la pulizia del corion opaco che ricopre le uova. Seguendo i metodi descritti in Trpiš (1970)21 e Kaiser et al. (2014)22, descriviamo i protocolli che usiamo per fissare gli embrioni, pur mantenendo l'integrità strutturale, e lo sbiancamento per eliminare l'endocorion che consente la visualizzazione microscopica e l'imaging.
Protocollo
1. Progettazione e costruzione di costrutti UAS
- Progettazione e assemblaggio di vettori per l'espressione genica candidata
- Determinare la sequenza da utilizzare per l'upregulation del gene candidato.
- Sequenziare il cDNA/gDNA dal ceppo di interesse e confrontarlo con la sequenza pubblicata per verificarne l'identità e identificare potenziali SNP e siti di restrizione per il digest diagnostico.
- Assicurarsi che il primer in avanti utilizzato per l'amplificazione genica copra la sequenza nativa di Kozak e il codone di inizio, se del caso. Un primer con ~10 bp di legame a monte del codone di partenza comprenderà la sequenza di Kozak.
- Includere il codone di arresto nel frammento amplificato dal primer inverso nella maggior parte delle circostanze. Utilizzare sequenze di terminazione 3' fornite nei vettori plasmidici descritti, o amplificare da sequenze genomiche geniche candidate.
- Ordina sequenze commerciali con polarizzazione codone specifica, se lo desideri.
- Utilizzare procedure standard di subclonazione per inserire cassette geniche in vettori plasmidici UAS, ad esempio pSL-attB-UAS14-gyp[3xP3-eYFP]15 (Figura 1) sia per l'upregulation che per i costrutti RNAi.
- Produrre zanzare transgeniche create utilizzando la ricombinazione mediata dalla ricombinazione ΦC311 scambio di cassette10,17,18,23.
- Determinare la sequenza da utilizzare per l'upregulation del gene candidato.
- Creazione di costrutti a forcina RNAi: amplificazione a passo singolo mediante PCR asimmetrica15,24
- Estrarre il DNA genomico (gDNA) dalla femmina adulta An. gambiae che trasporta il gene candidato desiderato utilizzando il metodo Livak25.
- Progettare il primer in avanti per legarsi all'esone bersaglio ai 5' del frammento desiderato diretto verso l'introne vicino. Progettare l'estremità 3' di un primer a ponte per legarsi all'estremità dell'esone precedente per amplificare l'introne. L'estremità 5' è complementare a un piccolo frammento dell'esone bersaglio immediatamente dopo l'introne.
- Eseguire una reazione PCR asimmetrica come descritto in Xiao (2006)24 (Figura 2).
- Clonare il prodotto PCR purificato in un vettore adatto che trasporta il promotore UAS (ad esempio, pSL-attB-UAS14-gyp[3xP3-eYFP]15).
NOTA: gli enzimi all'interno del sito di clonazione multipla che sono appropriati per la clonazione pSL-attB-UAS14-gyp[3xP3-eYFP]15 e i passaggi successivi richiesti sono indicati nella Figura 1. Il digest enzimatico singolo è essenziale in quanto viene aggiunto un solo sito di restrizione. La defosforilazione del plasmide migliorerà l'efficienza della clonazione.
- Estrarre il DNA genomico (gDNA) dalla femmina adulta An. gambiae che trasporta il gene candidato desiderato utilizzando il metodo Livak25.
- Costruzione di costrutti a forcina RNAi: Fusion PCR di cDNA e gDNA15
- Estrarre il DNA genomico (gDNA) dalla femmina adulta An. gambiae che trasporta il gene candidato desiderato utilizzando il metodo Livak25.
- Includere gDNA in una reazione PCR per amplificare l'area target dell'esone e sequenze di introne insieme (Figura 2).
- Progettare l'estremità 3' del primer in avanti per legarsi alla sequenza di esoni bersaglio inverso per amplificare verso la sequenza di introne target e l'estremità 5' per trasportare un sito di restrizione per facilitare la clonazione.
- Progettare il primer inverso (1) per legarsi all'estremità 5' dell'introne e la sporgenza dell'estremità 5' porta le prime basi della sequenza in avanti dell'esone vicino. Questa sporgenza è utilizzata nella PCR di fusione.
- Purificare il prodotto di reazione desiderato.
- Estrarre l'RNA, rimuovere il DNA usando la DNasi e preparare il cDNA dalla femmina adulta An. gambiae che trasporta il gene candidato desiderato seguendo i protocolli del produttore.
- Utilizzare il cDNA in una reazione PCR per amplificare solo l'area target dell'esone (Figura 2).
- Progettare il primer in avanti (2) in modo che l'estremità 3' si leghi all'estremità 3' della sequenza esonica target complementare e l'estremità 5' del primer porti un sito di restrizione per l'uso nella clonazione.
NOTA: il primer anteriore di 1.3.1.2 può essere riutilizzato in questa seconda reazione. Tuttavia, ciò significa che un singolo digest enzimatico è essenziale. L'utilizzo di un secondo primer in avanti con un sito di restrizione diverso consentirà un doppio digest che può aumentare l'efficienza della clonazione. - Primer inverso di progettazione (2) - l'estremità 3' si lega all'estremità 5' dell'esone vicino amplificando l'esone bersaglio. L'estremità 5' si lega all'estremità 3' del filo in avanti degli introni. Questa sporgenza è utilizzata nella PCR di fusione.
- Purificare il prodotto di reazione desiderato.
- Progettare il primer in avanti (2) in modo che l'estremità 3' si leghi all'estremità 3' della sequenza esonica target complementare e l'estremità 5' del primer porti un sito di restrizione per l'uso nella clonazione.
- Includere i prodotti dei livelli 1.3.1 e 1.3.2 come modelli per una reazione PCR di fusione utilizzando concentrazioni standard con i primer Forward 1 e 2. Purificare il prodotto desiderato.
- Digerire il prodotto purificato per generare le sporgenze per la clonazione. Clonare in un vettore adatto a valle del promotore UAS. Enzimi appropriati per la clonazione di pSL-attB-UAS14-gyp[3xP3-eYFP]15 e i passaggi successivi richiesti sono indicati nella Figura 1.
- Estrarre il DNA genomico (gDNA) dalla femmina adulta An. gambiae che trasporta il gene candidato desiderato utilizzando il metodo Livak25.

Figura 2 - Rappresentazione diagrammatica della creazione di costrutti RNAi per l'inserimento in pSL-attB-UAS14-gyp[3xP3-eYFP] con due metodi: (A) PCR asimmetrica a passo singolo (adattato da Xiao. Y H et al (2006) e (B) PCR di fusione a più fasi. Fare clic qui per visualizzare una versione più grande di questa figura.
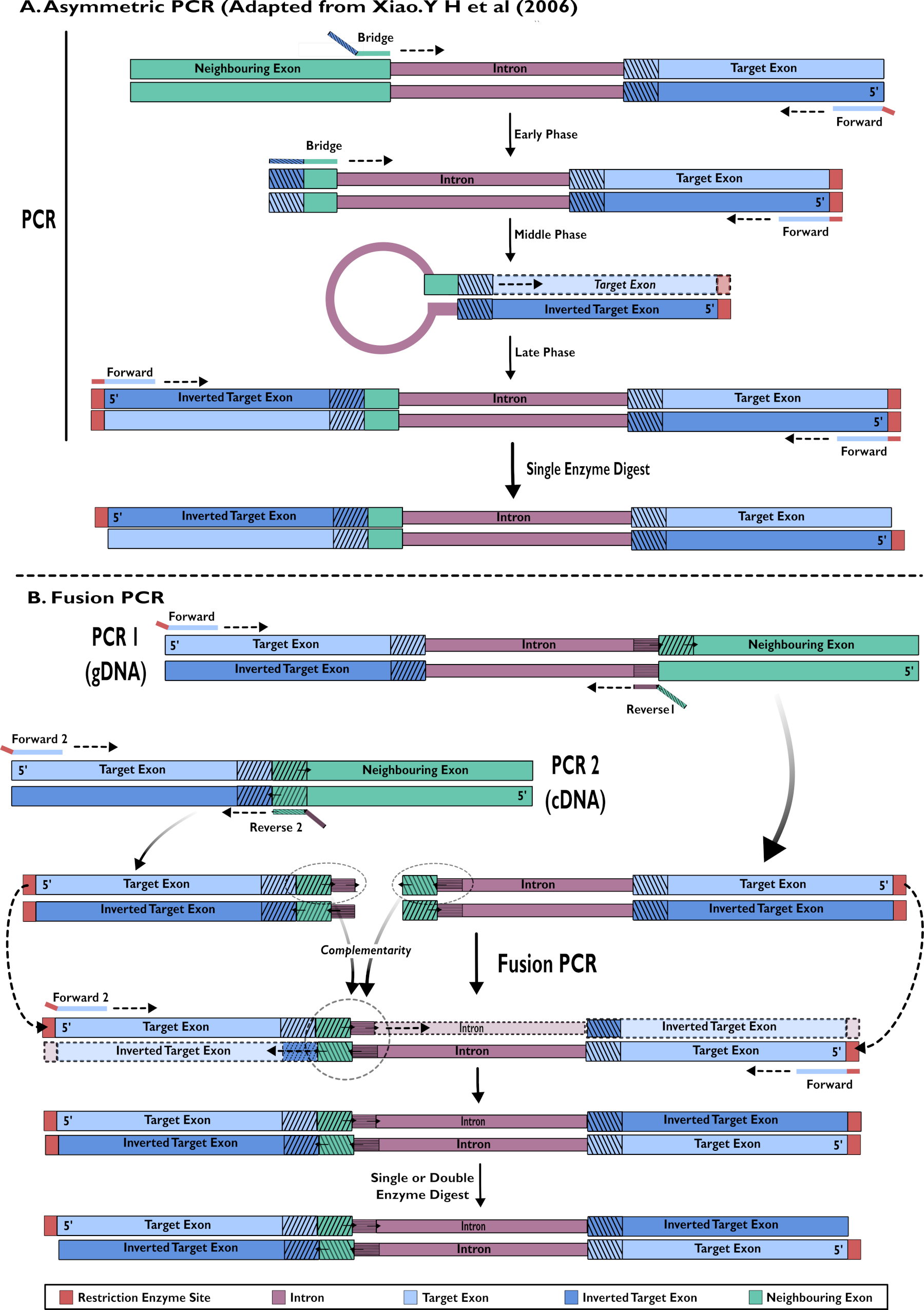{kind=link}
2. Screening delle pupe di An. gambiae
- Raccolta di pupe per la caratterizzazione microscopica
NOTA: In tutti questi protocolli l'acqua si riferisce all'acqua distillata integrata con lo 0,01% di sale di stagno.- Zanzare del Gambiae posteriore che utilizzano protocolli standard (ad esempio, MR426) allo stadio pupale.
ATTENZIONE: Fare attenzione a non ferire le pupe durante questo processo. - Raccogliere le pupe su una parabola piatta trasparente adatta all'uso con uno stereomicroscopio (ad esempio, una capsula di Petri in plastica da 100 x 15 mm, evitando i bordi).
NOTA: Per raccogliere le pupe utilizziamo una pipetta Pasteur in plastica da 3 ml con circa 10 mm di taglio dall'estremità per allargare l'estremità e prevenire lesioni alle zanzare. Lo screening e il sexing possono essere completati su individui, tuttavia, questo è molto lento. Si raccomanda di effettuare screening e sexing su gruppi di 50-200 pupe (la dimensione del gruppo possibile è limitata dalle dimensioni del piatto utilizzato ed è soggetta a preferenze personali). Se un gran numero viene sottoposto a screening, l'efficienza può essere aumentata allineando prima le pupe da 4 a 5 in profondità nelle linee e spostando le pupe bersaglio fuori da questa linea. - Usando una pipetta Pasteur, rimuovere con cura quasi tutta l'acqua intorno alle pupe. Lasciare abbastanza acqua intorno alle pupe in modo che siano effettivamente immotili ma possano essere spostate facilmente con un pennello fine. Se diventano difficili da spostare, aggiungi più acqua.
NOTA: Quando viene rimossa abbastanza acqua, le pupe giacciono su un fianco, consentendo la visualizzazione degli occhi per il rilevamento della fluorescenza e l'identificazione dei genitali dimorfici (Figura 4DE).
ATTENZIONE: Assicurarsi che le pupe non secchino. Se rimane solo un volume d'acqua molto piccolo, può ridursi ulteriormente con il calore della lampada del microscopio e quando diviso tra piscine di pupe. A volte è necessario aggiungere acqua aggiuntiva durante il processo utilizzando una pipetta Pasteur da 3 ml ai gruppi desiderati.
- Zanzare del Gambiae posteriore che utilizzano protocolli standard (ad esempio, MR426) allo stadio pupale.
- Identificazione di marcatori fluorescenti nelle pupe
NOTA: L'uso di uno stereoscopio a basso ingrandimento consente uno screening ad ampio campo, l'ordinamento può essere fatto su un microscopio composto invertito, ma deve essere fatto individualmente.- Quando si esegue lo screening per un marcatore fluorescente è innanzitutto fondamentale conoscere i modelli attesi di espressione ed ereditarietà. Considera quanto segue:
- Colore(i): determina quali filtri visualizzare l'espressione.
- Modello di espressione spazio-temporale: comprendi dove e in quale fase della vita ti aspetti di vedere l'espressione.
- Rapporto tra fenotipi diversi: stabilire quale percentuale della popolazione dovrebbe portare i marcatori di interesse.
- Condurre lo screening fluorescente al buio, poiché anche la scarsa illuminazione può interferire con la risoluzione della fluorescenza. Tuttavia, utilizzare una lampada accanto allo stereoscopio quando la luce è necessaria per altre manipolazioni.
ATTENZIONE: assicurarsi che l'area di lavoro attorno allo stereoscopio fluorescente sia libera prima di spegnere le luci. - Accendere la lampadina fluorescente e lasciare riscaldare per il periodo consigliato dal produttore (normalmente 10-15 min). Selezionare il filtro richiesto sullo stereoscopio fluorescente e verificare che sia visibile un fascio di luce colorato diretto al centro della piastra del palco. Se questo non è visibile o è molto debole, la lampadina fluorescente potrebbe non essersi completamente riscaldata, l'otturatore è chiuso o l'ottica del microscopio non è ben allineata.
- Usando la luce bianca, centra le pupe nel campo visivo e mettile a fuoco. Potrebbe essere necessario modificare questo ingrandimento quando si passa da un filtro all'altro a seconda dell'intensità della fluorescenza.
- L'uso di un pennello dettagliato fine assicura che le pupe esaminate non si sovrappongano.
- Spegni la luce bianca dello stereoscopio e usa la messa a fuoco fine per mettere a fuoco l'area delle pupe che trasportano il fenotipo di interesse. Il modello fluorescente dovrebbe essere visibile. Esempi di fluorescenza controllata dal promotore 3xP3 sono forniti nella Figura 3.
- Utilizzare l'ingrandimento più basso al quale il fenotipo fluorescente atteso può essere distinto in modo affidabile dagli individui senza fluorescene.
- Per i ceppi con fluorescenza brillante utilizzare anche una luce a campo luminoso a bassa intensità durante lo screening, se il segnale fluorescente è ancora chiaramente identificabile.
- Al termine dello screening primario, scansiona rapidamente le popolazioni sotto altri filtri per rilevare potenziali contaminazioni.
ATTENZIONE: Assicurarsi che vi sia una chiara distanza tra i gruppi di pupe selezionate per prevenire la contaminazione da movimento delle pupe. Tieni presente che le dimensioni dei gruppi cambieranno man mano che le pupe vengono sessuate e che le distanze possono apparire più grandi quando si guarda sotto ingrandimento. Prestare particolare attenzione quando le piscine non sono all'interno del campo visivo.
- Quando si esegue lo screening per un marcatore fluorescente è innanzitutto fondamentale conoscere i modelli attesi di espressione ed ereditarietà. Considera quanto segue:

Figura 3 - Anopheles gambiae pupae che esprimono marcatori fluorescenti guidati dal promotore 3xP3 (A) eYFP, (B) dsRed e (C) eCFP. Ingrandimento: A = 16X, B, C = 20X.
-
Sexing Pupae
- Raccogli pupe. Rimuovere l'acqua in eccesso, ma fornire sufficiente in modo che le pagaie anali si separino leggermente dai genitali per facilitare la visualizzazione e la caratterizzazione morfologica (Figura 4D,E).
- Se una pupa / e non è dalla loro parte, usa un pennello con dettagli fini per girare delicatamente la pupa e spostare le pagaie anali in modo che i genitali esterni possano essere identificati.
- Pupe separate basate su genitali esterni distintivi; i maschi hanno un lungo tubo che estrude dal segmento dorsale finale circa la metà della lunghezza delle pagaie anali (Figura 4B). I genitali esterni delle pupe femminili sono considerevolmente più corti e biforcati (Figura 4A).
NOTA: A volte, se il 4 ° esoscheletro larvale instar rimane attaccato o i genitali esterni sono danneggiati (Figura 4C), l'identificazione sicura del sesso è più difficile. Quando il sesso di una pupa non è chiaro, è buona norma scartarlo. Se l'individuo deve essere mantenuto, la pupa dovrebbe essere lasciata emergere in isolamento e il suo sesso determinato utilizzando caratteristiche morfologiche adulte. È probabile che se i suoi genitali sono danneggiati l'individuo potrebbe non accoppiarsi con successo. - Crea una piscina per ogni sesso all'estremità opposta del piatto alla piscina non sessuata, spostando le pupe identificate attraverso il piatto usando un pennello dettagliato. Etichettare la parte inferiore del piatto in cui verranno raccolte le due piscine per identificarle in seguito.
- Quando è richiesto sia il sexing che lo screening fluorescente, eseguire prima lo screening fluorescente, poiché è il processo più rapido dei due.

Figura 4 - Sexing Anopheles gambiae pupae. Pupe individuali che indicano i genitali esterni di (A) una femmina (B) un maschio e (C) un individuo che non può essere facilmente identificato a causa del distacco incompleto dell'esoscheletro larvale. Immagini ingrandite qui sotto evidenziando i genitali esterni. Pupe con ♀ (femmina) e ♂ (maschio) che indicano i genitali esterni delle pupe con (D) ~50% della pupa immersa nell'acqua e con (E) tutta l'acqua rimossa evidenziando la differenza nella facilità di visualizzazione dei genitali esterni. Ingrandimento: A, B, C = 40x, D, E = 30x. Fare clic qui per visualizzare una versione più grande di questa figura.
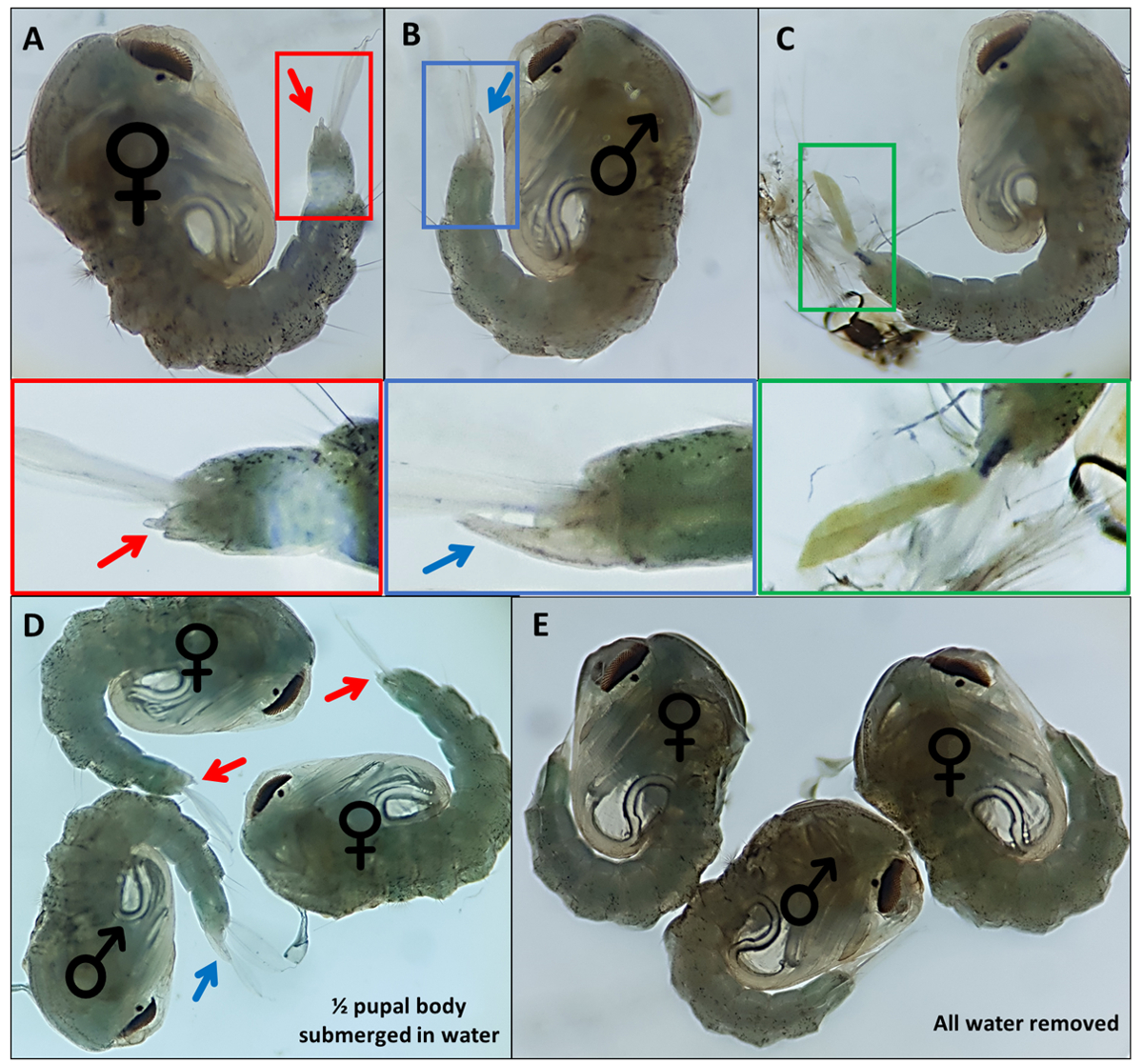{kind=link}
- Conferma del sesso da adulti
- Fino a quando non è stato dimostrato un tasso di errore molto basso, confermare il sesso delle pupe dalla morfologia adulta dopo l'emergenza. Separare le pupe sessuate in gruppi di 10 o meno in un tubo trasparente da 20 ml con pochi ml di acqua, sigillando con un batuffolo di cotone idrofilo, etichettato con il sesso previsto e consentire di emergere durante la notte.
NOTA: Poiché gli adulti vengono trasferiti la mattina seguente, non è necessario fornire cibo agli adulti emergenti. - Confermare il sesso degli adulti emersi utilizzando caratteristiche morfologiche il giorno seguente.
- Se alcuni maschi sono presenti nelle collezioni femminili, scartare le femmine, nel caso in cui l'accoppiamento sia già avvenuto.
- Se ci sono femmine presenti nella collezione maschile, rimuovere la femmina / i e tenere i maschi per l'incrocio.
- Fino a quando non è stato dimostrato un tasso di errore molto basso, confermare il sesso delle pupe dalla morfologia adulta dopo l'emergenza. Separare le pupe sessuate in gruppi di 10 o meno in un tubo trasparente da 20 ml con pochi ml di acqua, sigillando con un batuffolo di cotone idrofilo, etichettato con il sesso previsto e consentire di emergere durante la notte.
- Impostazione dei cross di sistema GAL4-UAS
- Aspirare il numero desiderato di adulti maschi e femmine dai tubi al punto 2.4 in una gabbia o in un piccolo secchio allestito nel modo standard per l'allevamento di An. gambiae.
NOTA: Fare attenzione a non danneggiare gli adulti durante questo trasferimento. - Utilizzare circa 50 femmine con un numero uguale di maschi, quando ~ 2000 adulti sono richiesti dalla progenie.
NOTA: Dove una croce deve essere alimentata più volte per generare più lotti fino a 200 di ogni sesso può essere impostato in gabbie di 30 cm x 30 cm x 30 cm. Quando solo un piccolo numero di femmine (<20) sono disponibili per la croce, aggiungiamo ~ 4 volte il numero di maschi per aumentare la probabilità di accoppiamento riuscito. - Il sangue alimenta le femmine incrociate e la progenie posteriore allo stadio appropriato, seguendo protocolli standard26, per condurre una valutazione fenotipica (ad esempio, resistenza agli insetticidi, capacità vettoriale e test dei costi di fitness).
- Laddove è probabile l'effetto materno dell'espressione transgenica, impostare incroci reciproci delle linee del conducente e del responder e testare il fenotipo atteso.
NOTA: Incroci che utilizzano popolazioni "eterozigoti" o miste di linee driver e responder, producono progenie con ciascuno dei 4 possibili genotipi. Ciò fornisce controlli wild type, solo UAS e gal4, nonché i transeterozigoti GAL4-UAS con cui analizzare il fenotipo. Se le popolazioni omozigoti sono incrociate, impostare ulteriori croci per fornire controlli appropriati per confrontare i fenotipi. La progenie deve essere sottoposta a screening come sopra separando la progenie che porta entrambi o solo uno dei due marcatori, così come i negativi, per la valutazione fenotipica.
- Aspirare il numero desiderato di adulti maschi e femmine dai tubi al punto 2.4 in una gabbia o in un piccolo secchio allestito nel modo standard per l'allevamento di An. gambiae.
- Stabilire popolazioni omozigoti da linee generate attraverso RCME che trasportano marcatori fluorescenti alternativi
NOTA: È essenziale che il marcatore fluorescente di entrambe le linee sia presente nella stessa posizione genomica e che siano completamente distinguibili.- Impostare un incrocio parentale di circa 200 adulti con un numero uguale di maschi marcati in modo differenziale di una linea e femmine dell'altra linea dopo lo screening per selezionare individui che mostrano fluorescenza e sesso corretti, come descritto sopra. Circa una settimana dopo il sangue alimenta la croce utilizzando protocolli stabiliti26.
- Allevare la progenie di F1 alle pupe utilizzando protocolli standard e raccogliere pupe come descritto in precedenza.
- Schermo per la fluorescenza selezionando quelli che portano entrambi i marcatori parentali (transeterozigoti). Crea un incrocio di F1 con queste pupe.
- Una settimana dopo, il sangue alimenta le femmine di F1 e la progenie posteriore allo stadio di pupa seguendo i protocolli standard.
- Scherma le pupe F2 selezionando quelle che visualizzano SOLO uno dei marcatori. Questi saranno omozigoti per l'inserimento. Solo il 25% della progenie sarà omozigote per ogni inserimento, quindi assicurati che sia allevata una progenie sufficiente per fornire una gabbia di riserva (400-500).
NOTA: La selezione della progenie transeterozigote deve essere del tutto rigorosa altrimenti il processo viene contaminato e l'omozigosi completa potrebbe non essere raggiunta. Ricontrolla tutta la progenie selezionata per l'intercross di F1.
- Impostare un incrocio parentale di circa 200 adulti con un numero uguale di maschi marcati in modo differenziale di una linea e femmine dell'altra linea dopo lo screening per selezionare individui che mostrano fluorescenza e sesso corretti, come descritto sopra. Circa una settimana dopo il sangue alimenta la croce utilizzando protocolli stabiliti26.
3. Protocollo di compensazione degli embrioni di An. gambiae
-
Alimentazione e manutenzione del sangue
- Zanzare del Gambiae posteriore agli adulti seguendo protocolli standard (ad esempio, MR4).
- Il sangue alimenta le femmine adulte di 5-7 giorni, assicurando che la maggior parte sia completamente ingorgata.
ATTENZIONE: In tutto questo protocollo lavorare rapidamente è essenziale per garantire che le uova non siano autorizzate a desiccare.
-
Deposizione indotta delle uova
- 3 giorni dopo l'alimentazione del sangue raccogliere le uova attraverso la deposizione indotta.
- Assemblare la camera di ovodeposizione.
- Riempire la pentola di ovodeposizione con acqua ad una profondità di circa 5 mm. Attaccare la pentola a un'estremità di un tubo di polipropilene da 50 ml, precedentemente tagliato con un seghetto in modo che entrambe le estremità siano aperte. (Usiamo un disco di plastica per una pentola (Figura 5); tuttavia, è possibile utilizzare il coperchio originale del tubo).
- Coprire l'altra estremità del tubo di polipropilene tagliato con materiale (tubo flessibile/collant) o sezioni di guanto di lattice fissate con una fascia elastica, in modo che gli adulti possano essere introdotti ma non possano sfuggire (Figura 5). Esistono altri progetti alternativi di camere di videposizione che possono essere utilizzati26.
- Introdurre con attenzione 10-15 femmine (sangue nutrito nel passaggio 3.1.2) nella camera di ovodeposizione. Coprire la camera di ovodeposizione per produrre oscurità e lasciare agire per 20 minuti.
ATTENZIONE: Evitare di spostare la pentola di deposizione delle uova una volta che le uova sono state deposte per evitare lo spiaggiamento e l'essiccazione delle uova. - Staccare con cura il tubo di polipropilene da 50 ml dal vaso di ovodeposizione, assicurandosi di non rilasciare le zanzare. Le uova bianche dovrebbero essere visibili. Verificare che ne siano stati deposti sufficienti per lo scopo proibito. Ripetere l'operazione se necessario.
- Coprire la pentola (per la protezione dalla polvere) e consentire alle uova di maturare fino alla fase di sviluppo di interesse.
- Usa un pennello dettagliato per raccogliere le uova dalla pentola e metterle sull'acqua in un blocco di vetro scavato di 40 mm2.

Figura 5 - Esempio di camera di ovodeposizione (A) smontata per evidenziare i componenti e (B) assemblata. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
-
Fissaggio dell'embrione
ATTENZIONE: Eseguire tutte le fasi di fissaggio (passaggio 3.3) in una cappa aspirante dovuta all'uso di formaldeide.- Preparare la soluzione FAA come descritto in Kaiser et al. (2014)22. La FAA comprende 3,6 milioni di formaldeide, 0,87 milioni di acido acetico e 8,5 milioni di etanolo assoluto prodotto in volume con acqua distillata (dH2O).
- Per 10 mL di FAA combinare 2,68 mL di 13,42 M di formaldeide, 4,96 mL di 17,14 M di etanolo e 0,5 mL di acido acetico 17,4 M con 1,86 mL di H2O distillato. Il fissativo può essere conservato per almeno 3 mesi in un contenitore di vetro ermeticamente sigillato, conservato in un armadio chimico designato.
- Rimuovere con cura l'acqua dal mattone di vetro con una micropipetta e coprire le uova in 500 μL di FAA e oscillare delicatamente (~ 25 RPM) su uno shaker orbitale a temperatura ambiente per 30 minuti. A questo punto non è visibile alcun cambiamento di colore.
- Risciacquare accuratamente le uova con acqua distillata. Eseguire il risciacquo 15 volte per rimuovere tutte le tracce di formaldeide. Utilizzando una micropipetta da 1000 μL, aggiungere e quindi rimuovere 1 mL di dH2O alla volta assicurandosi di non danneggiare le uova mentre lo si fa.
- Conservare le acque reflue dei risciacqui in un contenitore designato per scartare la formaldeide per lo smaltimento secondo le linee guida di sicurezza.
- A questo punto, le uova fisse possono essere conservate a 4 °C durante la notte in acqua per mantenerle idratate.
- Preparare la soluzione FAA come descritto in Kaiser et al. (2014)22. La FAA comprende 3,6 milioni di formaldeide, 0,87 milioni di acido acetico e 8,5 milioni di etanolo assoluto prodotto in volume con acqua distillata (dH2O).
-
Sbiancamento degli embrioni
ATTENZIONE: Eseguire tutte le fasi di sbiancamento (fase 4) in una cappa aspirante a causa del potenziale rilascio di gas di cloro quando l'ipoclorito di sodio e l'acido acetico sono combinati.- Preparare la soluzione sbiancante (soluzione di Trpiš - descritta in Trpiš (1970)21 e modificata secondo Kaiser et al. (2014)22). La soluzione di Trpiš è 0,59 M di ipoclorito di sodio e 0,35 M di acido acetico disciolto in H2O distillato.
- Per un volume di 10 mL di soluzione di Trpiš, combinare 2,68 mL di 2,2 M di ipoclorito di sodio e 0,2 mL di acido acetico 17,4 M con 7,12 mL di H2O distillato.
NOTA:La soluzione di Trpiš può essere conservata per almeno 3 mesi in un contenitore di vetro ermeticamente sigillato e conservata in un armadio chimico sicuro. La soluzione potrebbe dover essere vorticata dopo lo stoccaggio e deve essere sempre aperta in una cappa aspirante in caso di rilascio di gas di cloro.
- Per un volume di 10 mL di soluzione di Trpiš, combinare 2,68 mL di 2,2 M di ipoclorito di sodio e 0,2 mL di acido acetico 17,4 M con 7,12 mL di H2O distillato.
- Coprire le uova fisse con 1 mL di soluzione di Trpiš e incubare a temperatura ambiente per 30 minuti. Le uova inizieranno a sviluppare chiazze pallide dopo circa 5 minuti di incubazione, raggiungendo infine un colore bianco latte una volta eliminate.
- Risciacquare le uova come al punto 3.3.3 per rimuovere la soluzione di Trpiš.
- Conservare le acque reflue in un contenitore per rifiuti designato e smaltire con l'acqua in eccesso nello scarico.
- Preparare la soluzione sbiancante (soluzione di Trpiš - descritta in Trpiš (1970)21 e modificata secondo Kaiser et al. (2014)22). La soluzione di Trpiš è 0,59 M di ipoclorito di sodio e 0,35 M di acido acetico disciolto in H2O distillato.
-
Immagazzinamento
- Conservare in 500 μL di dH2O e conservare tra 2-8 °C per alcuni giorni. Rimuovere la maggior parte dell'acqua con attenzione prima della visualizzazione e dell'imaging sulla massa, ma evitare l'essiccazione delle uova lasciando un piccolo volume d'acqua nel vetro dell'orologio. Questo non disturberà fotografare le uova. Le singole uova possono essere posizionate sul vetrino del microscopio per immagini ad ingrandimento più elevato.
Risultati
L'espressione 3xP3 di eYFP, dsRed ed eCFP fornisce un'identificazione affidabile e facilmente distinguibile degli individui che possiedono i geni marcatori che producono espressione negli occhi e nei gangli ventrali di An. gambiae pupae (Figura 3). La morfologia differenziale osservata nei genitali esterni maschili e femminili utilizzati per il sexing e un esempio di pupe non identificabili sono evidenziati nella Figura 4. La rimozione di tutta...
Discussione
Comprendere la funzione del gene della zanzara è vitale per sviluppare nuovi approcci per controllare l'anofele e influenzare la trasmissione della malaria. Il sistema GAL4-UAS descritto è un sistema versatile e potente per l'analisi funzionale dei geni candidati e ad oggi abbiamo utilizzato il sistema per esaminare le basi genetiche della resistenza agli insetticidi17 e della produzione di idrocarburi cuticolari15,23, nonché per etichettare fluorescentemente diverse popolazioni di cel...
Divulgazioni
Gli autori non hanno nulla da rivelare.
Riconoscimenti
Riconosciamo con gratitudine i finanziamenti di LSTM e IVCC (Adriana Adolfi), BBSRC (New Investigator Award (AL), MRC (PhD studentship a BCP: MR / P016197 / 1), Wellcome (Sir Henry Wellcome Postdoctoral fellowship to LG: 215894 / Z / 19 / Z) che hanno incorporato l'analisi Gal4UAS nelle proposte.
Materiali
| Name | Company | Catalog Number | Comments |
| 100 x 15 mm plastic Petri dish | SLS | 2175546 | Pack of 10 |
| 1000 µL Gilson Pipette | Gilson | F144059P | |
| 20/25 mL Universal Tubes | Starlab | E1412-3020 | Pack of 400 |
| 3 mL Pasteur Pipettes | SLS | G612398 | Greiner Pasteur pipette 3 mL sterile individually wrapped |
| 50 mL Falcon Tubes | Fisher Scientific | 11512303 | |
| Absolute Ethanol | Fisher Scientific | BP2818-500 | 500 mL |
| Acetic Acid | SLS | 45726-1L-F | 1 L |
| Cages | SLS | E6099 | 30x30x30 with screen port |
| Fine Paint Brushes | Amazon | UKDPB66 | KOLAMOON 9 Pieces Detail Painting Brush Set Miniture Brushes for Watercolor, Acrylic Painting, Oil Painting (Wine Red) |
| Fish food | Amazon | Tetra Min Fish Food, Complete Food for All Tropical Fish for Health, Colour and Vitality, 10 L | |
| Formaldehyde Solution | Sigma Aldrich | F8775 | |
| Mouth Aspirator | John Hock | 612 | |
| Pond Salt | Amazon | Blagdon Guardian Pond Tonic Salt, for Fish Health, Water Quality, General Tonic, pH Buffer, 9.08 kg, treats 9,092 L | |
| Pupae Pots | Cater4you | SP8OZ | 250 pots with lids |
| Small Plastic Buckets | Amazon | 2.5 L White Plastic Pail Complete with White Lid (Pack of 10) | |
| Sodium Hypochlorite | Fisher Scientific | S25552 |
Riferimenti
- Brand, A. H., Perimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 118 (2), 401-415 (1993).
- Duffy, J. B. GAL4 system in drosophila: A fly geneticist's swiss army knife. Journal of Genetics and Development. 34 (1-2), 1-15 (2002).
- Dow, J. A. . ELS. , (2012).
- Edi, C. V., et al. CYP6 P450 Enzymes and ACE-1 Duplication Produce Extreme and Multiple Insecticide Resistance in the Malaria Mosquito Anopheles gambiae. PLoS Genetics. 10 (3), 1004236 (2014).
- Daborn, P. J., et al. Using Drosophila melanogaster to validate metabolism-based insecticide resistance from insect pests. Insect Biochemistry and Molecular Biology. 42 (12), 918-924 (2012).
- Riveron, J. M., et al. Genome-wide transcription and functional analyses reveal heterogeneous molecular mechanisms driving pyrethroids resistance in the major malaria vector Anopheles funestus across Africa. Genes Genomes Genetics. 7 (6), 1819-1832 (2017).
- Riveron, J. M., et al. A single mutation in the GSTe2 gene allows tracking of metabolically based insecticide resistance in a major malaria vector. Genome Biology. 15 (2), (2014).
- Lynd, A., Lycett, G. J. Development of the Bi-Partite Gal4-UAS System in the African Malaria Mosquito, Anopheles gambiae. PLoS ONE. 7 (2), 31552 (2012).
- Lynd, A., Lycett, G. J. Optimization of the Gal4-UAS system in an Anopheles gambiae cell line. Insect Molecular Biology. 20 (5), 599-608 (2011).
- Adolfi, A., Pondeville, E., Lynd, A., Bourgouin, C., Lycett, G. J. Multi-tissue GAL4-mediated gene expression in all Anopheles gambiae life stages using an endogenous polyubiquitin promoter. Insect Biochemistry and Molecular Biology. 96, 1-9 (2018).
- Kokoza, V. A., Raikhel, A. A. Targeted gene expression in the transgenic Aedes aegypti using the binary Gal4-UAS system. Insect Biochemistry and Molecular Biology. 41, 637-644 (2011).
- O'Brochta, D. A., Pilitt, K. L., Harrell, R. A., Aluvihare, C., Alford, R. T. Gal4-based Enhancer-Trapping in the Malaria Mosquito Anopheles stephensi. Genes Genomes Genetics. 2, 21305-21315 (2012).
- Zhao, B., et al. Regulation of the Gut-Specific Carboxypeptidase: A Study Using the Binary Gal4/UAS System in the Mosquito Aedes Aegypti. Insect Biochemistry and Molecular Biology. 54, 1-10 (2014).
- Imamura, M., et al. Targeted Gene Expression Using the GAL4/UAS System in the Silkworm Bombyx mori. Genetics. 165 (3), 1329-1340 (2003).
- Lynd, A., et al. Development of a functional genetic tool for Anopheles gambiae oenocyte characterisation: application to cuticular hydrocarbon synthesis. bioRxiv. , (2019).
- Pondeville, E., et al. Hemocyte-targeted gene expression in the female malaria mosquito using the hemolectin promoter from Drosophila. Insect Biochemistry and Molecular Biology. 120, 103339 (2020).
- Adolfi, A., et al. Functional genetic validation of key genes conferring insecticide resistance in the major African malaria vector, Anopheles gambiae. Proceedings of the National Academy of Sciences of the United States of America. 116 (51), 25764-25772 (2019).
- Pondeville, E., et al. Efficient integrase-mediated site-specific germline transformation of Anopheles gambiae. Nature Protocols. 9 (7), 1698-1712 (2014).
- Horn, C., Schmid, B. G. M., Pogoda, F. S., Wimmer, E. A. Fluorescent transformation markers for insect transgenesis. Insect Biochemistry and Molecular Biology. 32, 1221-1235 (2002).
- Clements, A. . A. Biology of Mosquitoes, Volume 1: Development, Nutrition and Reproduction. 1, (1992).
- Trpiš, M. A new bleaching and decalcifying method for general use in zoology. Canadian Journal of Zoology. 48, 892-893 (1970).
- Kaiser, M. L., Duncan, F. D., Brooke, B. D. Embryonic Development and Rates of Metabolic Activity in Early and Late Hatching Eggs of the Major Malaria Vector Anopheles gambiae. PLoS ONE. 9 (12), 114381 (2014).
- Grigoraki, L., Grau-Bové, X., Yates, H. C., Lycett, G. J., Ranson, H. Isolation and transcriptomic analysis of Anopheles gambiae oenocytes enables the delineation of hydrocarbon biosynthesis. eLife. 9, 58019 (2020).
- Xiao, Y. -. H., Yin, M. -. H., Hou, L., Pei, Y. Direct amplification of intron-containing hairpin RNA construct from genomic DNA. BioTechniques. 41 (5), 548-552 (2006).
- Livak, K. J. Organization and Mapping of a Sequence on the Drosophila melanogaster X and Y Chromosomes That Is Transcribed during Spermatogenesis. Genetics. 107 (4), 611-634 (1984).
- MR4, CDC, NEI & beiResources. . The MR4 Methods in Anopheles Research Laboratory Manual. 5th Edition. , (2015).
- Sik Lee, Y., Carthew, R. W. Making a better RNAi vector for Drosophila: use of intron spacers. Methods. 30 (4), 322-329 (2003).
- Cha-aim, K., Hoshida, H., Fukunaga, T., Akada, R., Peccoud, J. . Gene Synthesis: Methods and Protocols. , 97-110 (2012).
- Cavener, D. R. Comparison of the consensus sequence flanking translational start sites in Drosophila and vertebrates. Nucleic Acids Research. 15 (4), 1353-1361 (1987).
- Wang, Y., Wang, F., Wang, R., Zhao, P., Xia, Q. 2A self-cleaving peptide-based multi-gene expression system in the silkworm Bombyx mori. Scientific Reports. 5, (2015).
- Galizi, R., et al. A synthetic sex ratio distortion system for the control of the human malaria mosquito. Nature Communications. 5, 3977 (2014).
- Kondo, S., et al. Neurochemical organisation of the Drosophila Brain Visualised by Endogenously Tagged Neurotransmitter Receptors. Cell Reports. 30 (1), 284-297 (2020).
- Lee, P. -. T., et al. A gene-specific T2A-GAL4 library for Drosophila. eLife. 7, 35574 (2018).
- Marois, E., et al. High-throughput sorting of mosquito larvae for laboratory studies and for future vector control interventions. Malaria Journal. 11, 302 (2012).
- Crawford, J. E., et al. Efficient production of male Wolbachia-infected Aedes aegypti mosquitoes enables large-scale suppression of wild populations. Nature Biotechnology. 38 (4), 482-492 (2020).
- Goltsev, Y., et al. Developmental and evolutionary basis for drought tolerance of the Anopheles gambiae embryo. Developmental Biology. 330 (2), 462-470 (2009).
- Rezende, G. L., et al. Embryonic desiccation resistance in Aedes aegypti: presumptive role of the chitinized Serosal Cuticle. BMC Developmental Biology. 8 (1), 82 (2008).
- Vargas, H. C. M., Farnesi, L. C., Martins, A. J., Valle, D., Rezende, G. L. Serosal cuticle formation and distinct degrees of desiccation resistance in embryos of the mosquito vectors Aedes aegypti, Anopheles aquasalis and Culex quinquefasciatus. Journal of Insect Physiology. 62, 54-60 (2014).
- Chang, C. -. H., et al. The non-canonical Notch signaling is essential for the control of fertility in Aedes aegypti. PLOS Neglected Tropical Diseases. 12 (3), 0006307 (2018).
- Clemons, A., Flannery, E., Kast, K., Severson, D., Duman-Scheel, M. Immunohistochemical Analysis of Protein Expression during Aedes aegypti Development. Spring Harbor Protocols. 10, 1-4 (2010).
- Juhn, J., James, A. A. Hybridization in situ of Salivary Glands, Ovaries and Embryos of Vector Mosquitoes. Journal of Visualized Experiments. , e3709 (2012).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati