
Method Article
A 3D Cartographic Description of the Cell by Cryo Soft X-ray Tomography
In This Article
Summary
Here, a protocol describing the sample preparation and data collection steps required in cryo soft X-ray tomography (SXT) to image the ultrastructure of whole cryo-preserved cells at a resolution of 25 nm half pitch, is presented.
Abstract
Imaging techniques are fundamental in order to understand cell organization and machinery in biological research and the related fields. Among these techniques, cryo soft X-ray tomography (SXT) allows imaging whole cryo-preserved cells in the water window X-ray energy range (284-543 eV), in which carbon structures have intrinsically higher absorption than water, allowing the 3D reconstruction of the linear absorption coefficient of the material contained in each voxel. Quantitative structural information at the level of whole cells up to 10 µm thick is then achievable this way, with high throughput and spatial resolution down to 25-30 nm half-pitch. Cryo-SXT has proven itself relevant to current biomedical research, providing 3D information on cellular infection processes (virus, bacteria, or parasites), morphological changes due to diseases (such as recessive genetic diseases) and helping us understand drug action at the cellular level, or locating specific structures in the 3D cellular environment. In addition, by taking advantage of the tunable wavelength at synchrotron facilities, spectro-microscopy or its 3D counterpart, spectro-tomography, can also be used to image and quantify specific elements in the cell, such as calcium in biomineralization processes. Cryo-SXT provides complementary information to other biological imaging techniques such as electron microscopy, X-ray fluorescence or visible light fluorescence, and is generally used as a partner method for 2D or 3D correlative imaging at cryogenic conditions in order to link function, location, and morphology.
Introduction
Cryo-SXT can play a central role in biological imaging research as it provides 3D medium resolution (25-30 nm half pitch) volumes of hydrated whole cells1,2,3,4,5,6. In the water window energy range, between the carbon and the oxygen absorption K edges (4.4-2.3 nm), carbon-rich cellular structures absorb 10 times more than the oxygen-rich medium that permeates and surrounds them. In this energy range, vitrified cells up to 10 µm thickness can be imaged without the need for sectioning or staining, leading to quantitative high absorption contrast projections, which, combined with sample rotation capabilities, allow for the tomographic reconstruction of the cellular structure. Cryo-SXT fills a niche in terms of specimen dimensions and spatial resolution that is not easily accessible by any other imaging technique.
In brief, the absorption contrast of cryo-SXT is quantitative, as the attenuation of the photons through the specimen of thickness t obeys the Beer-Lambert law as follows:  , where I0 represents the incident intensity and µl the linear absorption coefficient, which depends on the wavelength λ and the imaginary part β of the refractive index of the specimen (
, where I0 represents the incident intensity and µl the linear absorption coefficient, which depends on the wavelength λ and the imaginary part β of the refractive index of the specimen ( ). The attenuation is a function of the biochemical composition and the thickness of the structures being imaged, with each biochemical component having a specific X-ray linear absorption coefficient µl (LAC). This means that each tomography voxel value depends on the chemical elements and their concentration in that voxel7. This allows for the natural discrimination of different organelles such as nuclei, nucleoli, lipid bodies or mitochondria, or different compaction states of chromatin solely based on their inherent LAC values reconstructed2,8,9.
). The attenuation is a function of the biochemical composition and the thickness of the structures being imaged, with each biochemical component having a specific X-ray linear absorption coefficient µl (LAC). This means that each tomography voxel value depends on the chemical elements and their concentration in that voxel7. This allows for the natural discrimination of different organelles such as nuclei, nucleoli, lipid bodies or mitochondria, or different compaction states of chromatin solely based on their inherent LAC values reconstructed2,8,9.
In addition, cryo-SXT is a high throughput technique with tomograms being collected in few minutes. This specifically enables mesoscale imaging of cell populations that can be captured at key time points such as division, differentiation, and apoptosis, but also at different response states such as those induced by chemical exposure to specific drug therapies or to pathogenic infections. Data collected at those key points will deliver 3D description of the system with a faithful record of the spatial organization of the different cellular organelles at those specific moments.
Usually, cryo-SXT is used in combination with other techniques following correlative approaches that allow locating specific features, events, or macromolecules within the 3D cellular environment4,10,11,12,13,14,15,16, or hard X-ray fluorescence data17,18. Correlative approaches at cryogenic conditions are of paramount importance in order to obtain the most complete and valuable picture of the system of interest. A succinct summary of the typical workflow at the Mistral (Alba) and B24 (Diamond) cryo-SXT beamlines is sketched in Figure 1.
Moreover, taking advantage of the wavelength tuning capability at synchrotron facilities, spectroscopic information can be obtained in addition to the structural one using the specific differential absorption of particular elements contained in the sample. An example of this would be the location of calcium in the study of biomineralization processes in cells19,20,21. By taking 2D images at different photon energies (spectra) or tomograms below and at the x-ray absorption edge of interest, the pixels or voxels containing the selected element can be identified. Spectra also permits differentiating chemical states (i.e., the evolution of amorphous calcium to hydroxyapatite as in the previous biomineralization example20). Quantification of different elements is possible in 2D and 3D. Spectroscopic imaging of vitrified cells is typically done in the water window, but is also possible at other energy ranges if the water content is low enough or if other sample preparation protocols, including dehydration, are used22. A detailed spectroscopy step-by-step protocol is beyond the focus of the protocol herein.
In what follows, the protocol focuses on briefly summarizing the major sample preparation steps, although each system might need individual refinement, followed by a detailed step-by-step data collection procedure for cryo soft X-ray tomography.
Protocol
1. Sample preparation
- Preparing the grid support
- Irradiate the grids with UV for 3 h with the carbon film facing upward for sterilization.
- Optional: In case of problems with cells not attaching to the grid, use one of the following steps.
- Hydrophilize the carbon support foil by plasma treatment of the grids to increase sample spreading and better cell adhesion.
- Place grids carbon side up in the glow discharge chamber equipment and expose the grid to the plasma for 30 s to 15 min (using Ar or/and O2) depending on the equipment.
- Functionalize the grids with Poly-L-lysine (PLL)
- Add individual drops of 60 µL PLL in a Petri dish and place the grid on top of the PLL drop with the carbon film facing down. Incubate for 30 min at 37 °C and blot the PLL using a filter paper.
- Functionalize the grids with fetal bovine serum (FBS).
- Submerge the grids in FBS overnight. Dip the grids in a buffer solution to wash and leave the grid on a filter paper to remove excess liquid.
- Hydrophilize the carbon support foil by plasma treatment of the grids to increase sample spreading and better cell adhesion.
- Growing adherent cells on grids
- Grow cells in a cell culture dish of 100 mm to reach 80%-90% confluency (Figure 2A).
- Seed 1-5 x 105 cells/mL (adjust value to system) on top of the Au grids in a P60 Petri dish (3 mL total).
NOTE: The addition of the cell suspension has to be done very carefully with the carbon film of the grid facing upward in a cell culture dish of 60 mm. Prepare several grids per condition, one condition per P60 Petri dish (Figure 2B). - Allow the cells to settle until the confluency on the grid reaches several cells (1 to 10 depending on the cell size) in each mesh square (depending on the cell line, this can take up to 24 h).
NOTE: Prior to freezing, the grids should be checked with a visible light microscope (VLM), evaluating the carbon film integrity as well as cell confluency in each grid (Figure 2C). Wait until the proper cell density on the grid is reached. If the grid is too confluent or the carbon foil is broken, start over.
- Deposition of cells in suspension on grids
- Pick a prepared Au or Cu grid using the tweezer from the plunge freezer (see section 1.6).
- Prepare a 1-5 x 105 cell suspension (optical density absorbance of 0.3 in case of bacteria) and add 4 µL of the prepared suspension to the grid.
- Incubate the grid with the drop for few minutes horizontally to allow deposition of the cells and then place the tweezer holding the grid inside the climate-controlled vitrification chamber set to the appropriate temperature and humidity conditions (step 1.6.1).
- Optional: fluorescent tagging of the samples
NOTE: It can be beneficial to fluorescently label some organelles of the sample. Depending on the interest, specific fluorophores or cells stably expressing a fluorescent protein or transfected for transient expression of a protein of interest can be used. This allows easy detection of cells using cryo-epifluorescence microscopy and aids finding cells of interest for subsequent X-ray imaging. In what follows, the protocol only details the use of fluorophores.- Prepare a working solution for the fluorophore (see manufacturer's recommendation) and follow the specific protocol.
- Add the fluorophores to the Petri dish containing the grids and mix gently.
- Leave to incubate in a dark environment and go directly to step 1.6 after the incubation is finished.
NOTE: It is important to be ready to vitrify once the incubation is finished to avoid unspecific tagging and consequently blurred fluorescent signal.
- Au nanoparticles (NPs) preparation for tomogram projection alignment
- Take one aliquot of 1mL of the Au fiducial stock solution (100 nm at Mistral or 250 nm at B24) and centrifuge at low speed (to avoid aggregation) for 1 min to allow the NPs to pellet.
NOTE: If possible, it is preferred to leave the fiducials to settle naturally overnight or more, to avoid aggregation. - Remove the supernatant. Immediately prior to freezing, re-suspend the NPs in 20 µL serum-free medium or buffer solution to obtain a homogeneous solution.
NOTE: Sonication and vortexing is recommended to help homogenize the solution.
- Take one aliquot of 1mL of the Au fiducial stock solution (100 nm at Mistral or 250 nm at B24) and centrifuge at low speed (to avoid aggregation) for 1 min to allow the NPs to pellet.
- Plunge-freezing grids
CAUTION: Liquid nitrogen can cause cold-burns and proper protective equipment should be worn (long lab coat, safety goggles, gloves, long pants, and closed shoes). Ethane is highly explosive and should be kept away from any sparks or open fire.- Follow the manufacturer's instructions to prepare and use the plunge freezer instrument.
NOTE: Humidity is usually set to 80%-90%; temperature will depend on cell type (yeast 30 °C maximum, mammalian cells 37 °C, insect cells 28 °C, etc.). - Take a grid with the mounting tweezers from the Petri dish by the rim, taking great care not to bend the grid and mount it on the plunge freezer device. Ensure that the cells face away from the blotting paper.
- Optional: wash the grid three times in buffer in the case of adherent cells.
- Add 1.5 µL of Au NP fiducials on top of the cells (through the hole on the right side of the chamber) and leave to settle for 30 s before blotting and plunging the grid.
NOTE: In the case of cells in suspension, add Au NP fiducials to the grid while it is still in horizontal position before mounting the tweezer and let them settle for 30 s. Blotting is crucial for high quality grids. Blotting distance needs to be calibrated before starting; flatness of blotting paper is important for reproducible blotting (follow instructions by the manufacturer). Blotting time needs to be assessed for each cell types. - Prepare several grids per condition. Transfer the grid and store at cryogenic temperatures to preserve vitrification.
- Follow the manufacturer's instructions to prepare and use the plunge freezer instrument.
- Screening grids with cryo visible light microscopy
- Transfer the grids under liquid nitrogen from the cryo-boxes to a standard cryo-cassette (three positions for 3 mm TEM grids) inside a pre-cooled cryo-stage (see instructions from the manufacturer).
- The cryo-cassette is placed onto the cryo-stage bridge and the cryo-stage is mounted on a wide field epifluorescence light microscope.
- Image the grids at -196.5 °C using a long working distance objective (10x, 50x, 100x) in order to locate suitable cells for cryo-SXT imaging and assess the grid quality (presence of thick ice, integrity of the carbon support film, presence of fluorescent signal, etc.).
- Localize the cells of interest using bright field or/and fluorescence imaging.
NOTE: At this stage, capture the images if a linked camera is present on the microscope. Use it for image correlation. - After screening, return the grids to the cryo-boxes in a liquid nitrogen storage dewar.
NOTE: If no good samples are found, repeat the sample preparation steps modifying any of the parameters. Usually, the parameters that require modification are blotting time, in case the ice is too thick or the confluency, if there are too many cells per mesh square.
2. Loading into the Transmission X-Ray Microscope (TXM)
- Cool the transfer chamber with liquid nitrogen until it reaches <100 K, cool the workstation (Figure 3A) and turn on the heater of the workstation rim. Wait until it stops boiling.
- Put the needed cryo-boxes containing grids into the corresponding locations in the workstation (Figure 3A). Pay attention to safely transfer them under cryo conditions.
- Load the selected grids into the sample holders previously cooled (Figure 3B); load the holders onto the shuttle and protect them with the covers (Figure 3C).
- Load the shuttle into the transfer chamber at <100 K and pump it down to low vacuum (Figure 3D).
- Attach the transfer chamber to the TXM (Figure 4A) and load the shuttle from the transfer chamber into the TXM following the vacuum procedure on the screen (Figure 4B).
- Once the shuttle is inside with the samples, the TXM robot arm can bring one sample holder at a time to the sample stage (Figure 4C).
3. Imaging using the TXM software
NOTE: Grids at the sample stage are first imaged using an on-line visible light microscope (VLM) to map the grid either in brightfield and/or fluorescence mode before being imaged with X-rays. Use the joystick icon corresponding to the Motion Control tab on the top left panel to open the Motion Control.
- Imaging the grid with the on-line VLM.
- Bright field mosaic acquisition
- Select the VLM camera (Magnifying lens > VLM) and turn on the VLM led source for bright field imaging (Microscope > Acquisition > Acquisition Settings > Source Settings and select Transmission).
- Rotate the sample to -60 degrees in order to face the VLM objective (Motion Control > Sample > Sample θ) and move the sample to the expected centered positions (Motion Control > Sample and change Sample X, Sample Y).
- While acquiring images in steps of 20 to 50 µm first to bring the grid roughly in focus (Microscope > Acquisition > Acquisition Settings > Acquisition Modes > Continuous > Start; Motion Control > Sample and change Sample Z).
- Refine the focus with smaller steps down to 5 µm until the cells and/or the holes of the carbon support film are in focus (Microscope > Acquisition > Acquisition Settings > Acquisition Modes > Continuous > Start; Motion Control > Sample and change Sample Z) and start the acquisition of a full mosaic map of the grid in bright field mode. Use the default values for the mosaic. (Microscope > Acquisition > Acquisition Settings > Acquisition Modes > Mosaic > Start).
NOTE: A mosaic map is made out of single images. The number of images (number of columns, rows, and the step size in X and Y) should be set to visualize the whole grid. The step size depends on the size of the field of view (FoV). The default values are used here. If the grid is not well centered in the X-Y plane with respect to the full mosaic FoV, stop the acquisition, correct the sample X and Y coordinates and repeat the acquisition.
- Fluorescence mode mosaic acquisition
- Turn off the VLM-led source for bright field imaging (Microscope > Acquisition > Acquisition Settings > Source Settings and de-select Transmission) and select the LED light source corresponding to the desired excitation wavelength (red, green, or blue) and the corresponding optical filter manually on the set-up.
- Refine the focus on the fluorescence image (Microscope > Acquisition > Acquisition Settings > Acquisition Modes > Continuous > Start; Motion Control > Sample and change Sample Z), and then acquire a mosaic map retaining the positional parameters (X and Y) from the bright field mosaic (Microscope > Acquisition > Acquisition Settings > Acquisition Modes > Mosaic > Start).
- Switch off the LED light-source
- Optional: Based on the bright field and fluorescence mosaic maps, annotate the regions of interest (ROI) identified previously (step 1.7.4) or new ROI (X-Y positions in the image).
- Bright field mosaic acquisition
- X-ray mosaic acquistion
- Select the CCD X-ray detector (Magnifying lens > select Pixis), bring the sample to 0 degree rotation (Motion Control > Sample and change Sample θ) and move the X-rays optics (Condenser and Zone Plate) to the aligned positions (Motion Control > Condenser > change Condenser z; Motion Control > Zone Plate and change Zone Plate Z).
NOTE: Cool down the CCD chip to -65 °C before imaging (Microscope > Camera temperature > Pixis and change set temperature to -65 °C and click on Apply). - Move the sample to the center of the mesh square of one of the selected ROI (Motion Control > Sample and change Sample X, Sample Y).
- Use binning 2 and exit slit at 5 µm to minimize irradiation and 1 s exposure at Mistral, and binning 1 and 60 µm at B24, adjust the focus using sample Z translation (Microscope > Acquisition > Acquisition Settings > Camera Settings and change binning; Motion Control > XS and change XS; Microscope > Acquisition > Acquisition Settings> Acquisition Modes > Continuous > Start; Motion Control > Sample and change Sample Z). Start in steps of 5 µm and refine it down to steps of 0.5 µm, until the cell or the carbon foil holes are well in focus.
- Acquire a mosaic map of the mesh square (Microscope > Acquisition > Acquisition Settings > Acquisition Mode > Mosaic > Start).
NOTE: Mosaic acquisition parameters can be determined in the same way as for the VLM mosaics (section 3.1.1.4). The step size depends on the magnification preselected by the beamline staff. - Move the sample to a Flat-Field (FF) position (an empty area within the grid, preferably a hole in the carbon support) (Motion Control > Sample and change Sample X, Sample Y).
- Set the exposure time to 1 s at Mistral and 0.5 s at B24 (Microscope > Acquisition > Acquisition Settings > Camera Settings and change Exposure Time) and acquire a single image (Microscope > Acquisition > Acquisition Modes > Single > Start).
- Normalize (divide) the acquired mosaic by the FF image to obtain the transmission (with values between 0 and 1) and save the normalized mosaic (click on the first right upper corner icon on the right side of the image to open the menu>choose Reference tab> click Single Reference and browse the specific FF).
- Select the CCD X-ray detector (Magnifying lens > select Pixis), bring the sample to 0 degree rotation (Motion Control > Sample and change Sample θ) and move the X-rays optics (Condenser and Zone Plate) to the aligned positions (Motion Control > Condenser > change Condenser z; Motion Control > Zone Plate and change Zone Plate Z).
- Preparing to collect an X-ray tilt-series
NOTE: Find the rotation axis for each region of interest being imaged.- Make a selection of areas within the mosaics to perform tomography, i.e., by placing a square shape (click on Tool on the left side of the image window) with the size of the FoV in the X-ray mosaic map.
NOTE: When selecting areas, consider the distance from the border (≥10 µm) and other cells, to avoid overlap of cells during rotation. In addition, check the condition of the cell (expected cell shape, ice thickness, success of vitrification, fiducial spread, etc.). - Sample alignment on the rotation axis
NOTE: The reported procedure is iterative. The iteration converges faster using ± 60° angles as maximum rotation angles (θM). If they are not accessible, use angles as close as possible to ± 60°.- Set the camera to binning 2, exposure time 1 s (Microscope > Acquisition > Acquisition Settings > Camera Settings and change Exposure Time; change Binning; Microscope > Acquisition > Acquisition Modes > Continuous > Start), set exit slit to 5 µm.
NOTE: Minimize the dose as much as possible by working with minimal aperture of exit slit. - With the rotation at 0°, move to the previously selected area using the sample X and sample Y translation and focus on the feature of the cell to put in the rotation axis using the sample Z translation (Motion Control > Sample and change Sample x; Sample y; Sample z).
- Rotate the sample to + θM (Motion Control > Sample and change Sample θ) and draw a line (L+) (click on the line tool button on the right side of the image window) on the feature of the cell to put on the rotation axis.
- Rotate to -θM (Motion Control > Sample and change Sample θ) and draw a line (L-) (click on the line tool button on the right side of the image window) on the feature of the cell you want to put on the rotation axis.
- While at +θM or -θM, use sample Z translation to move the selected feature to the center position between both lines (Motion Control > Sample and change Sample Z).
- Repeat the procedure from step 3.3.2.3 iteratively until a minimum distance L+ to L- is reached.
NOTE: The distance between the two lines L+ and L- should be smaller with respect to the previous iteration. - At sample θ = 0 (Motion Control > Sample and change Sample θ), move sample X twice the distance needed to put the selected feature at the center of both lines (Motion Control > Sample and change Sample X). Move the Zone Plate (ZP) X to bring the feature back to the center of the FoV (Motion Control > ZP and change ZP X). Do this step only once per grid.
- Re-optimize ZP Z position with respect to the new rotation axis by recording a ZP Z focal series (collections of images at different ZP Z positions, usually in steps of 0.3 µm) (Microscope > Acquisition > Acquisition Settings > Acquisition Modes > Focal Series > Start) and move the ZP Z to the position where the sample is in focus (Motion Control > Zone Plate and change Zone Plate z).
- Set the parameters for the tilt series acquisition.
NOTE: The maximum angular range is ultimately limited by the focal length of the ZP (±70 or ±65 degrees for a 40 nm or 25 nm ZP for a flat sample, respectively). Optimize the signal-to-noise ratio (S/N) and radiation damage to define the exposure time. For tomography, use different exposure times at different angular ranges. - Determine the highest rotation angular range, in case shadowing occurs before the maximum rotation angle is reached.
- Set the camera binning to 1 (Microscope > Acquisition > Acquisition Settings > Camera Settings and change Binning), open the exit slit to 15 µm at Mistral (Motion Control > XS and change XS) and set the rotation to 0° (Motion Control > Sample and change Sample θ).
- Acquire a single image with an exposure time of 1 s (Microscope > Acquisition > Acquisition Settings > Acquisition Modes > Single > Start) and estimate the exposure time required for each tilt angle of the tomography.
- Set the camera to binning 2, exposure time 1 s (Microscope > Acquisition > Acquisition Settings > Camera Settings and change Exposure Time; change Binning; Microscope > Acquisition > Acquisition Modes > Continuous > Start), set exit slit to 5 µm.
- Make a selection of areas within the mosaics to perform tomography, i.e., by placing a square shape (click on Tool on the left side of the image window) with the size of the FoV in the X-ray mosaic map.
- Tomography acquisition
- Move to the negative maximum angle +0.1 (for example, for ZP 25 nm go to -65.1°) (Motion Control > Sample and change Sample θ).
- Set the number of images as total number of angles (taking into account the image at angle 0) and the angular range (Microscope > Acquisition > Acquisition Settings > Acquisition Modes > Tomography and change the number of images; then, change Angle Start and Angle End).
- Set the defined exposure time and start the acquisition (Microscope > Acquisition > Acquisition Settings > Camera Settings and change Exposure Time and then click on Start).
- Move to the FF position (Motion Control > Sample and change Sample X; then, change Sample Y) and acquire 10 FF images (Microscope > Acquisition > Acquisition Modes > Average and change the number of images and then click on Start).
NOTE: At Mistral, spectromicroscopy or energy-dependent microscopy is possible when acquiring projections while scanning the energy across an absorption edge of interest. The final output is a stack of 2D projections, which will contain an X-ray absorption spectrum (XAS) in each pixel, i.e., images with chemical information. Extension to 3D combining spectroscopy with tomography is in principle possible. The total dose required might be a limitation and then specific strategies such as differential absorption imaging can be required.
4. Data analysis
NOTE: All data analysis is done with available open software and scripts developed with automated pipelines.
- At Mistral
- Pipeline converts tomography stacks from txrm extension (TXM software extension) to hdf5 (open source hierarchical data format) with all needed metadata, then normalizes the stack by the FF average and machine current, and finally deconvolves the stacks by the measured point spread function of the optical system for a specific Fresnel zone plate (ZP) lens and energy23,24. For deconvolution, find the proper k = 1/SNR value (depends on sample thickness). For the script developed at Mistral, type the command: txrm2deconv "input tomo" "input FF" -zp="ZP used" - e= " energy " - dx= " pixel size " - k= " 1/SNR " - t=-1.
- For the script developed at Mistral for automatic alignment, type the command: ctalignxcorr "normalized deconvolved stack.mrc" "normalized stack.hdf5".
NOTE: A number of software applications can be used for alignment of the projections to a common rotation axis using the Au NP fiducials25,26. Alignment of the projections requires subpixel accuracy. Automatic alignment can only be satisfactory when Au NP fiducials are enough (>7) and well spread on the field of view. Manual alignment is often needed a posteriori to correct automatic alignment for achieving the highest possible accuracy. - To reconstruct the aligned normalized stack, use any of the several algorithms available.
NOTE: The aligned stack can be reconstructed using Weighted Back Projection (WBP) or Simultaneous Iterative Reconstruction Techniques (SIRT) in few minutes. However, in order to preserve the linear absorption coefficients (LAC), Algebraic Reconstruction Techniques (ART) is preferred27. ART requires more computing time, so SIRT28 is done first for a fast automatic-aligned tilt series reconstruction (30 iterations in few minutes). Once the alignment is satisfactory, ART will be used.
- At B24
- A custom-built pipeline converts txrm data files to standard Tiffs with all the metadata needed and then sends them to a batch runtomo workflow that processes the data sets three ways: WBP, SIRT, and patch.
Results
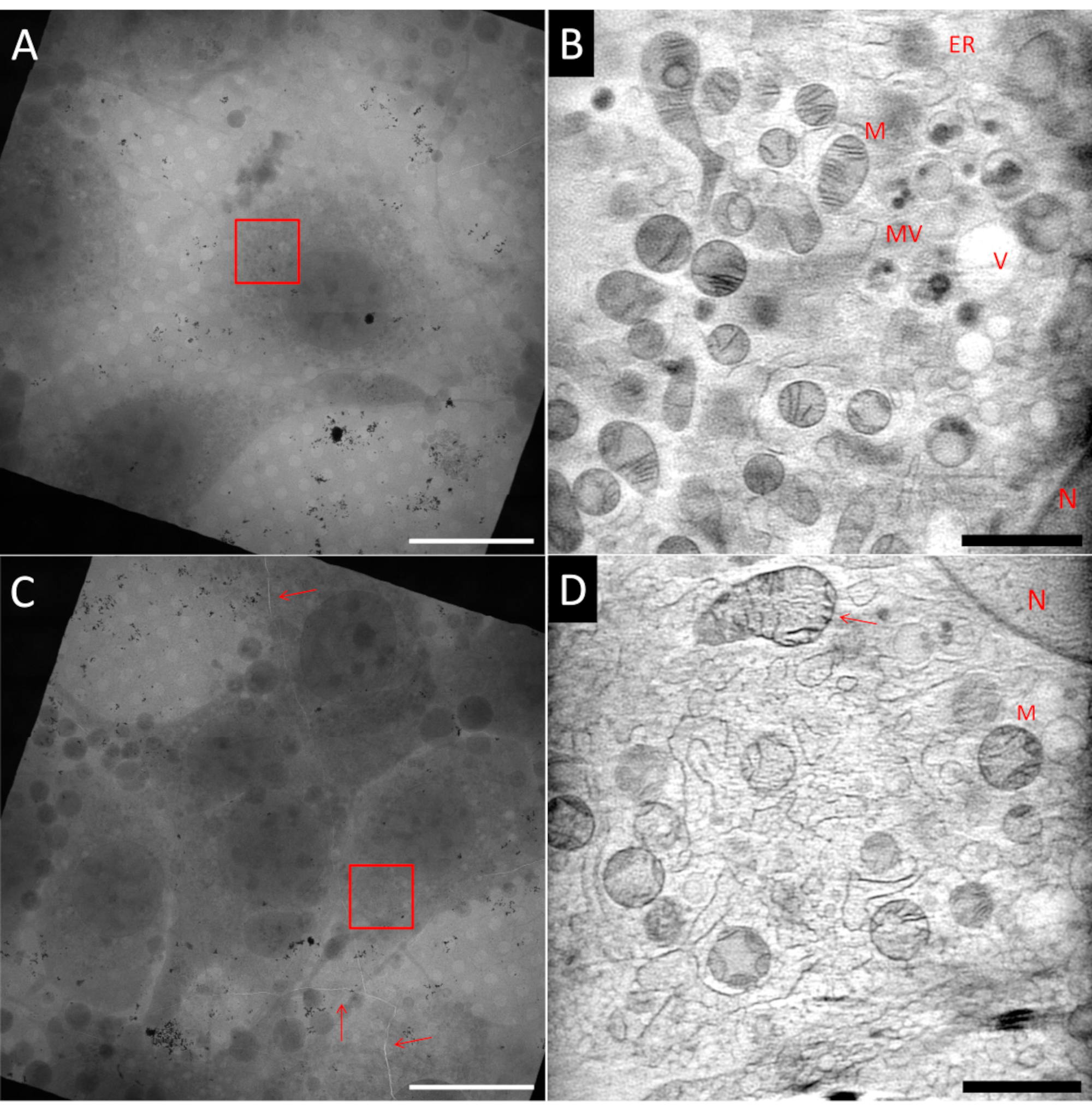
Preparing samples for cryo-SXT can be challenging. Even within the same sample grid, it is possible to have areas that are ideal, and areas that are non-ideal, as can be seen in Figure 5, which shows two squares from the same Au finder grid. The ideal sample should have single cells at the center of a square mesh, embedded in a thin layer of ice and surrounded by well-dispersed Au fiducial markers used for the alignment of tilt projections prior to tomography reconstruction. Figure 5A shows a fibroblast-like cell (NIH 3T3) that complies with many of these criteria. A single slice from a 3D reconstruction using ART27 of the area marked with the red box indicating the field of view (FoV) is shown in Figure 5B. Many different organelles such as mitochondria (M), endoplasmic reticulum (ER), vesicles (V) and the nucleus (N) can be distinguished thanks to the quantitative reconstruction of the LACs. In addition, the signal-to-noise ratio of the reconstruction is very high allowing to achieve high contrast of the cellular features. On the other hand, Figure 5C shows a square with higher cell density. Because of this, the blotting is usually less efficient, leading to a thicker ice layer, or even vitrification issues. In some cases, this can already be observed when screening the grid using epifluorescence mapping prior to the X-ray imaging, and those grids should be avoided at any cost. In Figure 5C, a crack within the grid and the vitrified ice can be observed going through the entire mesh square (marked by the red arrows). Any imaging near cracks should be avoided due to probable instability of the grid when exposed to the beam. In addition, cracks can be a sign of thick ice, as was the case in this area. A tilt series was recorded in the area marked with the red box. In Figure 5D, a single slice from the corresponding 3D reconstruction is shown. Even though some larger structures can be recognized, fine details are lost within the noise and grainy texture due to the poor vitrification quality of the thick ice, as can be seen specifically, for instance, on the upper mitochondria pointed by the arrow.

Figure 1: Workflow. Schematic workflow followed prior to cryo-SXT data collection. Please click here to view a larger version of this figure.
{kind=link}

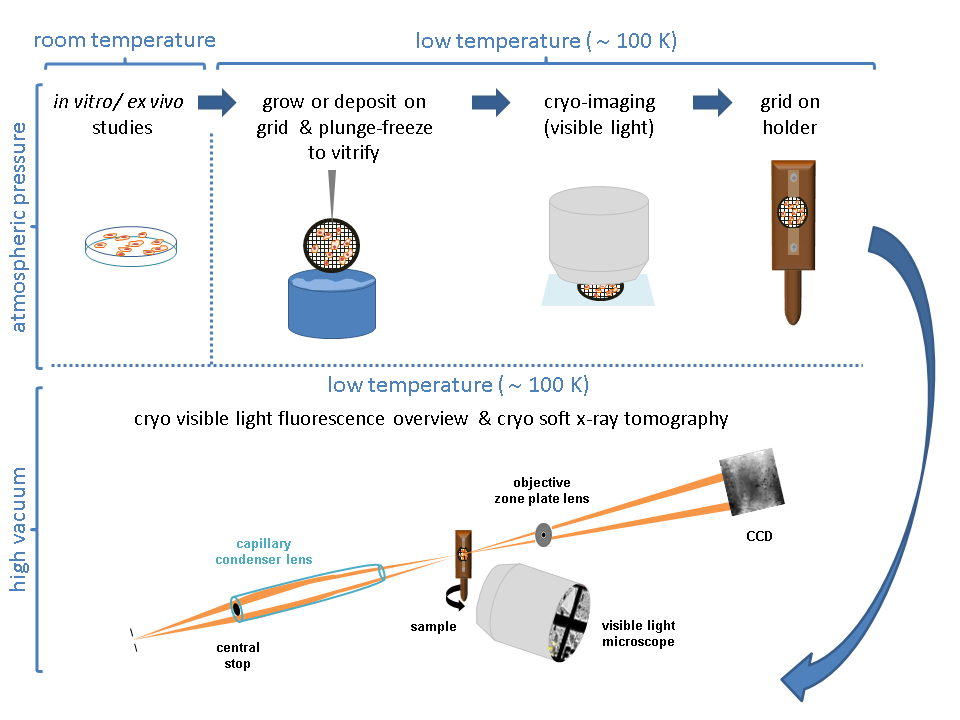
Figure 2: Growing cells on grids. (A) Cells growing in a P100 Petri dish with a confluence around 80%-90%. (B) P60 Petri dish with several grids after seeding the cells. (C) Cells growing on top of a grid after 24 h. Scale bars: 100 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Loading grids on the sample holders and into the transfer chamber. (A) Workstation filled with liquid nitrogen with the shuttle and the cryoboxes ready for loading the grids. (B) Sample holder inserted into the loader with the grid loaded. (C) Shuttle with the sample holder in position 3 without the cover. (D) Workstation with the transfer chamber attached. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Loading samples into the TXM (A) Attaching the transfer chamber to the TXM. (B) Shuttle inside the TXM. (C) TXM robot arm inserting the sample holder into the sample stage. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Example of cryo soft X-ray tomograms. Upper row: ideal sample, (A) 2D mosaic view of a grid square showing an isolated cell at the center. (B) One slice from the reconstructed 3D volume showing the marked area with the red box (A). Compared to (D) the image is much smoother and more details are visible. Lower row: non-ideal samples, (C) 2D mosaic view of a grid square showing too high cell confluency and cracks in the ice and grid foil (red arrows). (D) One slice from the reconstructed 3D volume showing the area marked with the red box in (C). The poor or suboptimal vitrification can be identified by the grainy texture of the image. N: Nucleus; M: Mitochondria; ER: Endoplasmic Reticulum; MV: Multivesicular bodies; V: Vacuole; Scale bars: A & C 20 µm; B & D 2 µm.VPlease click here to view a larger version of this figure.
{kind=link}
Discussion
Sample preparation is a critical step to obtain high quality soft X-ray tomograms, as their quality directly depends on the quality of the sample vitrification and the ice layer thickness in which the cell is embedded. Projections with high signal-to-noise ratio will be collected in regions with thin ice layer, allowing to minimize the radiation dose required to achieve the highest possible resolution. In addition, the cell confluency will also affect the final tomogram quality, since one should avoid having neighboring cells entering the FoV upon rotation. Finally, the right dispersion of Au fiducial markers will determine the accuracy of the projection alignment and then ultimately determine the quality of the final 3D reconstructed volume. Note that a proper spread of Au fiducials on the grid enables automatization of the projection alignment step, without which a high expertise is needed for such a critical step.
The protocol herein only depicts one possible sample preparation strategy, which has similarities with the ones used in cryo electron tomography (cryo-ET). In both cases, protocols improving the demanding sample preparation for better reproducibility will be fundamental for the success of these techniques, and efforts are being made toward this goal29. It is worth mentioning that in addition to imaging isolated cells, sections of tissue can also be visualized provided the transmission signal through the section will be enough at high tilt angles. Typically, this will imply sections of few microns (below 10 µm).
To image a specific structure or event inside a cell, one needs to make sure this particular feature is inside the FoV of the tilt series. As the FoV in cryo-SXT is limited to 10 x 10 µm2 to 15 x 15 µm2 depending on the lens and accounting for a pixel oversampling of the resolution to at least a factor of 2, it is often smaller than the full cell extension (see the red squares indicated in Figure 5). Therefore, the ROI must be found and properly labeled. This is usually done by means of fluorescent tags and visible light correlative approaches. 2D strategies combining epifluorescence are straightforward as the soft X-ray transmission microscope has an integrated on-line visible light fluorescence microscope, but other approaches for high resolution 2D or 3D fluorescence signal are also available4,12,13,15,16. In those cases, the grid needs to be imaged first in specific instruments such as super resolution microscopes. Note that the most efficient correlative approaches are those involving data collection at cryogenic conditions. This is because the time lapse between room temperature (RT), visible light fluorescence imaging, and sample vitrification, for instance, will hinder catching the right cellular event on time; in addition, the vitrification procedure might detach the cell of interest that has been imaged at RT from the grid. Even if most correlative imaging approaches might imply that the sample grids have to be manipulated and transported from one instrument to the other, and despite the increased risk of grid contamination or damage this poses, the reward is clear: to be able to pinpoint specific events or molecules within the cellular landscape.
When whole cell imaging is required, stitching different tomograms is possible provided the total dose applied does not exceed the radiation damage limit. Usually, the deposited dose for collecting few tomograms on the same cell is well below the limit at the achievable resolution (109 Gy) and, therefore, no specific strategy is required to lower the dose, although this is sample- and experiment-dependent. In the case of intensive data collection such as spectro-tomography, minimizing the dose would indeed be required and convenient data collection and specific processing strategies would need to be applied.
Cryo-SXT has several limitations, which should be mentioned here. The first one is the well-known missing wedge, which is intrinsic to using flat sample supports. Capillary sample supports allowing 180-degree rotation have been used in the past and are still used at some facilities, but they also present drawbacks such as an impoverished contrast due to the glass absorption and the restriction of using cells in suspension. A way to diminish the effect of the missing wedge is by performing dual tilt tomography. This is indeed possible at the Mistral beamline nowadays. The second limitation is set by the Fresnel zone plate lens used in such microscopes. This lens sets the ultimate resolution achievable and the depth of field (DoF), both being tightly related. This implies that increasing the resolution will diminish the DoF while the thickness of the cell will often be larger. For example, a 40 nm lens will have in theory a DoF of 3 µm and a resolution of 24.4 nm half pitch. The compromise between resolution and DoF is therefore strategic and the choice of the lens will depend on the type of the cell30,31. Finally, operational TXMs worldwide are far from being ideal microscopes and efforts are being made to improve the optical systems to reach the theoretical expectations. Finally, the visualization and segmentation of the reconstructed volumes can be carried out with specific software tools25,32,33,34.
In summary, cryo-SXT allows imaging cells quantitatively at medium resolution (25-30 nm half pitch) and in statistical numbers (few tens of tomograms per day). This allows obtaining the organization, distribution, and dimension of organelles at specific conditions, for instance, during pathogen infection or diseases, at precise time points or after particular treatments. It is, therefore, a useful complementary biological imaging technique to the more common electron and visible light microscopies, each of them tackling a specific range of sample dimensions and resolution. Cryo-SXT is frequently used in correlative approaches involving visible light fluorescence, but other cryo correlative strategies are also possible.
Disclosures
The authors declare no conflict of interest.
Acknowledgements
This project has received funding from the European Commission Horizon 2020 iNEXT-Discovery project and the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No 75439.
Materials
| Name | Company | Catalog Number | Comments |
| Amira | Thermo Fisher | Software for segmentation | |
| Au Holey Carbon Films finder grids | (Quantifoil Micro Tools Gmb | R 2/2 Au G200F1 | Au Holey Carbon Films finder grids |
| Au nanoparticles | BBI Group, Cardiff, UK | Au nanoparticles 100nm | 100 nm Au nanoparticles (NPs) at Mistral (Alba) |
| Au nanoparticles | BBI Group, Cardiff, UK | Au nanoparticles 250nm | 250 nm Au nanoparticles (NPs) at B24 (Diamond) |
| Axio Scope A1 | Zeiss | 430035 9060 | Fluorescence microscope |
| Blotting No.1 filter paper | Whatman | WHA10010155 | Blotting filter |
| Bsoft | Software for projection alignment, reconstruction and visualization (Heymann et al., 2008) | ||
| Chimera | Software for segmentation (Pettersen et al. 2004) | ||
| Cryo-EM Glow Discharge Set | PELCO easiGlow | 91000S | Glow Discharge Cleaning System |
| Cu Holey Carbon Films finder grids | (Quantifoil Micro Tools Gmb | R 2/2Cu G200F1 | Cu Holey Carbon Films finder grids |
| Fetal calf serum | Sigma | F9665 | Heat Inactivated, sterile-filtered, suitable for cell culture |
| ImageJ | Software for image processing and analysis in Java (NIH & LOCI University of Wisconsin) | ||
| IMOD | Software for projection alignment, reconstruction and visualization (Kremer et al., 1996) | ||
| Leica EM GP Grid Plunger | Leica | 16706401 | Automatic Plunge Freezer EM |
| LINKAM cryo-stage | Linkam Scientific Instruments | CMS 196 | Cryo-Correlative Microscopy Stage |
| MIB | Software for segmentation (Belevich et al. 2016) | ||
| P100 Petri dish | Sigma | P6106 | Treated for cell culture and sterile |
| P60 Petri dish | Sigma | D8054 | Treated for cell culture and sterile |
| Polylysin | Sigma | P4707 | Poly-L-lysine solution 0.01%, sterile-filtered |
| Soft X-Ray microscope 0.25-1.2keV | Xradia | NCT-SB | Transmission soft X-Ray microscope |
| SURVOS | Software for segmentation (Luengo et al. 2017) | ||
| Tomo3d | Software for reconstruction (SIRT, WBP) (Agulleiro et al. 2011) | ||
| TomoJ | Software for reconstruction (ART) (Messaoudi et al., 2007) | ||
| XM Data Explorer | Zeiss | TXM software |
References
- Carrascosa, J. L., et al. Cryo X-ray tomography of vaccinia virus membranes and inner compartments. Journal of Structural Biology. 168, 234-239 (2009).
- Uchida, M., et al. Soft X-ray tomography of phenotypic switching and the cellular response to antifungal peptoids in Candida albicans. PNAS. 106, 19375-19380 (2009).
- Schneider, G., et al. Three-dimensional cellular ultrastructure resolved by X-ray microscopy. Nature Methods. 10, 1-3 (2010).
- Chichón, F. J., et al. Cryo nano-tomography of vaccinia virus infected cells. Journal of Structural Biology. 177, 202-211 (2012).
- Groen, J., Conesa, J. J., Valcárcel, R., Pereiro, E. The cellular landscape by soft X-ray tomography. Biophysical Reviews. 11, 611-619 (2019).
- Kepsutlu, B., et al. Cells undergo major changes in the quantity of cytoplasmic organelles after uptake of gold nanoparticles with biologically relevant surface coatings. ACS Nano. 14, 2248-2264 (2020).
- Natterer, F. . The mathematics of computerized tomography. , (1986).
- Weiss, D., et al. Computed tomography of cryogenic biological specimens based on X-ray microscopic images. Ultramicroscopy. 84, 185-197 (2000).
- Clowney, E. J., et al. Nuclear aggregation of olfactory receptor genes governs their monogenic expression. Cell. 151, 724-737 (2012).
- Duke, E. M. H., et al. Imaging endosomes and autophagosomes in whole mammalian cells using correlative cryo-fluorescence and cryo-soft X-ray microscopy (cryo-CLXM). Ultramicroscopy. 143, 77-87 (2014).
- Cinquin, B., et al. Putting molecules in their place. Journal of Cellular Biochemistry. 115, 209-216 (2014).
- Hagen, C., et al. Multimodal nanoparticles as alignment and correlation markers in fluorescence/soft X-ray cryo-microscopy/tomography of nucleoplasmic reticulum and apoptosis in mammalian cells. Ultramicroscopy. 146, 46-54 (2014).
- Pérez-Berná, A. J., et al. Structural changes in cells images by soft X-ray cryo-tomography during Hepatitis C virus infection. ACS Nano. 10, 6597-6611 (2016).
- Chiappi, M., et al. Cryo-soft X-ray tomography as a quantitative three-dimensional tool to model nanoparticle:cell interaction. Journal of Nanobiotechnology. 14, 15 (2016).
- Varsano, N., et al. Development of correlative cryo-soft X-ray tomography and stochastic reconstruction microscopy. A study of cholesterol crystal early formation in cells. Journal of the American Chemical Society. 138, 14931-14940 (2016).
- Kounatidis, I., et al. Correlative cryo-structured illumination fluorescence microscopy and soft X-ray tomography elucidates reovirus intracellular release pathway. Cell. 182, 1-16 (2020).
- Kapishnikov, S., et al. Mode of action of quinoline antimalarial drugs in red blood cells infected by Plasmodium falciparum revealed in vivo. PNAS. 116 (46), 22946-22952 (2019).
- Conesa, J. J., et al. Unambiguous intracellular localization and quantification of a potent iridium anticancer compound by correlative 3D cryo X-ray imaging. Angewandte Chemie International Edition. 59, 1270-1278 (2020).
- Gal, A., et al. Native-state imaging of calcifying and noncalcifying microalgae reveals similarities in their calcium storage organelles. PNAS. 115 (43), 11000-11005 (2018).
- Procopio, A., et al. Chemical fingerprint of Zn-hydroxyapatite in the early stages of osteogenic differentiation. ACS Central Science. 5, 1449-1460 (2019).
- Kahil, K., et al. Cellular pathways of calcium transport and concentration toward mineral formation in sea urchin larvae. PNAS. 117 (49), 30957-30965 (2020).
- Conesa, J. J., et al. Intracellular nanoparticles mass quantification by near-edge absorption soft X-ray nanotomography. Scientific Reports. 6, 22354 (2016).
- Otón, J., Sorzano, C. O. S., Maribini, R., Pereiro, E., Carazo, J. M. Measurement of the modulation transfer function of an X-ray microscope based on multiple Fourier orders analysis of a Siemens star. Optics Express. 23 (8), 9567-9572 (2015).
- Otón, J., et al. Characterization of transfer function, resolution and depth of field of a soft X-ray microscope applied to tomography enhancement by Wiener deconvolution. Biomedical Optics Express. 7, 5092-5103 (2016).
- Kremer, J. R., Mastronarde, D. N., McIntosh, J. R. Computer visualization of three-dimensional image data using IMOD. Journal of Structural Biology. 116, 71-76 (1996).
- Heymann, J. B., Cardone, G., Winkler, D. C., Steven, A. C. Computational resources for cryo-electron tomography in Bsoft. Journal of Structural Biology. 161, 232-242 (2008).
- Messaoudi, C., et al. Three-dimensional chemical mapping by EFTEM-TomoJ including improvement of SNR by PCA and ART reconstruction of volume by noise supression. Microscopy Microanalysis. 19, 1669-1677 (2013).
- Agulleiro, J. I., Fernández, J. J. Fast tomographic reconstruction on multicore computers. Bioinformatics. 27, 582-583 (2011).
- Toro-Nahuelpan, M., et al. Tailoring cryo-electron microscopy grids by photo-micropatterning for in-cell structural studies. Nature Methods. 17, 50-54 (2020).
- Attwood, D. . Soft X-rays and Extreme Ultraviolet Radiation. Principles and Applications. , (2000).
- Howells, M., Jacobsen, C., Warwick, T. . Principles and Applications of Zone Plate X-ray. Microscopes. Science of Microscopy. , (2007).
- Pettersen, E. F., et al. UCSF Chimera--a visualization system for exploratory research and analysis. Journal of Computational Chemistry. 25, 1605-1612 (2004).
- Belevich, I., Joensuu, M., Kumar, D., Vihinen, H., Jokitalo, E. Microscopy image browser: a platform for segmentation and analysis of multidimensional datasets. PLOS Biology. 14 (1), 1002340 (2016).
- Luengo, I., et al. SuRVoS: Super-region volume segmentation workbench. Journal of Structural Biology. 198, 43-53 (2017).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved