
Method Article
Cryo-Structured Illumination Microscopic Data Collection from Cryogenically Preserved Cells
* These authors contributed equally
In This Article
Summary
This protocol demonstrates how to image biological cryo-preserved samples using cryo-structured illumination microscopy. We demonstrate the methodology by imaging the cytoskeleton of U2OS cells.
Abstract
Three-dimensional (3D) structured illumination microscopy (SIM) allows imaging of fluorescently labelled cellular structures at higher resolution than conventional fluorescence microscopy. This super-resolution (SR) technique enables visualization of molecular processes in whole cells and has the potential to be used in conjunction with electron microscopy and X-ray tomography to correlate structural and functional information. A SIM microscope for cryogenically preserved samples (cryoSIM) has recently been commissioned at the correlative cryo-imaging beamline B24 at the UK synchrotron.
It was designed specifically for 3D imaging of biological samples at cryogenic temperatures in a manner compatible with subsequent imaging of the same samples by X-ray microscopy methods such as cryo-soft X-ray tomography. This video article provides detailed methods and protocols for successful imaging using the cryoSIM. In addition to instructions on the operation of the cryoSIM microscope, recommendations have been included regarding the choice of samples, fluorophores, and parameter settings. The protocol is demonstrated in U2OS cell samples whose mitochondria and tubulin have been fluorescently labelled.
Introduction
SR imaging techniques have become widely accessible to biologists over the last decade1. They allow high-resolution imaging of fluorescently tagged samples beyond the diffraction limit. However, it has been challenging to adapt SR microscopy methods to work with samples at cryogenic temperatures2. This would be advantageous for correlative imaging in combination with electron or X-ray tomography. Recently, SIM has been adapted for use with cryogenic samples and has successfully been shown to enable correlative studies of biological cells in conjunction with soft X-ray tomography (SXT)3 at the correlative cryo-imaging beamline B24 at the Diamond Light Source Synchrotron (https://www.diamond.ac.uk/Instruments/Biological-Cryo-Imaging/B24.html). SIM can double the resolution of conventional wide-field microscopy by illuminating the sample with striped patterns of light (Moiré fringes) at three angles and in five phases. The interference between these light patterns and the sample fluorescence can be used to computationally uncover extra information about sub-diffraction structures4,5.
There are several advantages of SIM over other SR techniques for cryogenic applications. First, it can work without specially designed blinking fluorophores; conventional fluorophores can be used, giving access to a wider range of potential fluorescent tagging agents6. In addition, it only requires 15 images per z slice (in 3D; 9 images for 2D), whereas other SR methods take approximately 1000 images per slice, increasing the chance of the sample being heated and therefore increasing the risk of ice crystal formation, which can cause artefacts. Finally, this technique can image thicker biological samples of over 10 µm, allowing whole cells to be imaged in their near-native state6. The cryoSIM has been built using standard optical components and with open-access software for imaging, making it easy to document and duplicate if desired6. The cryoSIM has a 100x/0.9 numerical aperture objective (see the Table of Materials); further information on its optical components, design parameters, and performance has been described by Phillips et al.6 Here, this protocol demonstrates how to use the cryoSIM microscope including how to load and unload samples on the cryogenic stage, how to collect data on the microscope, and how to reconstruct the SIM images.
Protocol
NOTE: This protocol pertains to samples containing cells grown or deposited on transmission electron microscopy (TEM) 3 mm flat gold grids with a holey carbon support film that have been vitrified by plunge freezing or high-pressure freezing. This protocol assumes that samples have already been imaged using a conventional epifluorescence and brightfield microscope to map locations of interest for imaging in cryoSIM. See Figure 1 for an overview of the entire protocol.
1. Preparation of the cryo-stage
- Prepare ice-free liquid nitrogen (LN2) by passing LN2 through a funnel lined with any standard scientific dry paper wipes.
NOTE: As LN2 causes burns, wear appropriate personal protective equipment, including cryo-protective gloves and goggles, when handling it. Such liquids may displace oxygen and cause rapid suffocation and should be handled in a properly ventilated area. - Remove surface dust from the cryo-stage with pressurized air. Expel liquid from the pressurized air container before using it.
- Ensure the sample-loading cartridge is in place with the appropriate chambered sample holder (check that there is no grid left in the sample holder from a previous experiment) (Figure 2).
- Remove the lid from the external dewar of the cryo-stage, and pour filtered LN2 until approximately 1/4th full. Wait until initial boiling subsides before pouring more; fill the vessel to about 2/3rd full. Replace the lid carefully, pointing the nozzle away from the handler and the stage/optics while LN2 boils out from the outlet.
- Once LN2 has stopped coming out of the outlet, place the outlet pipe over the stage dewar on the cryo-stage.
- Plug-in the power source of the stage, and connect the USB cable to the cryo-stage. Ensure the heated sample chamber lid is plugged in. Plug in the external dewar to the stage.
NOTE: Do not plug in the external dewar until the outlet pipe has been positioned over the stage dewar on the cryo-stage. If LN2 overflows (or to prevent it from overflowing), pull out the USB cable for ~10 s, allow it to equilibrate, and reconnect the USB cable to reactivate the sensor. - After LN2 has been delivered into the stage dewar, press the release button on the cryo-stage to allow LN2 to enter the sample chamber.
NOTE: Do not leave the system unattended while LN2 is filling the chamber. - Wait for ~30-45 min to allow the system to cool and stabilize before commencing image acquisition. Periodically check the external dewar, and top-up with filtered LN2 if it is less than a quarter full (approximately every hour).
2. Transfer of the sample storage box into the cryo-stage
NOTE: Immerse the sample storage box, holder, and the tips of any instruments (e.g., forceps) inside filtered LN2 to cool them before touching any cold surfaces such as the sample or any objects inside the sample chamber. Wear a laboratory coat and gloves when handling biological samples.
- Ensure vitrified samples are in a cryo-compatible container, and bring them to the microscope. Press the corresponding button on the cryo-stage to turn on the light in the sample chamber.
- Use the hex key on the cassette tool to open the two plates of the sample transfer cassette. Open the plates wide enough to drop in the grid between the two plates, but avoid opening to the maximum open position to prevent the grid falling through the other side.
- Use long forceps to lift the sample grid box out of the LN2, turn it where the notch aligns with the position of the storage position inside the stage, and place it onto the stage. Use the appropriate device (e.g., a screwdriver) to open the storage box lid to the correct sample position.
- Using inverted forceps (or any fine-tip surgical forceps), remove the TEM grid from the sample holder, immerse it inside the LN2, and drop it into position in the sample transfer cassette, keeping close to the LN2 during the transfer process. Ensure the carbon film side is placed so that it will ultimately be facing the objective on the sample bridge.
- Close the sample cartridge using the hex key on the cassette tool. Close and remove the storage box along with any remaining samples.
- Use the magnet point on the cassette tool to lift and mount the cartridge containing the grid onto the sample bridge. Keep it immersed/close to the LN2 during the transfer process. Ensure the orientation is appropriate for the bridge (the two magnets will make contact with the magnets on the bridge). Place the cassette flat within the positioning pins of the bridge, and gently nudge to ensure it is fixed.
NOTE: A sample cassette for clipped grids that have been prepared for subsequent focused ion beam milling is also available at the CryoSIM facility at beamline B24.
3. Stage docking and focusing
- Move the cryo-stage lid opening to the imaging position, and turn off the sample chamber light. Slide the stage towards the optics to align it under the objective lens. Gently drop the objective into position using the lever, ensuring that it rests within the lid of the cryo-stage, but does not touch it.
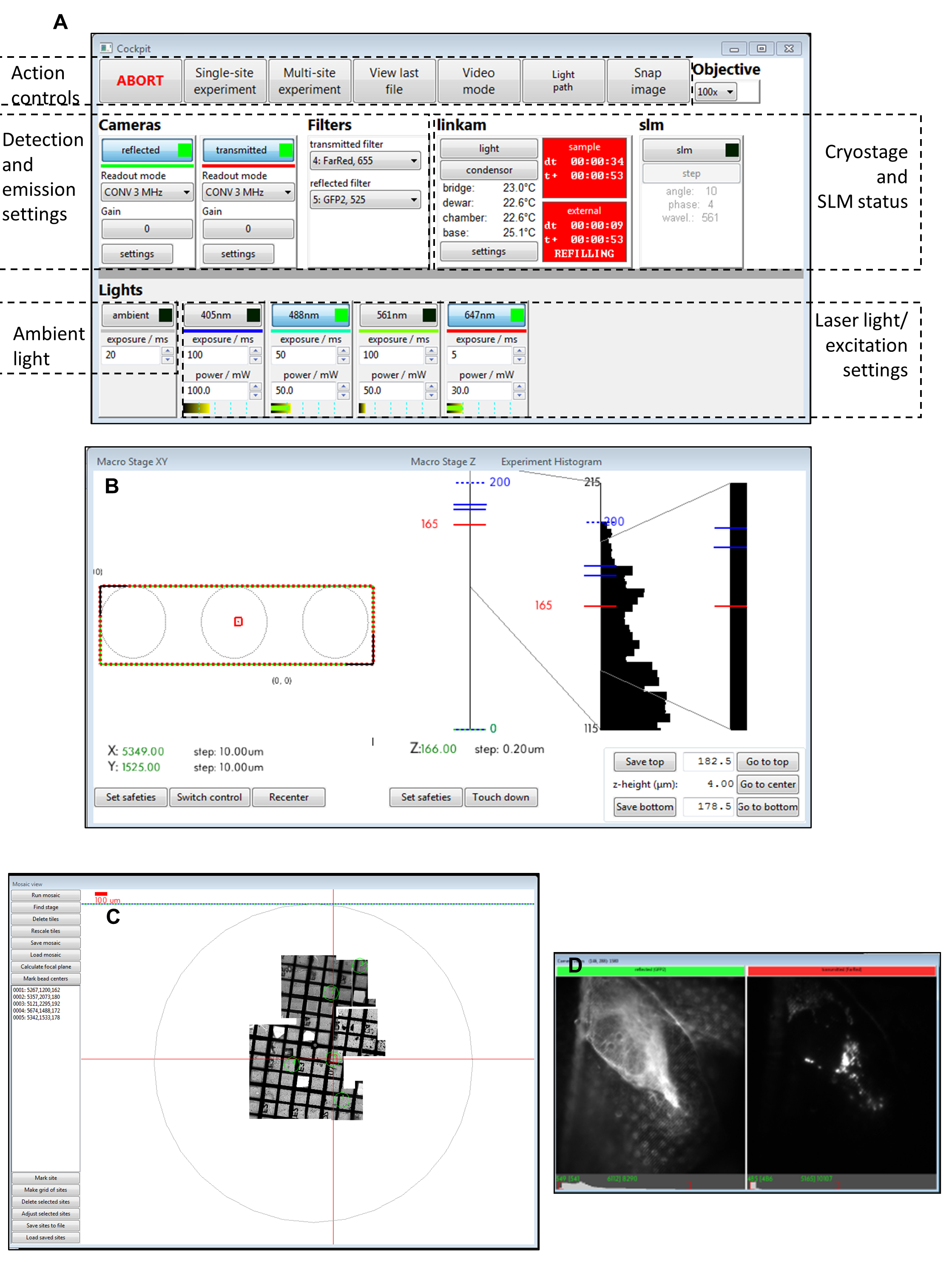
NOTE: Ensure the external Dewar's outlet pipe does not touch the stage dewar on the cryo-stage at any point during data collection. - Cover the stage and optics with an opaque black curtain. Start the control software Cockpit on the cryoSIM PC (Figure 3 and Figure 4).
- Click on the readout mode button for each camera, and set it to CONV 3 MHz. Check that the temperature of each camera is -80 °C, and the camera fan is off.
- Turn on the reflected camera. Under Lights, choose ambient (exposure 20 ms), and under linkam, click on condenser. Click on the Video mode button.
- In the Mosaic view window, zoom out (mouse scroll) to see the grid outline. Click on Find stage if it cannot be seen. Center the grid by double-left-clicking in the middle of the circle.
- Focus the sample until the grid support film (or any other relevant sample feature) is in focus, using the up and down keys to move the cryo-stage up and down, and using the 9 and 3 keys on the numerical pad to change the z-step (set it to 20 µm for initial focusing).
NOTE: If the user runs out of travel in z, the stage can be manually moved up or down using the wheel under the cryo-stage. Turn it by one notch at a time, and check if the sample is within the range of view. Alter the direction again if focusing worsens.
4. Brightfield mosaic acquisition
- Once the stage is centered, turn off video mode, and collect a visible light mosaic (click on Run mosaic in the Mosaic view to produce tiles of visible light images that spiral outward from the center). If the grid is bent, try different positions on the grid (double left click within Mosaic view), re-focus, and click on Run mosaic again to collect partial mosaics. Alternatively, drop-in a focused image on top of the mosaic (Figure 3).
- Save the view by clicking on Save mosaic. Give it a short filename containing information on the storage box and respective grid number (a timestamp will be appended automatically to the file name).
5. Identification of areas of interest
- Inspect the brightfield mosaic alongside any previous fluorescence "map" images for where cells or biological features of interest have previously been located. Check if those areas produce suitable fluorescence.
- Turn off the ambient light and condenser as well as the video mode if active. Turn on the required excitation laser (405, 488, 561, or 647), and choose the corresponding camera and filter, initially at 50 mW, for 50 ms exposure time.
NOTE: Increase/decrease these camera and filter settings depending on the fluorescence signal. - Press 0 to snap an image and * to auto-contrast. Alternatively, manually adjust the contrast by using the slider at the bottom of the image.
NOTE: Switch on the Laser on sign for the room. - Once biologically interesting cells with suitable fluorescence have been found, mark their positions using the Mark site button in Mosaic view.
NOTE: These marked sites will appear in the enclosed list and can be accessed by double-clicking on the coordinates. When returning to a marked site, zoom into the area before double-clicking. - Continue marking all potential sites before commencing image acquisition. Re-save the mosaic with the marked sites; click on Save sites to file.
- To stitch the mosaic images together using the StitchM software (developed inhouse at beamline B24), drag and drop the .txt mosaic file into the StitchM file with extension .bat and save the combined tiff image of the mosaic tiles in the same folder. To save an image with the marked sites, drag and drop the mosaic.txt file and the markers.txt file into the icon at the same time.
NOTE: Balance data collection against the needs of the project (e.g., if correlative imaging will be done, check how many images can be taken with the partner-imaging modality, and choose the appropriate number of sites for cryoSIM imaging). In addition, if the external dewar requires refilling with LN2 , the cryo-stage system will change position, and the marked sites will likely not return to the same locations; therefore consider this when choosing the number of sites to be imaged.
- To stitch the mosaic images together using the StitchM software (developed inhouse at beamline B24), drag and drop the .txt mosaic file into the StitchM file with extension .bat and save the combined tiff image of the mosaic tiles in the same folder. To save an image with the marked sites, drag and drop the mosaic.txt file and the markers.txt file into the icon at the same time.
- Turn off the ambient light and condenser as well as the video mode if active. Turn on the required excitation laser (405, 488, 561, or 647), and choose the corresponding camera and filter, initially at 50 mW, for 50 ms exposure time.
6. Data collection strategy
- Set the laser exposure time based on the counts in the dynamic range in the fluorescence image (at the bottom of the camera views window). Choose which filter to apply, and optimize these settings for each wavelength of excitation light to be used, turning each laser on separately.
NOTE: Although 10,000-20,000 counts are optimal, lower counts are acceptable if there is a good contrast. Ideally, check filter settings before each image stack is acquired because cells at different areas of the grid could have variable fluorescence levels.
7. Data collection
- Click on both cameras to turn them on. Return to one of the marked sites, and focus on the desired depth again. Once in focus in an area of interest, move out of focus upwards (↑ key) in the XY window in the Macro Stage to choose the height of the z stack to acquire, and click on Save top. Move out of focus downwards (↓ key), click on Save bottom and then on Go to the centre, and check that the image is still in focus.
NOTE: The sample height will be shown in the window. - Right-click on slm (spatial light modulator) in the Cockpit window, and make sure the angle is set at 0.41. In the Cockpit window, select Single-site experiment.
NOTE: Do not click the slm on; Cockpit will automatically turn it on during image acquisition.- From the dropdown list, select Structured Illumination. Alter the Stack height so that it equals the z-height + 1 µm.
NOTE: The addition of 1 µm ensures the capture of the entire sample in z and minimizes reconstruction artefacts. - Enter the exposure times (ms) for the 405 nm and 488 nm lasers in the upper row (for the reflected camera) and the exposure times for the 561 nm and 647 nm lasers in the lower row (for the transmitted camera).
NOTE: The values for exposure time should match the values decided on previously and shown in the main Cockpit window. - Input a file name (naming convention: box number_grid area_filters_FL) (FL (fluorescence) or BF (brightfield) depending on what type of imaging is being done). Click on Update to produce a new file containing the date and time without overwriting previous files. Then, click on Start.
- From the dropdown list, select Structured Illumination. Alter the Stack height so that it equals the z-height + 1 µm.
- Check the Camera view while data is being collected. Re-take the images if there is any xy displacement. If the LN2 dewar refills the cryo-stage during image acquisition, abort the process by clicking on the Abort button in the Cockpit software.
- Once the external dewar has finished refilling the stage dewar, repeat the experiment because the refill displaces the sample vertically. Refocus the image to re-center it in z, and repeat the Single-site experiment. Repeat the Single-site experiment for all combinations of lasers and filters needed.
NOTE: It takes approximately 30 s-1 min to finish refilling. During the imaging of one grid, refilling will happen ~4-8x.
- Once the external dewar has finished refilling the stage dewar, repeat the experiment because the refill displaces the sample vertically. Refocus the image to re-center it in z, and repeat the Single-site experiment. Repeat the Single-site experiment for all combinations of lasers and filters needed.
- At each position, collect a z stack using visible light.
- Switch off the lasers, and switch on Ambient light and condenser. Under Single-site experiment, select Z-stack, set the Ambient light to 20 ms exposure, and keep z height as above.
- Repeat steps 7.2 to 7.4 for all sites marked on the grid.
- Before moving to another sample, delete the mosaic by clicking on Delete tiles.
- Draw a square around all the tiles to delete them. Delete the markers as well by selecting all of them in the list and then clicking on Delete selected sites.
- Turn off the ambient light and condenser before proceeding to change the grid. Follow the steps in section 4 in reverse order to undock the stage from under the objective and change the grid.
8. After imaging
- After imaging is finished, undock the stage, and remove all the samples. Turn off the sample chamber light. Unplug the external dewar, and decant any remaining LN2 into another cryo-compatible container, allowing the dewar to safely return to a normal temperature.
- Put the lid plug on the cryo-stage. Wait until the option to bake-out the cryo-stage display becomes available after no more LN2 remains in the stage dewar. Press the bake-out button to enter the heating mode, and remove any moisture from the cryo-stage to avoid ice formation.
9. Reconstruction
- Transfer raw SIM data files to the appropriate workstation for reconstruction. Run processing in batches through the Processing Task builder window using channel-specific optical transfer functions (OTFS) (calculated from multi-fluorescent beads point spread functions) and K0 angles (0.29278, -1.8028, 2.3786) with a constant Wiener filter for all channels of 0.004 and a bias offset of 200.
- In the Additional options panel, ensure that negative intensities are discarded by keeping other options unchecked. Save the SIR images into a folder named "processed".
NOTE: Commercial SIM reconstruction software usually produces reconstructed SIM data and retains the file name, but appends SIR.dv at the end. A log file is also created that contains the processing protocol, steps, and statistical information on reconstruction success.
10. Chromatic shift correction
- Download the software Chromagnon7 to correct for the chromatic shift.
- Use the chromatic shift reference matrix that corresponds to the fluorescence wavelengths of the data collected (provided by the beamline).
NOTE: The reference file is a 'chromagnon.csv' file, which contains the alignment parameters and has been obtained from calibration images using multi-fluorescent nanoparticles. It can be used to batch-process multiple data sets at once.- Choose the appropriate reference file which matches the laser wavelength and filter used for imaging the sample, and add it to the reference field. Add the reconstructed SIR data to be aligned in the source field, and click on Run.
- Check that the fluorescence signal is now aligned in the images. For batch-processing, place all SIR images to be aligned in the source field and the reference file in the reference field, and press Run all.
Results
A sample containing U2OS cells was stained with a mixture of green microtubule cytoskeleton dye and red mitochondria dye, resulting in the staining of the microtubule component of the cytoskeleton (green) and the mitochondria (red). Subsequent imaging showed the localization of mitochondria within the cell as well as the arrangement of the microtubules, highlighting the structural framework that they provide to the cell and the assembly of the cytoskeleton around organelles such as the nucleus. The resolution in cryoSIM is significantly higher than that in standard epifluorescence microscopy (Figure 5). Figure 6 demonstrates how the fluorescent "map" from a conventional epifluorescence microscope can be used to locate areas of interest for imaging and the corresponding cryoSIM-reconstructed image from a location on the grid.

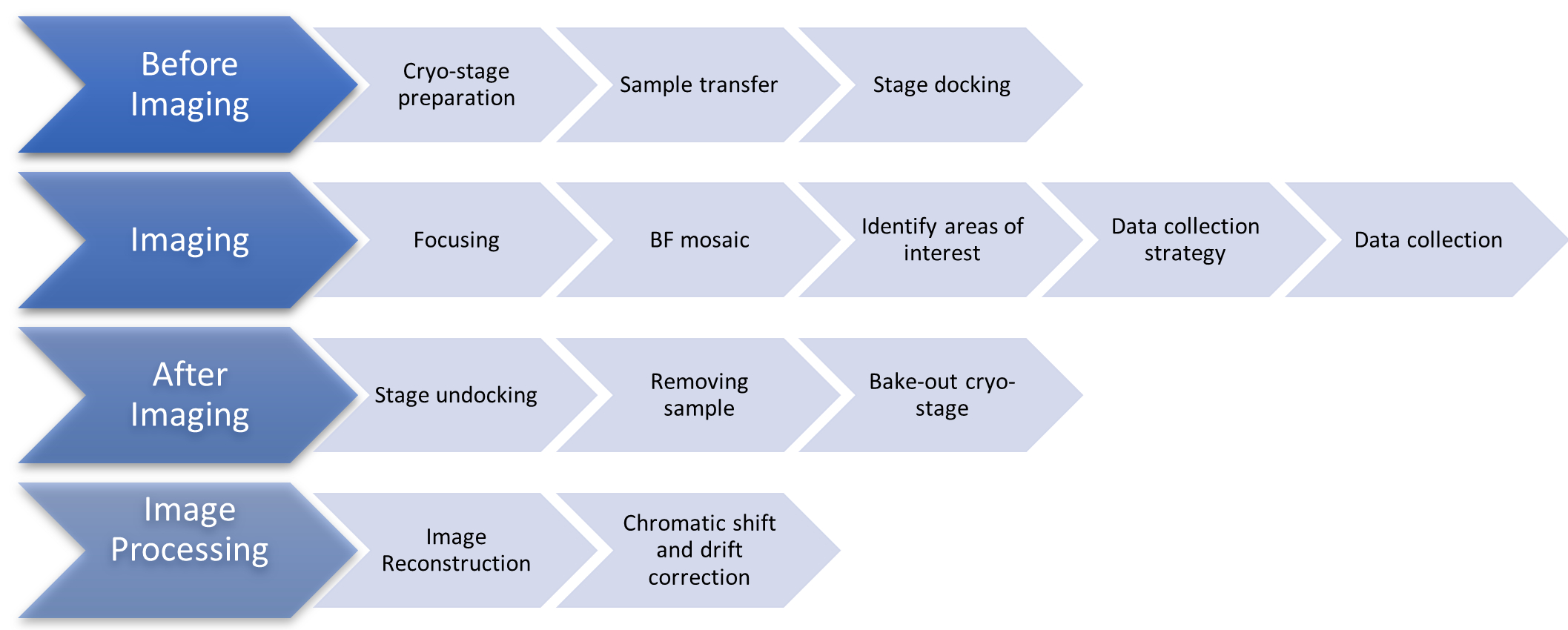
Figure 1: Flow chart showing the stages of the cryoSIM imaging protocol. Abbreviation: cryoSIM = Cryo-structured illumination microscopy. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: The cryo-stage. (A) The cryo-stage setup. (B) A sample grid shown held by inverted forceps. (C) Components of the cryo-stage. The connection ports are labelled, with the colors corresponding to orange: power supply, yellow: heated stage lid, blue: external dewar, green: connection to PC. Abbreviations: PC = personal computer; LN2 = liquid nitrogen. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Views of the Cockpit software panels. (A) Main panel, (B) macro stage XY, (C) mosaic view, (D) camera views. Abbreviation: SLM = spatial light modulator. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Views of the Cockpit software panels. (A) Z stack single site experiment. (B) SI single site experiment. (C) Keyboard shortcuts for the cockpit software used during image acquisition. (D) The cryoSIM microscope is on-site at beamline B24 at the Diamond Light Source synchrotron. Abbreviation: cryoSIM = Cryo-structured illumination microscopy. Please click here to view a larger version of this figure.
{kind=link}

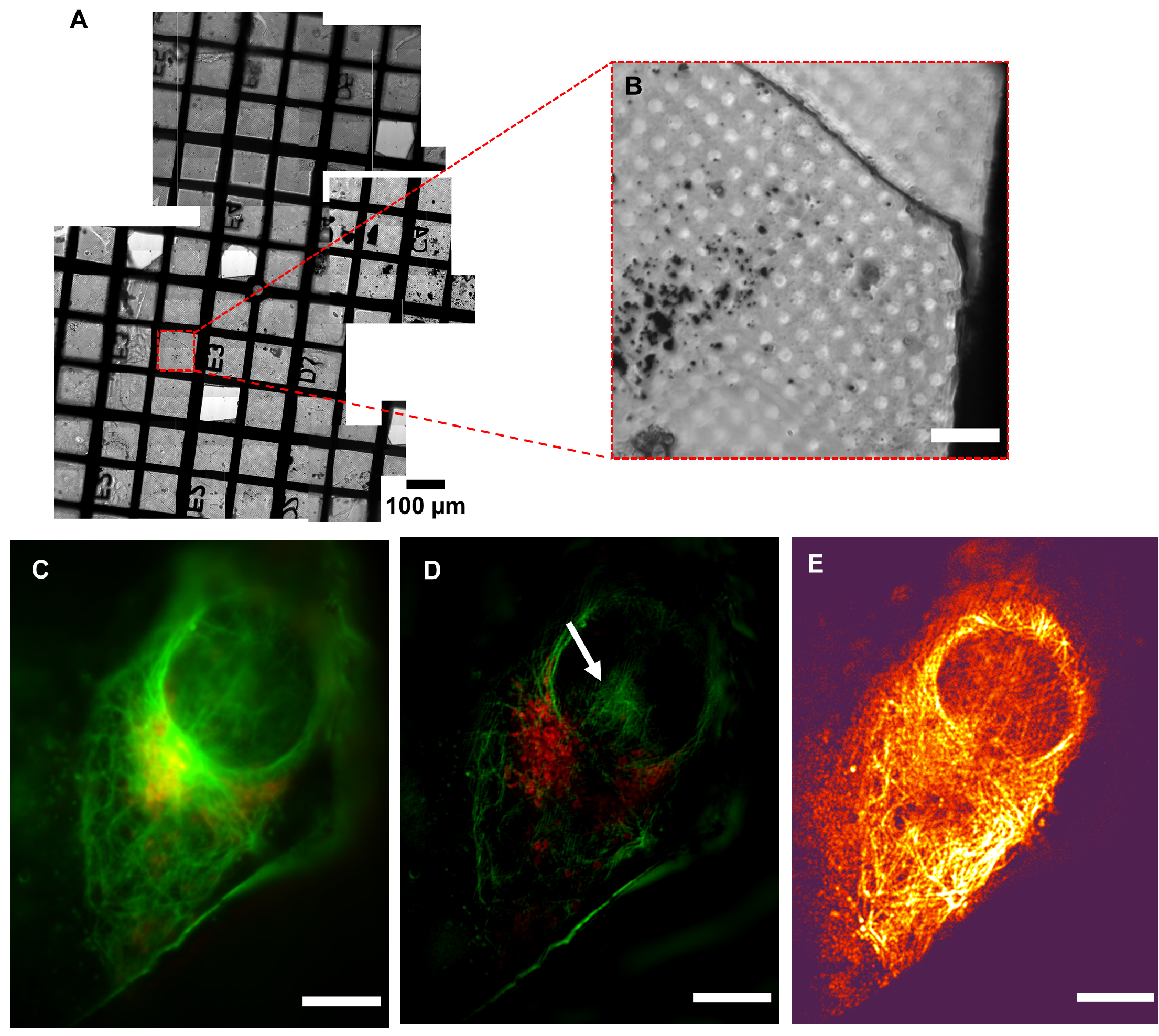
Figure 5: Resolution of cryoSIM. (A) Mosaic view of a grid under examination. (B) Brightfield image of an Area of Interest (AOI). (C) Pseudo-widefield image compared to its (D) SIM image showing the increase in resolution. The white arrow indicates SIM reconstruction artefacts. (E) Modulation contrast map combining the pixel intensity information of the reconstructed image with the color information of the respective modulation contrast-to-noise ratio (MCNR) values of the raw data generated by SIMCheck2. The bright and dark regions show high and low contrast, respectively. Scale bar = 10 µm. CryoSIM imaging settings: excitation/emission wavelengths: 488/525 nm, 50 mW laser power, 50 ms exposure time and 647/655 nm, 20 mW laser power, 5 ms exposure time. Abbreviation: cryoSIM = Cryo-structured illumination microscopy. Please click here to view a larger version of this figure.
{kind=link}

Figure 6: Image reconstruction in cryoSIM from a location on the grid in a fluorescence map from a conventional epifluorescence map. (A,B) Overlay of brightfield and fluorescence image maps generated with a conventional epifluorescence microscope. This map is used to locate regions of interest to subsequently image in cryoSIM. (C) The reconstructed cryoSIM image obtained at the location shown in (B). Abbreviation: cryoSIM = Cryo-structured illumination microscopy. Please click here to view a larger version of this figure.
{kind=link}
Discussion
3D SIM at cryogenic temperatures has many advantages over other SR imaging techniques for imaging vitrified biological material. It requires significantly fewer images per z slice compared to other SR methods, resulting in less irradiation and a lower chance of ice crystal formation for vitrified samples. It is also able to image whole cells and can be correlated with X-ray tomography to match structure with function. Interestingly, most commercially available fluorophores and fluorescence tags bleach less under cryogenic conditions than at room temperature. However, given the high quantum yield of most common fluorophores at room temperature (more than 80% in some cases), the absolute gain detected in photons is not due to changes in quantum yield, but due to a reduction in the complex bleaching processes. More information on the yield of fluorophores at cryogenic temperatures can be found in 8.
It is critical that samples arriving at the cryoSIM have been premapped using a conventional cryofluorescence microscope with brightfield capability to produce a grid "map" that includes highlighting of all potential AOIs for further imaging (Figure 5). Access time at the cryoSIM is allocated via a competitive process that involves the submission of a proposal, which is subsequently evaluated for technique feasibility and biomedical impact. Time at the equipment, therefore, is always "at a premium", and premapped grids allow the most efficient use of an allocation. It is also essential that the sample is kept vitrified, especially during sample transfer from the sample holder to the imaging platform, to minimize the formation of ice crystals and subsequent sample damage. The sample should be of good quality to produce the best SIM images. A well-prepared sample will be characterized by the following features: (a) it will have no ice crystal contamination, (b) the grid used will be a finder grid, (c) the carrier will be flat, (d) the grid mesh and substrate surface will not be auto-fluorescent, and (e) there will be no breaks in the support membrane. These prerequisites can be achieved by careful sample handling and ensuring that samples always stay vitrified.
It is important to check in advance whether proposed sample fluorophores will give enough signal in the cryoSIM microscope. Tools such as SPEKCheck9 can aid with choosing the optimal fluorophore and filter combinations. If there are issues with the raw data collection or the reconstruction process, artefacts can appear in the images after reconstruction. Examples of various artefacts have been documented by Demmerle et al.10 The image reconstruction parameters can be reviewed in the SoftWoRx log file if the reconstruction is not optimal by opening the reconstruction summary file. There should be consistent line spacing across angles in a given channel and relatively consistent amplitude. Variation of more than 30% and values significantly above 1 (if bead size compensation is applied) should be more closely investigated and are likely to indicate failed reconstructions. In addition, the SIMcheck2 software in Fiji can also be used to perform various checks on the raw and reconstructed data to diagnose the cause of errors in the imaging or reconstruction parameter settings. SIM-check and its modulation contrast map can also aid in the assessment of the quality of reconstructed data by interpreting which areas of an image are likely to be real structures versus artefacts.
Low modulation contrast (shown by dark color, in Figure 5E) within the nuclear area means that this region is going to be more susceptible to reconstruction artifacts, therefore implying that the hash patterns shown in the nucleus could be classified (Figure 5D) as an artefact. Strong fluorescence signal areas are more likely to accurately reflect native structures in the processed data. In areas of weak signal where fluorophores are distributed over wider areas, such as the total surface of a vesicle, it is likely that real signal coexists with processing artefacts, and care should be taken in the interpretation of that data. After inspection of the full-range reconstructed data to ensure there are no strange artifacts, and that the background is generally Gaussian and centered near zero, the data is generally clipped at zero, or the modal value-the peak of the background signal-should be very near zero. This ensures that the dynamic range of the displayed image is not dominated by negative background artifacts. When a weaker signal is expected, extra care should be taken in analyzing the features and ensuring they are real structures rather than reconstruction artefacts.
There are some limitations of the imaging system. Because the sample stage is flat, samples with variable thickness or grids that are not flat are not ideal subjects for imaging. Additionally, if correlative imaging will be done using soft X-ray tomography, cells near grid boundaries should not be imaged as these will not be visible in the X-ray microscope during tilt series acquisition. Finally, the amount of blotting of the sample before plunge freezing has a significant impact on the imaging quality; too little blotting results in samples that are too thick, giving suboptimal SIM images with high background noise, while too much blotting can cause cells to become misshaped and therefore more susceptible to heat damage from the incident laser beam. In summary, cryoSIM is a powerful fluorescence microscopy tool for imaging biological samples in 3D in a near-native stage and has wide-ranging applications in many areas.
Disclosures
The authors declare no competing financial interests.
Acknowledgements
This project has received funding from the European Commission Horizon 2020 iNEXT-Discovery project. I. M. Dobbie acknowledges funding from the Wellcome Trust (107457/Z/15/Z). This work was carried out with the support of the Diamond Light Source, instrument B24 (proposal BI25512). Our thanks to the staff at Micron and all our excellent users and collaborators for helping us establish the cryoSIM and its correlative potential.
Materials
| Name | Company | Catalog Number | Comments |
| Auto grids | FEI | ||
| Autogrids | Thermo Fisher Scientific | 1036173 | |
| BioTracker 488 Green Microtubule Cytoskeleton Dye | Sigma-Aldrich | SCT142 | |
| Cockpit Software | Oxford University | https://github.com/MicronOxford/cockpit | |
| cryo compatible polyurethane container | Jena bioscience | CC-FD-800 | |
| Cryo TEM grid storage box | Thermo Fisher Scientific | Model#AutoGrid | |
| cryo-SIM microscope | Custom made | N/A | Custom made, see following reference for the design: Michael A. Phillips, Maria Harkiolaki, David Miguel Susano Pinto, Richard M. Parton, Ana Palanca, Manuel Garcia-Moreno, Ilias Kounatidis, John W. Sedat, David I. Stuart, Alfredo Castello, Martin J. Booth, Ilan Davis, and Ian M. Dobbie, "CryoSIM: super-resolution 3D structured illumination cryogenic fluorescence microscopy for correlated ultrastructural imaging," Optica 7, 802-812 (2020). Has a Nikon TU Plan Apo 100x/0.9 NA. |
| Cryostage system | Linkam Scientific Instruments | CMS196 | |
| Fine tip surgical forceps | Ted Pella | 38125 | |
| MitoTracker Deep Red FM | Thermo Fisher Scientific | M22426 | |
| Python Microscope Software | Oxford University | https://www.python-microscope.org/ | |
| Scientific dry paper wipes | Kimberly-Clark 7551 | 2 | |
| SIM Reconstruction Software | softWoRx, GE Healthcare | Version 6.5.2 | |
| StitchM Software | Diamond Light Source | https://github.com/DiamondLightSource/StitchM | |
| TEM grids for samples | Quantifoil Micro Tools | G200F1 |
References
- Schermelleh, L., et al. Super-resolution microscopy demystified. Nature Cell Biology. 21 (1), 72-84 (2019).
- Ball, G., Demmerle, J., Kaufmann, R., Davis, I., Dobbie, I. M., Schermelleh, L. SIMcheck: A toolbox for successful super-resolution structured illumination microscopy. Scientific Reports. 5 (1), 15915 (2015).
- Kounatidis, I., et al. 3D Correlative cryo-structured illumination fluorescence and soft X-ray microscopy elucidates reovirus intracellular release pathway. Cell. 182 (2), 515-530 (2020).
- Gustafsson, M. G. L. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. Journal of Microscopy. 198 (2), 82-87 (2000).
- Rego, E. H., Shao, L., Rego, E. H. Practical structured illumination microscopy. Methods in Molecular Biology. 1251, 1251 (2015).
- Phillips, M. A., et al. CryoSIM: super-resolution 3D structured illumination cryogenic fluorescence microscopy for correlated ultrastructural imaging. Optica. 7 (7), 802 (2020).
- Matsuda, A., Schermelleh, L., Hirano, Y., Haraguchi, T., Hiraoka, Y. Accurate and fiducial-marker-free correction for three-dimensional chromatic shift in biological fluorescence microscopy. Scientific Reports. 8, 7583 (2018).
- Kaufmann, R., Hagen, C., Grünewald, K. Fluorescence cryo-microscopy: current challenges and prospects. Current Opinion in Chemical Biology. 20, 86-91 (2014).
- Phillips, M. A., Pinto, D. M. S., Dobbie, I. M. SPEKcheck-fluorescence microscopy spectral visualisation and optimisation: a web application, javascript library, and data resource. Wellcome Open Research. 3, 92 (2018).
- Demmerle, J., et al. Strategic and practical guidelines for successful structured illumination microscopy. Nature Protocols. 12 (5), 988-1010 (2017).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved