Method Article
Preparation and Cryo-FIB micromachining of Saccharomyces cerevisiae for Cryo-Electron Tomography
In This Article
Summary
We present a protocol for lamella preparation of plunge frozen biological specimens by cryo-focused ion beam micromachining for high-resolution structural studies of macromolecules in situ with cryo-electron tomography. The presented protocol provides guidelines for the preparation of high-quality lamellae with high reproducibility for structural characterization of macromolecules inside the Saccharomyces cerevisiae.
Abstract
Today, cryo-electron tomography (cryo-ET) is the only technique that can provide near-atomic resolution structural data on macromolecular complexes in situ. Owing to the strong interaction of an electron with the matter, high-resolution cryo-ET studies are limited to specimens with a thickness of less than 200 nm, which restricts the applicability of cryo-ET only to the peripheral regions of a cell. A complex workflow that comprises the preparation of thin cellular cross-sections by cryo-focused ion beam micromachining (cryo-FIBM) was introduced during the last decade to enable the acquisition of cryo-ET data from the interior of larger cells. We present a protocol for the preparation of cellular lamellae from a sample vitrified by plunge freezing utilizing Saccharomyces cerevisiae as a prototypical example of a eukaryotic cell with wide utilization in cellular and molecular biology research. We describe protocols for vitrification of S. cerevisiae into isolated patches of a few cells or a continuous monolayer of the cells on a TEM grid and provide a protocol for lamella preparation by cryo-FIB for these two samples.
Introduction
Recent technological and software developments have made electron cryo-microscopy (cryo-EM) of vitrified biological specimens one of the key techniques in structural biology research in the last decade1,2. The preparation of a specimen for cryo-EM usually consists of the application of a purified protein or a complex of protein with nucleic acid onto the sample carrier (TEM grid), followed by removal of most of the liquid with a filter paper, and plunge freezing of the grid with the residual thin layer of a sample into liquid ethane or propane3. The sample is thus fixed in a thin layer (typically <80 nm) of amorphous buffer in a fully hydrated state, at near-native conditions, and without need of any chemical fixation or heavy metal contrasting. Imaging of the structurally homogeneous specimen in the transmission electron microscope then results in data that can be used for the determination of the three-dimensional structure of the macromolecule at near-atomic resolution using a single particle analysis protocol2. Such an in vitro structure corresponds to the representation of the macromolecule under the conditions and treatment utilized during sample preparation. Although the structures determined under the in vitro conditions usually correspond to the fully functional state of the macromolecule, the capacity to image spatial relationships among various macromolecules inside the cell would provide an additional functional context to the structural data.
Cryo-electron tomography (cryo-ET) is used to reconstruct 3D volumes of pleomorphic objects or macromolecular complexes in situ4,5. The advantage of cryo-ET is that the three-dimensional information is obtained by imaging a single entity. However, the resolution at which individual macromolecular complexes or organelles are observed is very limited. Therefore, averaging of the macromolecules (sub-tomogram averaging, STA) with the same structure from a larger number of tomograms is necessary to reach 4-8 Å resolution models from the cryo-ET data6,7. It has recently been shown that cryo-ET and STA can also be applied to determine high-resolution structures of macromolecular machines such as ribosomes in the context of the cellular environment7. However, the utilization of transmission electron microscopy is limited by the specimen thickness. In general, this is not a problem for single-particle cryo-EM where optimization of the vitrification conditions can eventually result in the embedding of the sample in a thin layer of ice. On the other hand, most of the cells are not in fact electron transparent for the 300 keV electron beam. The inelastic mean free path in the vitrified biological specimens for the 300 keV electrons is approximately 395 nm8, which means that cryo-ET studies are limited to the cellular periphery for most of the cells.
Different techniques were developed to thin the sample down to a sufficient thickness for cryo-ET. Cryo-ultramicrotomy utilizes mechanical slicing of the sample with a diamond knife at the liquid nitrogen temperature (-196 °C) to provide 60-80 nm thick sections suitable for cryo-ET9,10,11. Multiple sections can be prepared from a single cell and the data analysis can eventually produce 3D structural information for the larger part of the cell. However, the mechanical slicing can cause several artifacts such as curved sections, crevasses, or sample compression, which may influence the resulting structure and bias the cryo-ET data10,11,12. Cryo-focused ion beam micromachining (cryo-FIBM) represents an alternative approach where a thin cellular section is prepared by gradual ablation of the specimen using a focused beam of Ga+ ions (FIB) in a multi-step process, which can result in 80–300 nm thick cellular cross-section (lamella)13,14,15. In contrast to cryo-ultramicrotomy, only one lamella is prepared from a single cell, which represents ~0.3–3% of its volume, and micromachining of multiple cells is usually necessary to find a region of interest in the milled cross-section. In addition, the throughput of the whole workflow is nowadays still fairly low, often limited to 6–8 high-quality lamellae from an 8-hour cryo-FIBM session. On the other hand, the cryo-FIBM cross-sections are devoid of any compression artifacts and provide suitable input for high-resolution cryo-ET. Moreover, the transfer of the lamella to the sample carrier for cryo-ET is not necessary as the sample is retained on the TEM grid during the whole lamella preparation process and the same grid can be subsequently transferred to TEM. We expect that the throughput of the cryo-FIBM will be significantly improved soon, primarily from availability of software for unsupervised lamella milling16,17 and utilization of FIBs operating on the principle of charge-couple plasma, which will afford faster material ablation.
Saccharomyces cerevisiae (yeast) are eukaryotic cells of spherical shape and diameter of ~2-5 µm. Thanks to its size, accessibility, genetics, generation time, and simple manipulation, the yeast is extensively studied as a eukaryotic model organism in cellular and molecular biology research similar to Escherichia coli, which is well studied as a prokaryotic model organism in bacteriology. The yeast can be easily cultured in suspension and a high quantity of cells is generated in a short time (doubling time 1 – 2 hours). More importantly, yeast shares a complex internal cellular structure with animal and plant cells while retaining a small genome comprised of a low content of non-coding DNA. Structural characterization of the yeast proteome from the high-resolution in situ data can thus help to provide a mechanistic description for the extensive amount of functional data available in the literature.
Herein, we provide a comprehensive protocol for the acquisition of in situ cryo-ET data on the yeast sample, which covers all steps from the sample cultivation down to the cryo-FIBM lamella preparation, and the specimen transfer to TEM for cryo-ET data acquisition.
Protocol
1. Cultivation and preparation of Saccharomyces cerevisiae cells for vitrification
- Prepare liquid growth medium for Saccharomyces cerevisiae.
- Autoclave a 500 mL glass bottle for the preparation of growth medium.
- Weigh 2.2 g of yeast extract (1.1%) and 4.4 g of Peptone (2.2%) and mix in 200 mL of water.
- Sterilize by autoclaving for 15 min at 121 °C.
- Weigh 10 g of glucose powder and mix in 50 mL of water to get 20% glucose solution. Pass the solution through a 0.2 µm filter and store it at 4 °C.
- Prepare solid medium for Saccharomyces cerevisiae.
- Weigh 4 g of agar powder and mix with 200 mL of growth medium.
- Sterilize in an autoclave for 15 min at 121 °C.
- Cool the medium to 40-50 °C and add 20 mL of 20% sterile glucose (prepared in the previous step). Pour ~20 mL of the complete medium into the Petri dish and let it solidify at ambient temperature.
- Wrap the agar plates in parafilm to protect from drying and store at 4 °C.
- Culture Saccharomyces cerevisiae in suspension
NOTE: The protocol is optimized for the preparation of a sample for cryo-FIBM from a suspension of the cell line Saccharomyces cerevisiae strain BY4741 [ATCC 4040002] or similar strains.- Autoclave a 50 mL Erlenmeyer (or similar) flask.
- Work in a hood or laminar flow box. Pipette 10 mL of the growth medium to a sterile 50 mL Erlenmeyer flask.
- Supplement the medium with 1 mL of filtered 20% glucose. Pick one colony of yeast from an agar plate with a sterile, disposable inoculation loop (1-10 μL).
- Place the Erlenmeyer flask in the incubator and culture at 30 °C with agitation (150-200 rpm) until the exponential phase is reached (approximately 7 h).
NOTE: We have observed that the exponential phase is reached after ~15 h when colonies are picked from agar plates that have been cultured at ambient temperature for 4 weeks. See also Note below.
- Culture Saccharomyces cerevisiae on an agar plate from glycerol stock.
- Use a new agar plate from the 4 °C storage.
- Take the S. cerevisiae stock from the -80 °C deep freezer and place it into a freezing stand to avoid complete thawing of the stock.
- Scrape off and transfer a small culture with a sterile inoculation loop (1-10 μL) to 50 μL of growth medium. Mix properly.
- Transfer the whole volume of mixed S. cerevisiae culture and disperse with a sterile spreading stick over the surface of the agar plate.
- Incubate at 30 °C for approximately 48 hours until 1.5-2 mm diameter colonies are formed.
NOTE: It is advisable to culture the colonies freshly before the experiment. Colonies older than 1 week will require a prolonged period for cultivation in liquid media to reach the exponential growth phase.
- Culture Saccharomyces cerevisiae on an agar plate.
- Use prepared agar plates with grown S. cerevisiae colonies.
- Pipette 10 mL of sterile growth medium and 1 mL of filtered 20% glucose to a 50 mL Erlenmeyer flask.
- Pick one colony of S. cerevisiae culture with a sterile inoculation loop and mix with a growth medium in a flask.
- Incubate 50 minutes at 30 °C with agitation (150-200 rpm).
- Dilute the suspension culture ten times with the growth medium and disperse 50 μL of suspension on an agar plate with a sterile spreading stick.
- Incubate at 30 °C for approximately 48 hours until 1.5-2 mm colonies are observed.
- Wrap the edges of the Petri dish with parafilm to prevent it from drying. Store at room temperature and use for a maximum of 4 weeks.
- Prepare Saccharomyces cerevisiae for plunge freezing in cell clusters.
- Prepare the S. cerevisiae cell culture according to the protocol in the section Cultivation of Saccharomyces cerevisiae in suspension (Section 1.3) and incubate ~7 h at 30 °C with agitation (150-200 rpm).
- Measure the OD of S. cerevisiae cell suspension culture at 600 nm using a UV/Vis spectrophotometer.
- Concentrate the cell suspension to OD600 = 1.
- Prepare S. cerevisiae for plunge freezing in a cell monolayer.
- Prepare S. cerevisiae cell culture according to the protocol in the section Cultivation of Saccharomyces cerevisiae suspension cell culture and incubate ~7 h at 30 °C with agitation (150-200 rpm).
- Measure the OD of S. cerevisiae cell suspension culture at 600 nm using an UV/Vis spectrophotometer.
- Transfer the cells in medium to the centrifuge tube and gently spin for 2 minutes at a relative centrifugal force (900 x g).
- Discard the medium from the cell pellet by pipetting.
- Add fresh medium to the cell pellet. Calculate the volume of medium to get a cell suspension of OD600 equaling 30 to 60.
- Add glycerol (50% stock solution) to the cell suspension to a final concentration of 5% shortly before vitrification.
NOTE: Glycerol works as a protective agent, which improves the quality of the ice in the regions between the cells. Glycerol is added to cells only shortly before vitrification to minimize its uptake into the cell.
2. Vitrification of the Saccharomyces cerevisiae specimen
- Glow discharge TEM grids with the carbon film side facing up for 30-45 s (pressure: 6-9 Pa, current: 7 mA).
- Set the vitrification robot to the following parameters: temperature: 18 °C, humidity: 100%, blot time: 6 s, wait time: 5 s, blotting cycle: 1x, and blot force: 5.
NOTE: Blot force is an instrument-specific value and optimal blot force values may differ among different machines. The optimal value for a particular plunger must be experimentally confirmed. - Prepare liquid ethane for vitrification.
- Mount the non-absorbent surface pad to the blotting pad facing the sample. Use the filter paper for the other blotting pad.
- Pick the glow discharged grid with the tweezers and mount the tweezers to the plunge freezing instrument.
- Apply 3.5 μL of S. cerevisiae suspension to the carbon side of the grid inside the plunger climate chamber. Mix properly before every application on the grid.
- Plunge freeze the grid into the liquid ethane.
- Transfer the grid from liquid ethane to LN2. Store grids with vitrified cells under LN2 conditions or mount them into the TEM grid cartridge for loading into the FIB-SEM microscope.
3. Mounting TEM grids into the grid cartridge
NOTE: The workflow described here utilizes the TEM grids mounted into the grid cartridge to facilitate sample handling and transfer between SEM and TEM microscopes. The cartridge assembly consists of a C-ring, a TEM grid, and a C-clip ring. Other options are available when working with instrumentation from other microscope manufacturers. The grid cartridge assembly workstation is filled with LN2. The LN2 level covers the grid box with the TEM grids, but the mounting of the TEM grids into the cartridge is performed in LN2 vapors. It is highly recommended to wear a protective facemask or shield during the vitrification procedure to prevent contamination from breathing to the specimen. Do not work with the tools that have accumulated ice contamination.
- Put the grid cartridge assembly workstation together and prepare dry tools for clipping.
- Cool the assembly workstation down with LN2. Cool the clipping tools to the LN2 temperature.
- Place a grid box with a vitrified sample into the assembly workstation.
- Place a TEM grid with the specimen with cells facing down to the C-ring and secure with a C-clip.
- Place the clipped cartridges into the grid box and close properly.
- Store cartridges in the LN2 Dewar or load them into the FIB-SEM microscope.
4. Loading and manipulation of the sample to the FIB-SEM microscope
NOTE: The instructions were written for the utilization of the dual-beam microscope Versa 3D equipped with the PP3010 cryo-FIB/SEM preparation system. Alternative solutions may require different specific parameters; however, the general concept of the workflow should be still valid.
- Cool the microscope down to the liquid nitrogen temperature.
- Pump the microscope chamber and preparation chamber (if present) to a high vacuum before the start of cooling down (< 4 x 10-4 Pa) to prevent contamination growth.
- Set the nitrogen gas flow to 5 L/min for the preparation chamber, microscope stage, and microscope anticontaminator. Wait until all components reach a temperature of < -180 °C.
NOTE: The FIB-SEM microscope setup used in this study utilizes cooled nitrogen gas to cool down its stage and anticontaminator. Other systems may use a different method for stage cooling. The chamber pressure reaches ~3 x 10-5 Pa once the stage and anticontaminator are at -190 °C on the instrument used here. The growth rate of the hydrocarbon contaminating layer on the surface of the lamella with the experimental setup used in this study is ~15 nm/hour.
- Load the grid cartridge with the specimen to the microscope.
- Assemble and cool the loading station to the LN2 temperature.
- Place the shuttle (sample holder), the grid box with the specimen, the grid box opener, and tweezers into the cooled loading station.
- Carefully transfer the grid cartridge with the specimen facing up into the shuttle.
- Flip the shuttle inside of the loading station to the loading position.
- Load into the microscope.
- Optionally, coat the specimen with metal protective layers.
NOTE: A strong charging effect can be observed when imaging frozen hydrated biological material SEM. In addition, imaging biological samples with FIB (even at low currents) induces fast sample damage. Therefore, an additional coating of the sample might performed inside the FIB-SEM microscope, to protect the specimen surface.- Deposit the protective layer with organometallic platinum by the gas injection system (GIS).
- Set the sample to the eucentric height (coincidence point for imaging with electrons and ions).
- Tilt the stage back to 45° (sample tilted 90° relative to electron beam).
- Move the stage in the z-axis 4 mm below the eucentric height.
- Set the GIS needle to 26–30 °C.
- Deposit ~300-1000 nm of the organometallic platinum layer to the grid with the biological specimen (corresponds to 30–120 s of the GIS deposition).
NOTE: We usually apply GIS for 30 s for samples with small cellular clusters and 45 s for samples with a monolayer of the cells.
- Sputter coat the specimen surface with a conductive metal layer.
- Deposit ~10 nm of the metal layer (Ir, Au, Pt) to the grid with a biological specimen.
- Deposit the protective layer with organometallic platinum by the gas injection system (GIS).
- Set microscope parameters for the lamella preparation
- For FIB, use the following parameters: high voltage = 30 kV, current = 10 pA (imaging), 10 pA–3 nA (FIB-milling)
- For SEM, use the following parameters: high voltage = 2–5 kV, spot size = 4.5, current = 8–27 pA.
- Set the scan rotation to 180° for both beams.
- Set the stage tilt: milling angle 6-11° (corresponds to stage tilt of 13°–18° for the sample shuttle with pre-tilt of 45° and FIB/SEM microscope with 52° angle between SEM and FIB column).
5. Preparation of the Saccharomyces cerevisiae lamella
- Check the grid quality and select a suitable cluster of Saccharomyces cerevisiae.
- Check that the TEM grid is properly blotted from both sides without additional water around the cell cluster or at the back side of the grid.
NOTE: In order to check the backside of the grid, rotate the stage to -10° and image the grid with FIB. - Select the optimal cell clusters for lamella preparation according to the following recommendations.
- Position the cell clusters in the grid center. The milling area should not extend outside of the square with dimensions 1100 x 1100 μm positioned in the center of the grid (550 μm in each direction from the grid center).
- Position the cell clusters in the central part of the grid square with no overlap to the grid bar.
- Position the cell clusters in the grid square with compact holey carbon foil without cracks.
- Ensure that the cluster is not surrounded by ice contamination.
- Check that the TEM grid is properly blotted from both sides without additional water around the cell cluster or at the back side of the grid.
- Select the optimal milling position in the Saccharomyces cerevisiae monolayer.
- Ensure that the grid bars surrounding the cell monolayer on the selected grid square are visible.
- Select the optimal position in the cellular monolayer according to the following recommendations. The selected areas for milling should satisfy the following criteria:
- Have the region of interest in the grid center. The milling area should not extend outside the square of dimensions 1100 x 1100 μm positioned in the grid center (550 μm in each direction from the center of the grid).
- Have the region of interest in the central part of the grid square with no overlap with the grid bar.
- Do not surround the cell monolayer with ice contamination.
- Prepare S. cerevisiae lamella with cryo-FIB.
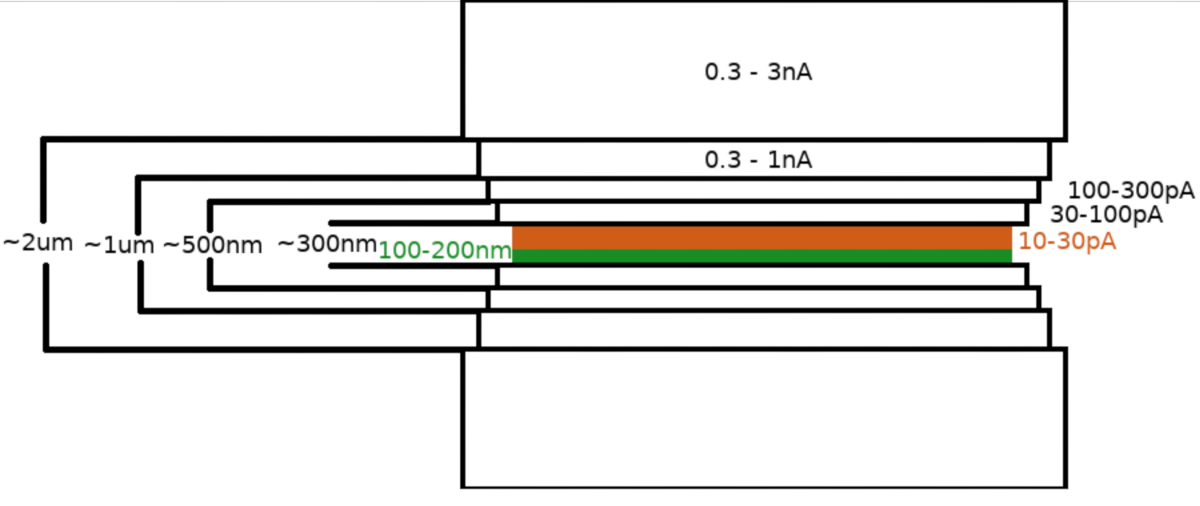
NOTE: The milling pattern is generated and centered relative to the region of interest. Cryo-FIBM is performed sequentially with multiple milling steps performed at different FIB settings. The lamella with roughly 2 μm thickness is initially milled using high current (0.3–3 nA). The lamella is then gradually thinned to 500 nm. The fine-milling step at low currents (10-30 pA) is used to finalize the lamella to ~100–200 nm thickness.- Set a region of interest (ROI) to the eucentric height and save this position.
NOTE: The coincidence point needs to be determined and saved for every position separately. The eucentric height is set by tilting the stage to 0° and centering on the ROI by moving the stage in the x and y direction. The stage is then tilted to 25° and the ROI is brought back to the center of the scanned area by changing the stage z-axis position. Finally, the stage is tilted back to 13°–18° for milling. - Define a rectangular milling pattern above the ROI with a scan direction of Top to Bottom.
- Define a rectangular milling pattern below the ROI with a scan direction of Bottom to Top.
- Define the inactive rectangular milling pattern covering the region of interest for a rough estimation of the lamella thickness. This pattern is not milled during lamella preparation.
- Mark all patterns and set the lamella width (x dimension). The width of the milling pattern should not exceed 2/3 of the cluster width. This corresponds to 8–15 μm in most cases.
- Set parameters for rough milling steps
- FIB current: 0.3–3.0 nA ; final lamella thickness: 1.5–2 μm; width of the FIBM area: 8-12 um; stage-tilt: 13-17°; duration: 8 minutes; active milling patterns: upper and lower.
- FIB current: 0.3–1.0 nA; final lamella thickness: 1 μm; width of the FIBM area: 7.5-11.5 μm; stage-tilt: 13-17°; duration: 8 minutes; active milling patterns: upper and lower.
- FIB current: 100–300 pA; final lamella thickness: 0.5 μm ; width of the FIBM area: 7.5-11.5 μm; stage-tilt: 13-17°; duration: 8 minutes; active milling patterns: upper and lower.
- FIB current: 30–100 pA; final lamella thickness: 0.3 μm ; width of the FIBM area: 7.5-11.5 μm; stage-tilt: 13-17°; duration: 8 minutes; active milling patterns: upper and lower.
- Set parameters for the fine milling step:
- FIB current: 10–30 pA; final lamella thickness: <0.2 um; width of the FIBM area: 7-11 μm; stage-tilt: 13-17° (+1°); duration: 12 minutes; active milling patterns: upper.
NOTE: The rough milling steps (5.3.6) are carried out sequentially for each lamella. In contrast, the fine milling step (5.3.7) does not directly follow the rough milling, but fine milling steps are carried out sequentially for all the lamellae at the end of the session to minimize the hydrocarbon contamination on the lamella surface. An extra +1° stage tilt is used during fine milling step to increase the uniformity of the lamella thickness across its length.
- FIB current: 10–30 pA; final lamella thickness: <0.2 um; width of the FIBM area: 7-11 μm; stage-tilt: 13-17° (+1°); duration: 12 minutes; active milling patterns: upper.
- Set a region of interest (ROI) to the eucentric height and save this position.
6. Transfer of Saccharomyces cerevisiae lamella to cryo-TEM
- Prepare a properly dried Dewar and fill it with LN2.
- Unload the samples with lamellae from the FIB/SEM microscope under cryo-conditions, transfer them to a grid box, and store them in a LN2 storage Dewar for long-term storage. Alternatively, load the grids directly into cryo-TEM.
- Correct orientation of the lamella relative to the cryo-TEM stage tilt axis is important (see accompanying video at 8:10 for more details). Ensure that the milling direction of prepared lamellae is perpendicular to the cryo-TEM stage tilt axis.
- Pre-tilt the cryo-TEM stage to compensate for the lamella tilt relative to the grid plane and collect the tilt-series using dose-symmetric scheme19.
NOTE: The magnitude of the pre-tilt (typically 6–8°) is determined by the angle between the TEM grid plane and the FIB direction during micromachining. The position of the lamella front edge in the image in cryo-TEM can be used to determine the sign of the pre-tilt angle. For that, the sense of the microscope stage rotation must be known. In our experimental setup, the position of the front edge of the lamella on the right side of the image taken in nanoprobe SA mode corresponds to negative pre-tilt.
Results
The Saccharomyces cerevisiae culture was harvested in the middle of the exponential growth phase. We prepared two types of specimens in which the cells were either distributed as small clusters of several cells over the surface of the TEM grid (Figure 1A,C) or formed a continuous monolayer over individual grid squares of the TEM grid (Figure 1B,D). The discriminative factor for the preparation of the sample with either distinct cell islets or the cellular monolayer is the concentration of the cell culture applied to the TEM grid. The harvested cell culture was concentrated to OD600 = 1.0 for the former case, or to OD600 = 30 to 60 for the latter case, respectively. The sample for the preparation of the cellular monolayer was further supplemented with 5% v/v glycerol prior to vitrification. The glycerol is critical for the vitrification of the buffer solution, which fills the space in between the cells (Figure 2) as the reflections from the crystalline buffer can be detrimental for proper positional tracking and focusing during cryo-ET data collection.
In addition, the yeast suspension culture was blotted against the non-absorbent material such as the PTFE blotting pad or the custom 3D printed pad made of FlexFill 98A material. The blotting paper was positioned only on the backside of the grid with respect to the sample application (back-blotting). The back-blotting strategy is recommended for the suspension culture plunge freezing as blotting with the filter paper from both sides results in adhesion of the cells to the blotting paper (Figure 1E).
The protocol described here utilizes TEM grids clipped in the grid cartridge, which forms stable support for the grid and facilitates sample handling of the sample after the vitrification. This enforces a necessity that other sample holders and shuttles in FIB/SEM and TEM microscope can accept such a grid cartridge.
Upon transfer of the sample into the the FIB/SEM microscope, the specimen was first coated with a 0.3–1.0 μm layer of methylcyclopentadienyl platinum using the microscope gas injection system (GIS). An additional layer of the inorganic iridium was sputtered on the sample surface to harden the GIS layer and render the surface conductive. The lamellae were milled in multiple steps (Figure 3) where (I) the milling current, (II) the lamella width, and (III) the distance of the milling area above and below the samples were decreased in a stepwise manner. The final milling step (“polishing”) was carried out at low current (10-30 pA) only from the top side of the lamella and with the sample inclined by an additional 1° towards the Ga+ beam. Utilization of the described protocol has on average resulted in 8-10 lamellae prepared on two TEM grids within one 6-8 hour session.
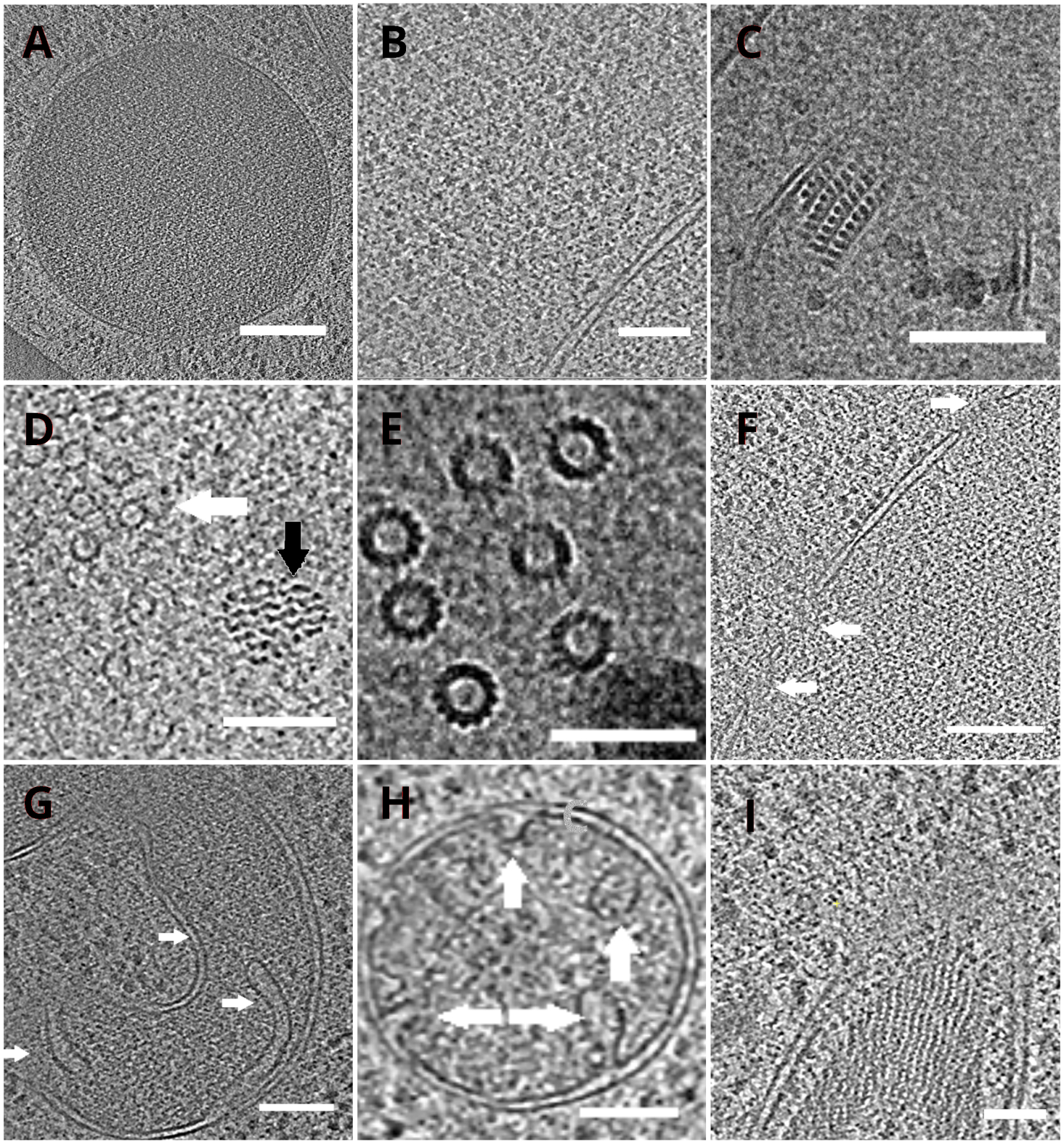
The TEM grids with the lamellae were subsequently transferred into a transmission electron microscope. The lamellae were first screened and only those which showed minimal curtaining (artefacts stemming from uneven milling across the lamella surface), low surface contamination level, and good cellular contrast (usually observed for lamellae with <200 nm thickness) were selected for the acquisition of the cryo-ET data. In addition, lamellae containing cracks across the whole length were discarded from data collection. In general, about 50% of the lamellae transferred to TEM were suitable for data acquisition. Tilt series were collected on the post-GIF K2 direct electron detector with the energy-selecting slit set to 20 eV. The data collection was carried out in SerialEM software18 and the tilt series were collected using a dose symmetric scheme19 with the tilt range of ±60° and the increment of 3°. The data was acquired at the magnification corresponding to the pixel size of 3.47 A/px. The overall dose of 65 e/Å2 was uniformly distributed over the individual sub-frames. The tilt images were collected as a set of three frames, which were subsequently corrected for the motion and radiation damage during data acquisition using MotionCor220 program. Parameters of the contrast transfer function were estimated using Ctffind421. The tilt series were processed in eTomo18. The patch tracking routine was used to align the images. The tomogram was reconstructed using a weighted back projection algorithm after 2x binning of the images, and subsequently filtered using SIRT-like filter (set to 8 iterations) in IMOD18. The tomogram segmentation was carried out manually in Amira software22. The reconstructed tomograms provide a high-resolution representation of the yeast cellular interior and enable us to observe organelles such as vacuoles or mitochondria at a high level of detail or study macromolecular complexes such as microtubules, or nuclear pore complexes in situ and under near-native conditions (Figure 4).

Figure 1: FIB and SEM images of vitrified S. cerevisiae
FIB (A) and SEM (C) images of the small yeast clusters vitrified on the TEM grid. FIB (B) and SEM (D) images of the yeast forming a continuous monolayer on the grid surface. The sample was coated with GIS and Irridium layer before imaging. The scale bars in panels A-B corresponds to 10 μm. The yeast sample is blotted against non-absorbent material such as PTFE or FlexFill 98A (green) and with the blotting paper positioned from the backside of the grid (white, E). Please click here to view a larger version of this figure.
{kind=link}

Figure 2: S. cerevisiae lamellae
A TEM image of a lamella micromachined from the sample with continuous monolayer yeast over the grid surface. The reflections observed between the cells contained improperly vitrified medium/buffer (A, highlighted with red circles). A TEM image of lamella generated on the yeast vitrified into a continuous monolayer with the addition of 5% glycerol into the medium/buffer (B). Scale bar corresponds to 2 μm. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Cryo-FIBM workflow
Schematic depiction of the lamella milling process. The initial rough milling steps are performed at high FIB currents from both sides of the tentative lamella position (highlighted in green) whereas the final polishing step is performed only from the top side and at low FIB current (highlighted in orange, see accompanying video at 6:33). Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Yeast organelles and macromolecular complexes depicted by cryo-ET
Slices of the reconstructed tomograms depicting a vacuole (A, scale bar: 200 nm), ribosomes (B, scale bar: 200 nm), a paracrystalline core of peroxisome (C, scale bar: 100 nm), microtubule (white arrow) in the proximity of unidentified fibrous structure (black arrow, D, scale bar: 100 nm), details of multiple microtubules (E, scale bar: 50 nm), a nuclear membrane with pores indicated by arrows (F, scale bar 200nm), mitochondrion (G,H, scale bar: 100 nm, the arrows indicate individual cristae), a bundle of unidentified filamentous structures (I, scale bar: 100 nm). Panels B, C, D, E, G contain a section of tomograms prepared from small clusters of cells whereas the sections of tomograms collected on lamellae from a monolayer of the cells are shown in panels A, F, H, I. Please click here to view a larger version of this figure.
{kind=link}
Discussion
The preparation of the cellular samples for cryo-ET is a complex workflow that requires utilization of several high-end instruments. The sample quality can be compromised during each preparation step that influences the throughput of the whole protocol. In addition, the necessity of the sample transfer between individual instruments poses an additional risk of sample contamination or devitrification. Therefore, optimization of individual steps in the sample preparation workflow is of high importance to increase the throughput and reproducibility of the lamella preparation workflow. The protocol presented here describes the optimized preparation of Saccharomyces cerevisiae for structural characterization of macromolecular complexes in situ by cryo-ET.
The protocol describes the preparation of two types of yeast specimens that mainly differ in the concentration of the cells on the TEM grid. Both yeast samples yielded high-quality lamellae for cryo-ET and selection of the specimen type can be made in accordance with the goals of the particular study. The yeast forms isolated clusters of few cells randomly scattered over the grid surface in the first case, whereas a continuous monolayer of cells is present on the TEM grid surface for the second sample type. The former is suitable for fast lamella preparation thanks to the small volume of the material that must be milled away. The final lamella is fairly short, and, therefore, contains only 2-4 cellular cross-sections. The areas suitable for sample preparation are randomly distributed over the grid surface including the grid squares, which may partially restrict the automation of the lamella preparation workflow. The latter type of the specimen requires utilization of larger currents during the initial milling phase to retain the overall milling time. In addition, this type of sample is more prone to artifacts that stem from uneven milling (curtaining). Therefore, GIS is sputtered onto the sample surface for a 50% longer period than in the case of the sample with small cellular clusters to form a thicker protective layer. Next, the sample is sputtered with an additional layer of Iridium (alternatively platinum or gold) to cure the GIS layer, render it stiffer, and increase the sample surface conductivity. FIBM of additional areas on each side of the lamella (~2-5 μm from the lamella edge) during the first step of the rough lamella milling was found beneficial to diminish the number of broken lamellae most probably due to decreased tension in the final cross-section23. The final lamella is long and contains ~10 cellular cross-sections, which increases the number of regions suitable for cryo-ET. The improper vitrification of the medium or buffer between the cells can be easily attenuated by the addition of the cryo-protectant to the buffer solution (5% glycerol used in this study). Since most of the squares are suitable for lamella preparation, the sample with cells organized into a continuous monolayer is well suited for unsupervised lamella preparation.
Another important aspect in the lamella preparation workflow is the transfer to the transmission electron microscope and proper positioning of the lamella to the microscope stage tilt axis. Optimally, the lamella main axis is perpendicular to the tilt axis of the microscope which affords tracking and focusing at the height of the imaged region and prevents the lamella edges from shielding the field of view at high tilt angles. When collecting the cryo-ET data using the dose-symmetric scheme,18 the sample should be initially rotated in the microscope to compensate for the tilt of the lamella with respect to the grid plane.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was funded by the Technology Agency of the Czech Republic, grant number TN01000008, projects Instruct-Ultra (Grant 731005), iNEXT-Discovery (Grant 871037) funded by Horizon 2020 program of the European Commission, and the CIISB research infrastructure, an Instruct-ERIC Centre (LM2018127). We acknowledge the support obtained from Thermo Fisher Scientific Brno.
Materials
| Name | Company | Catalog Number | Comments |
| Agar | Himedia | MB053 | |
| Glucose | PENTA | 12020-31000 | |
| Glycerol | Merck | G5516-1L | |
| ethane | Messer | 1007 | |
| LN2 | Lineq | LN2-1L | |
| Peptone | Merck | P5905-1KG | |
| Saccharomyces cerevisiae | ATCC | 201388 | strain BY4741 |
| Tweezers | Dumont | T539 | |
| Yeast extract | Duchefa Biochemie | Y1333.1000 | |
| Disposable | |||
| Blotting papers | Ted Pella | 47000-10 | |
| C-clip | ThermoScientific | 9432 909 97551 | |
| C-clip ring | ThermoScientific | 9432 909 97561 | |
| Spreading sticks | Merck | Z376779-1PAK | |
| Sterile inoculation loops | BRAND | BR452201-1000EA | |
| Sterile plastic Petri dishes | Sigma | SIAL0166 | |
| TEM grids | Quantifoil | 4420G-XA | |
| Equipment | |||
| Autoclave | Systec | 101291545 | |
| balances | BEL | M124A | |
| Cryo-FIB/SEM microscope | ThermoScientific | 1006123 | |
| Cryo-TEM microscope | ThermoScientific | 9432 057 03301 | |
| Laminar flow box | Telstar | AH5 | |
| Plasma cleaner | Gatan | 955.82001 | |
| Shaking incubator | New Brunswick | M1282-0002 | |
| UV/VIS spectrophotometer | WPA | S800 | |
| Vitrification robot | ThermoScientific | 9432 053 50621 |
References
- McMullan, G., Faruqi, A., Clare, D., Henderson, R. Comparison of optimal performance at 300keV of three direct electron detectors for use in low dose electron microscopy. Ultramicroscopy. 147, 156-163 (2014).
- Zivanov, J., et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife. 7, (2018).
- Dubochet, J., McDowall, A. W. Vitrification of pure water for electron microscopy. Journal of Microscopy. 124, 3-4 (1981).
- Villa, E., Schaffer, M., Plitzko, J. M., Baumeister, W. Opening windows into the cell: focused-ion-beam milling for cryo-electron tomography. Current Opinion in Structural Biology. 23, 771-777 (2013).
- Mahamid, J., et al. Visualizing the molecular sociology at the HeLa cell nuclear periphery. Science. 351 (6276), 969-972 (2016).
- Schur, F. K. Toward high-resolution in situ structural biology with cryo-electron tomography and subtomogram averaging. Current Opinion in Structural Biology. 58, 1-9 (2019).
- O'Reilly, F. J., et al. In-cell architecture of an actively transcribing-translating expressome. Science. 369, 554-557 (2020).
- Rice, W. J., et al. Routine determination of ice thickness for cryo-EM grids. Journal of Structural Biology. 204, 38-44 (2018).
- Al-Amoudi, A., Norlen, L. P. O., Dubochet, J. Cryo-electron microscopy of vitreous sections of native biological cells and tissues. Journal of Structural Biology. 148, 131-135 (2004).
- Al-Amoudi, A., Studer, D., Dubochet, J. Cutting artefacts and cutting process in vitreous sections for cryo-electron microscopy. Journal of Structural Biology. 150, 109-121 (2005).
- Pierson, J., et al. Improving the technique of vitreous cryo-sectioning for cryo-electron tomography: electrostatic charging for section attachment and implementation of an anti-contamination glove box. Journal of Structural Biology. 169, 219-225 (2010).
- Dubochet, J., et al. How to "read" a vitreous section. Methods in Cell Biology. 79, 385-406 (2007).
- Rigort, A., et al. Focused ion beam micromachining of eukaryotic cells for cryoelectron tomography. Proceedings of the National Academy of Sciences. 109, 4449-4454 (2012).
- Schaffer, M., et al. Cryo-focused Ion Beam Sample Preparation for Imaging Vitreous Cells by Cryo-electron Tomography. Bio-protocol. 5, (2015).
- Wagner, F. R., et al. Preparing samples from whole cells using focused-ion-beam milling for cryo-electron tomography. Nature Protocols. 15 (6), 2041-2070 (2020).
- Buckley, G., et al. Automated cryo-lamella preparation for high-throughput in-situ structural Biology. Journal of Structural Biology. 210 (2), 107488 (2020).
- Zachs, T., et al. Fully automated, sequential focused ion beam milling for cryo-eletron tomography. eLife. e52286, (2020).
- Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. Journal of Structural Biology. 152, 36-51 (2005).
- Hagen, W. J., Wan, W., Briggs, J. A. Implementation of a cryo-electron tomography tilt-scheme optimized for high resolution subtomogram averaging. Journal of Structural Biology. 197, 191-198 (2017).
- Zheng, S. Q., et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nature Methods. 14, 331-332 (2017).
- Rohou, A., Grigorieff, N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. Journal of Structural Biology. 192, 216-221 (2015).
- Stalling, D., Westerhoff, M., Hege, H. -. C. Amira: A highly interactive system for visual data analysis. The Visualization Handbook. , 749-767 (2005).
- Wolff, G., et al. Mind the gap: Micro-expansion joints drastically decrease the bending of FIB-milled cryo-lamellae. Journal of Structural Biology. 208, 107389 (2019).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved