
Method Article
Efficient and Scalable Production of Full-length Human Huntingtin Variants in Mammalian Cells using a Transient Expression System
In This Article
Summary
We provide scalable protocols covering construct design, transient transfection, and expression and purification of full-length human huntingtin protein variants in HEK293 cells.
Abstract
Full-length huntingtin (FL HTT) is a large (aa 1-3,144), ubiquitously expressed, polyglutamine (polyQ)-containing protein with a mass of approximately 350 kDa. While the cellular function of FL HTT is not entirely understood, a mutant expansion of the polyQ tract above ~36 repeats is associated with Huntington's disease (HD), with the polyQ length correlating roughly with the age of onset. To better understand the effect of structure on the function of mutant HTT (mHTT), large quantities of the protein are required. Submilligram production of FL HTT in mammalian cells was achieved using doxycycline-inducible stable cell line expression. However, protein production from stable cell lines has limitations that can be overcome with transient transfection methods.
This paper presents a robust method for low-milligram quantity production of FL HTT and its variants from codon-optimized plasmids by transient transfection using polyethylenimine (PEI). The method is scalable (>10 mg) and consistently yields 1-2 mg/L of cell culture of highly purified FL HTT. Consistent with previous reports, the purified solution state of FL HTT was found to be highly dynamic; the protein has a propensity to form dimers and high-order oligomers. A key to slowing oligomer formation is working quickly to isolate the monomeric fractions from the dimeric and high-order oligomeric fractions during size exclusion chromatography.
Size exclusion chromatography with multiangle light scattering (SEC-MALS) was used to analyze the dimer and higher-order oligomeric content of purified HTT. No correlation was observed between FL HTT polyQ length (Q23, Q48, and Q73) and oligomer content. The exon1-deleted construct (aa 91-3,144) showed comparable oligomerization propensity to FL HTT (aa 1-3,144). Production, purification, and characterization methods by SEC/MALS-refractive index (RI), sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE), western blot, Native PAGE, and Blue Native PAGE are described herein.
Introduction
Huntington's disease (HD) is a rare neurodegenerative disease primarily characterized by unsteady and involuntary motor movement, as well as cognitive and psychiatric alterations, such as personality changes and apathy1,2. HD is associated with an expansion of the CAG repeat tract located in exon 1 of the huntingtin gene (HTT) to more than 35 repeats, with a higher number of CAG repeats correlating with an earlier onset of the disease3,4. The translational product of HTT, the huntingtin protein (HTT), is implicated in neuronal viability and brain development5,6,7,8,9.
HTT is a scaffolding protein reported to participate in a wide range of cellular processes, vesicle transport, cell division, ciliogenesis, and autophagy10,11. However, the molecular pathogenesis of HD is not entirely clear, and the identification of key protein interactors mediating the pathological impact of polyQ-expanded mHTT is lacking. Some research suggests a gain of toxic function from mHTT driven by the oligomerization propensity of the expanded HTT protein, as HTT aggregates have been identified in neurons and glia in HD patients and animal models of the disease12,13,14,15,16,17. To fuel the investigation of the function and structure of FL HTT and mHTT variants and supply researchers with high-quality protein standards for assay development, a robust and scalable supply of homogenous recombinant protein is needed.
Due to its size (aa 1-3,144, numbering based on polyQ length Q23), proteolytic instability, and propensity to aggregate, FL HTT has proven difficult to express and isolate as a soluble protein. Previously, the exon 1 region (aa 2-90) of HTT has been expressed and purified at a large scale using various tags that can increase the solubility of the protein in Escherichia coli18,19,20. FL HTT was first expressed and purified in an insect cell expression system using baculovirus21,22, and low-resolution 30 Å electron microscopy (EM) structures of chemically crosslinked FL Q23-HTT and Q78-HTT were reported23. The investigation of HTT structure was further advanced when the production of FL Q17, Q46, and Q128-HTT with native posttranslational modifications (PTMs) was achieved in human cells using stable cell lines or adenovirus expression systems24. These studies suggest that although purified HTT mainly exists in the monomeric state, it also tends to form high-order oligomers and aggregates.
Analytical ultracentrifugation of FL Q128-HTT, with a highly expanded polyQ region, afforded more oligomeric and aggregate fractions than the protein with the non-expanded polyQ region24. Using a stable cell line, a strategy has been successfully adapted to stabilize FL HTT by co-expression with the interaction partner HAP40. A cryo-EM structure of the FL HTT and HAP40 complex has been solved at an average 4 Å resolution using the purified protein complex (PDB:6EZ8)25. This co-expression strategy has been adapted successfully to a baculovirus system, and a series of high-quality HTT variants with different polyQ lengths have been expressed and purified from insect cells26. Since then, more cryo-EM structures of the complex of HTT with variable polyQ lengths and HAP40 and higher resolution structures were solved and deposited in the Protein Data Base27,28 (PDB: 7DXK, 7DXH, 6X9O).
We optimized a transfection and expression method in HEK293 cells, using polyethylenimine (PEI), for rapid transient expression of FL HTT. As a proof-of-principle, FL HTT variants containing 23 glutamines (FL Q23-HTT) were first purified and characterized using a modification of a purification method described previously24. This transient transfection method is convenient, highly efficient, and scalable; it can produce purified HTT with yields of 1-2 mg/L, comparable to the stable cell line method reported24. Because the protein is produced in a human cell line, the HTT produced is more likely to have native human PTMs when subjected to mass spectrometry proteomics analysis11,29,30,31. Milligram quantities of the FL Q48-HTT, FL Q73-HTT, and exon1-deleted (ΔExon1-HTT) variants of FL HTT were produced, demonstrating that the transient expression method is especially useful in rapidly producing alternative variants of HTT without depending on the time-consuming effort required to establish stable cell lines for production.
The following protocol exemplifies the standard method used in these authors' laboratory for cell culture, transfection, protein purification, and postpurification protein characterization to produce FL Q23-HTT from a 2 L cell culture. The protocol can be scaled up to larger cultures or adapted to purify other HTT variants. Up to 10 L cell cultures of FL HTT and various site or truncation mutations of HTT and HTT homologs have been performed successfully in the laboratory using the same protocol. Purified FL HTT contains a high percentage of monomers along with dimers and higher-order oligomers. The same aggregate profile is observed among the variants produced (Q23, Q48, Q73, and deleted Exon1). As aggregation can occur when proper care is not taken, a formulation and freeze-thaw stability study was conducted to identify the best conditions for protein handling. Methods, such as Blue Native PAGE and SEC/MALS-RI, are also described to analyze the HTT oligomer content as part of the quality control process. To benefit the HD research community, plasmids and HTT proteins described in this study are also deposited in the HD Community Repository at the Coriell Institute (www.coriell.org/1/CHDI).
Protocol
1. Design and production of constructs for FLAG-tagged HTT mammalian expression
- Retrieve the full-length human HTT protein sequence (P42858) from National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/).
NOTE: Researchers need to be familiar with HTT's domain organizations and maintain HTT core 3D structure when designing constructs for HTT mutants. - Request a gene synthesis service to perform codon optimization for human cell expression based on the sequence of P42858. Change the polyQ number from Q16 to the desired Q length (Q23 was chosen as the first construct here) and synthesize the full-length HTT gene.
NOTE: The synthesized codon-optimized full-length Q23-HTT construct was delivered as an insert in the pUC18 plasmid in this study. - Optional: Add features to facilitate the cloning of different Q lengths and purification in the constructs.
NOTE: A tobacco etch virus (TEV) cleavage site and FLAG purification tag (AAAENLYFQGDYKDDDDK) were added to the C-terminal end of the constructs. Two HindIII sites were designed in the constructs to encompass the polyQ region (translated protein sequence is not changed by introducing HindIII sites). This allows the researcher to change HTT's Q length by restriction enzyme digestion and ligation without resynthesizing the full HTT gene.
2. Clone the synthesized HTT constructs into pcDNA3.1.
- Digest 5 µg of pUC18-Q23-HTT and 5 µg of pcDNA3.1 using 2 µL of NheI and PmeI each at 37 °C for 2 h.
- Run a 0.5% w/v agarose gel and purify the Q23-HTT fragment and the digested pcDNA3.1 vector using an agarose gel extraction kit. Quantitate the concentrations of purified DNA by OD280 using a UV spectrometer that can measure microliters of samples.
NOTE: OD260/280 ranging from 1.8 to 2.0 is typically observed. The synthesized FL HTT is supplied as an insert with NheI and PmeI at both ends in a pUC18 plasmid. Use other restriction enzymes if HTT is synthesized differently. - Use 10 ng of digested pcDNA3.1 vector in the reaction. Ligate the purified DNAs at 1:1 (HTT:pcDNA3.1) molar ratio in a 10 µL reaction at room temperature for 5 min using T4 DNA ligase.
- Transform the ligated product into competent E. coli cells (see the Table of Materials) using the protocol specified by the ligase manufacturer.
- Pick up 6 single colonies and make overnight cultures in 4-6 mL of LB supplemented with 100 µg/mL carbenicillin at 37 °C.
- Allocate 1 mL from each overnight culture. Add glycerol to 25% v/v and save the glycerol stock at -80 °C. Purify the remaining overnight culture using a mini-prep kit according to the steps specified in the user's manual.
- Sequence all plasmids using sequencing primers spanning across the transcription region of the plasmid. Choose one glycerol stock that has the correct sequence as the master glycerol stock and discard the rest.
- Optional: Request a gene synthesis service to synthesize DNA with the different Q lengths (Q48, Q73, and Exon1) that span the two HindIII sites in the pcDNA3.1-Q23-HTT plasmid. Digest pcDNA3.1-Q23-HTT and the newly synthesized DNAs using HindIII, and religate them with T4 ligase as in steps 2.2-2.7 to make FL HTT with different polyQ lengths in the pcDNA3.1 plasmid.
NOTE: Plasmid constructs used in this study are also available directly from the HD Community Repository at the Coriell Institute (www.coriell.org/1/CHDI); see the Table of Materials.
3. GIGA prep endotoxin-free plasmid DNA for large scale transfection
- Streak the bacterial glycerol stocks of pcDNA3.1-Q23-HTT-TEV-FLAG on an LB agar plate with carbenicillin (100 µg/mL). Incubate the plate at 37 °C for 16-24 h until single colonies appear.
- Pick up a single colony, inoculate a 5 mL starter culture in a rich medium formulated for plasmid amplification with carbenicillin (100 µg/mL), and grow at 37 °C for 8 h.
- Choose an endotoxin-free GIGA plasmid purification kit. Follow the steps outlined in the manual of the plasmid GIGA kit to purify the pcDNA3.1-Q23-HTT-TEV-FLAG plasmid.
- Measure the plasmid endotoxin levels using a limulus amoebocyte lysate (LAL)-based endotoxin quantification kit. Follow the procedure specified in the manufacturer's manual.
NOTE: A high-quality, low-endotoxin level plasmid purification is essential to obtain good transfection efficiency. Using this protocol, 20-40 mg of plasmid (supercoiled form >80%) can be obtained per L of bacterial culture at plasmid concentrations > 4 mg/mL. A properly purified plasmid should have an endotoxin level < 30 EU/mg. OD260/280 ranging from 1.8 to 2.0 is typically observed.
4. Large-scale transfection of 2 L of HEK293 cells by polyethyleneimine (PEI)
- Add 1 g of PEI 25K to 1 L of endotoxin-free water with stirring. Adjust the pH to 2.0 using 100 mM HCl and stir until all the PEI 25K dissolves. Adjust the pH to 7.0 using 100 mM NaOH solution and filter through a 0.2 µm filter. Aliquot and store at -20 °C for up to a year.
NOTE: Aliquots of PEI can be kept at 4 °C for up to two weeks but should never be refrozen after thawing. - Propagate HEK293 cells in the growth medium (see the Table of Materials) supplemented with penicillin-streptomycin (final concentration at 5 U/mL for penicillin and 5 µg/mL for streptomycin) in a humidified shaker incubator at 37 °C, 90 rpm, 5% CO2 for 18-24 h. Dilute the cells to 2 L at a density of ~1.2 × 106 cells/mL using the growth medium in 5 L Erlenmeyer flasks one day prior to transfection.
- Continue to grow the cells at 37 °C, 90 rpm, 5% CO2 for 18-24 h. Measure the cell parameters using an auto cell counter capable of measuring cell density and viability following the user's manual.
NOTE: The cell density should double, and viability should be >95%. The cell density before the transfection should be approximately 2.0 × 106-2.4 × 106 cells/mL. Dilute the cells to the desired density before transfection when necessary. - Calculate the amounts of plasmid and PEI required for the transfection; use 1 mg of plasmid and 3 mg of PEI for transfection of each liter of cell culture. Allocate 2 mg of plasmid and 6 mg of PEI needed for a 2 L transfection.
- Dilute the plasmid and PEI individually into a volume of phosphate-buffered saline equal to 1/20th of the total volume of cell culture (100 mL each for a 2 L transfection) and incubate at room temperature for 5 min. Mix the diluted plasmid and PEI by gentle swirling and incubate the mixture at room temperature for 30 min.
NOTE: The mixture will appear slightly cloudy after incubation. - Add the mixture to the cell culture and swirl gently to mix them.
- Grow the cells at 37 °C, 5% CO2, 90 rpm for 24 h.
- Add 2 M sodium butyrate solution to a final concentration of 2 mM. Add 1:1000 (v/v) anticlumping agent and 1:1000 (v/v) antifoam to the culture.
- Move the flask to a humidified shaker incubator at 32 °C, 90 rpm, 5% CO2, and continue to grow for 48 h.
- Measure the cell parameters, including cell density and viability, using the auto cell counter following the user's manual.
- Transfer 2.0 × 106 cells (Vol = 2.0 × 106/cell density) in a microcentrifuge tube. Pellet the cells at 2,000 × g for 1 min in a centrifuge for western blotting in section 5.
- Harvest the cells by centrifugation at 2,000 × g for 30 min and store the cell pellet at -80 °C before purification.
5. SDS-PAGE and western blot of HEK293 cell lysate to estimate HTT expression level
- Take an aliquot of 2.0 × 106 cells previously frozen (step 4.11) from the large-scale transfection of HEK293 cell culture. Add 250 µL of Tris-buffered saline (TBS) supplemented with 50 µg/mL digitonin, 5 mM EDTA, and 1x protease inhibitor cocktail, and re-suspend the cell pellet by aspirating several times using a pipette.
- Rotate the tubes gently for 30 min at 4 °C using a minirotator to lyse the cells. Pellet the insoluble material by centrifuging at 17,000 × g for 5 min.
- Add 1/3rd the volume of 4x reducing lithium dodecylsulfate (LDS) loading buffer to the supernatant and heat at 70 °C for 10 min.
- Load 5-20 µL of cell lysate onto a precast 3-8% Tris-acetate PAGE gel. Using the gel-compatible 1x Tris-acetate SDS running buffer, run the gel in a constant voltage mode at 150 V for 60 min.
NOTE: Tris-acetate SDS-PAGE was used for FL HTT analysis because it generates higher resolution than other types of SDS-PAGE for the proteins with molecular weight above 300 kDa. Proteins used in this study are also available directly from the HD Community Repository at the Coriell Institute (www.coriell.org/1/CHDI); see the Table of Materials. - To perform western blotting, assemble a transfer sandwich using a transfer buffer-equilibrated thick transfer paper, a methanol-activated polyvinylidene fluoride (PVDF) membrane, and an SDS-PAGE gel. Transfer the proteins to the PVDF membrane using a semi-dry western blotter according to the manufacturer's user manual.
NOTE: Typically, 20-30 min at 135 mA is sufficient for a 10 cm x 10 cm membrane. - Disassemble the transfer sandwich and block the membrane in TBST (20 mM Tris pH 7.4, 150 mM NaCl, and 0.1% v/v Tween-20) supplemented with 5% w/v non-fat milk.
- Incubate the membrane on a rocker for 1 h at room temperature with 15 mL of primary antibody (1:2,500 dilution for anti-FLAG antibody monoclonal antibody and 1:2,000 for all other primary antibodies).
NOTE: Primary antibodies used in this study are anti-FLAG M2, MAB5492, MAB5490, MAB2166, MAB3E10, MAB4E10, MAB2168, MAB8A4 (see the Table of Materials). - Wash the membrane 3 x 5 min using 30-50 mL of TBST.
- Incubate the membrane on a rocker with a fluorescent dye-conjugated goat anti-mouse IgG secondary antibody at 1:15,000, room temperature, in 15 mL of TBST containing 5% w/v dry milk.
- Visualize the western blot bands on a fluorescent imager using the wavelength specific to the secondary antibody. Quantitate the band signal using the software accompanying the imager per the user's manual.
NOTE: Quantitative western blotting can be performed using purified HTT as the standard. A linear standard range of HTT is instrument-specific and was established in this laboratory from 25 ng to 250 ng of HTT per lane using an anti-FLAG antibody. The western blot of HTT should be free of degradation; a total HTT expression level of 2-4 pg/cell is typically observed. Refer to a previously published protocol32 for details of how to perform a quantitative western blot.
6. Fast protein liquid chromatography (FPLC) purification of HTT using anti-FLAG column and SEC
- Anti-FLAG purification
- Estimate the quantity of FLAG resin needed for purification (typically, 12 mL of anti-FLAG M2 affinity resin for purification of 2-4 L of transfected cell culture). Pack 12-25 mL of anti-FLAG resin onto an empty column (see the Table of Materials) using FPLC at a flow rate of 4 mL/min using Buffer A (Table 1). Adjust the height of the plunger, so there is no gap between the end of the plunger and the bed of resin.
- Using a ratio of 10 mL of Lysis Buffer per 1 g of cell pellet, thaw and suspend the cell pellet in cold Lysis Buffer (Table 1).
- Pass the cell suspension once through a high-shear homogenizer at 10,000 psi. Clarify the lysate by centrifugation at 20,000 × g for 1 h in a centrifuge equipped with a compatible fixed-angle rotor.
- Program the FPLC (see the Table of Materials for the software used in the study) and run the following sequences.
- Load the clarified lysate via the sample pump.
- Wash with 4 column volumes (CVs) of Buffer A (Table 1).
- Wash with 4 CVs of Buffer B (Table 1).
- Wash with 8 CVs of Buffer C (Table 1).
- Wash with 3 CVs of Buffer D (Table 1).
- Wash with 3 CVs of Elution Buffer (Table 1).
- Analyze 10 µL of the peak fractions using SDS-PAGE. Collect and combine the peak fractions with the desired purity. Save ~50 µL of the combined eluates for SDS-PAGE analysis.
NOTE: Normally, a single peak will appear, and all fractions eluted in the peak contain ~90% pure HTT. - Regenerate an anti-FLAG column using 5 CVs of Regeneration Buffer (Table 1) and re-equilibrate the column using 5 CVs of Buffer A.
NOTE: Anti-FLAG resin can be reused up to five times or until the relative yield/liter drops to 50% of the first purification.
- Size exclusion column (SEC) purification using an SEC column
- Preequilibrate an SEC column that allows separation of proteins with molecular weight (MW) > 500 kDa (see the Table of Materials for the column used) using 2 × CV of SEC Buffer (Table 1).
- Directly load the anti-FLAG eluate (from step 6.1.5) via a 50 mL superloop. Run 1.2 × CV of SEC buffer per injection. Run the SEC separation overnight at 4 °C.
NOTE: A maximum of 5 mL or 15 mL of protein sample can be loaded on the SEC columns selected in this study. Program the FPLC so that multiple injections can be performed automatically. Sample method scripts are also included as Supplemental File 1 and Supplemental File 2. - Compare the elution profile with the standard HTT elution profile to distinguish the monomer, dimer, and higher-ordered oligomeric peaks. Pool the monomeric HTT fractions based on the elution profile of the SEC column. If desired, pool the higher-ordered oligomeric and dimeric HTT fractions separately.
- Concentrate the pooled HTT protein using a 100 kDa centrifugal concentrator at 4 °C. Calculate the protein concentrations by dividing their OD280 values by the respective extinction coefficients (the theoretical extinction coefficients of Q23-HTT, Q48-HTT, Q73-HTT, and ΔExon1-HTT are 0.776, 0.769, 0.762, and 0.798 (mg/mL)-1cm-1, respectively, for the calculation). Maintain the HTT concentration ≤ 1.0 mg/mL.
NOTE: It is essential to monitor the concentrating process as overconcentration will result in aggregation. - Aliquot the purified HTT protein in cryo-safe microcentrifuge tubes in a volume < 100 µL. Flash-freeze the aliquots using liquid nitrogen and store them at -80 °C.
7. Analytical HPLC SEC-MALS-dRI to analyze HTT polydispersity
- Perform all analytical SEC-MALS at 4 °C on a high-performance liquid chromatography (HPLC) system coupled with a UV detector, a multiangle light scattering detector, and a differential refractive index (dRI) detector.
- Before connecting the UHPLC column to the system, purge the pump and the detectors with filtered (0.1 µm) HPLC-grade water.
- Connect the UHPLC column (see the Table of Materials for the column used) to the system. Equilibrate the column with filtered (0.1 µm) water and then SEC-MALS buffer (Table 1) until all the detector signals reach baseline.
- Inject 2 µL of 6 mg/mL bovine serum albumin (BSA) at a flow rate of 0.3 mL/min for 15 min per injection and inspect the data quality. Perform normalization, peak alignment, and band broadening correction based on the BSA profile, and create a template for the following HTT sample runs.
- Quickly thaw a vial of the FL Q23-HTT sample in a room-temperature water bath using a float. Filter the HTT through a 0.1 µm spin filter. Inject 2-4 µL of the HTT sample and run for 15 min at 4 °C at a flow rate of 0.3 mL/min.
- Analyze the chromatographic and light-scattering data using accompanying software (see the Table of Materials). Use the dRI detector as a concentration detector and use 0.185 as the refractive index increment (dn/dc) for HTT. Generate a Zimm plot to determine the weight-averaged molecular mass for each peak33,34.
NOTE: Refractive index increment of HTT is calculated as 0.185 using program SEDFIT software35 and primary amino acid sequence of HTT as input.
NOTE: HTT monomer MW is determined by SEC-MALS at ~370 kDa ± 30 kDa. Purified HTT typically has monomer content between 60 and 75% (in this laboratory). Low monomer content may indicate that more care needs to be taken in handling to prevent aggregation.
8. Blue Native PAGE to analyze HTT polydispersity
- Prepare 1 L of Anode Buffer by mixing 50 mL of 20x Blue Native PAGE running buffer (see the Table of Materials) with 950 mL of H2O. Prepare 2 L of dark blue cathode buffer by mixing 100 mL of 20x Blue Native PAGE running buffer and 100 mL of Blue Native PAGE cathode additive (20x) with 1,800 mL of H2O. Chill the buffers to 4 °C before use.
- Quickly thaw a vial of FL Q23-HTT sample in a room-temperature water bath using a float. Keep the thawed protein on ice before use.
- Mix 5 µg of FL Q23-HTT (~1 mg/mL), 1 µL of 0.5% G250 additive, 2.5 µL of 4x Blue Native PAGE sample buffer, and water to bring the final volume to 10 µL.
- Load the mixed FL Q23-HTT sample on a 3-12% precast Bis-Tris gel. Load 7.5 µL of the unstained protein standard in the same gel as the standard.
- Fill the front of the tank with Dark Blue Cathode buffer and the back of the tank with Anode buffer.
NOTE: Fill the buffers after the sample is loaded to allow easy visualization when loading the samples. - Run the gel at 150 V for 120 min in a cold room.
- Destain the gel with Destaining Solution (Table 1) until bands are observed; transfer the gel to water. Visualize and document the gel on an imaging station.
NOTE: Blue Native PAGE was originally designed to analyze membrane proteins. It was adapted in this laboratory as an alternative method to estimate HTT's monomeric content. It binds to the hydrophobic regions of HTT and prevents it from forming aggregates under buffer conditions lacking detergent. Traditional Native PAGE without using Coomassie blue G250 causes HTT to form soluble oligomers and aggregates, likely due to the many hydrophobic pockets existing in HTT.
9. SDS PAGE followed by Coomassie or silver staining to analyze HTT purity
- Add 4x LDS sample buffer and 10x reducing reagent to purified FL Q23-HTT to make the final concentration of loading buffer and reducing reagent to be 1x.
- Heat the sample on a dry heating block at 70 °C for 10 min.
- Load a maximum of 1 µg of protein per well onto a 3-8% Tris acetate gel and run at 150 V for 1 h using Tris-acetate SDS running buffer.
NOTE: Proteins used in this study are also available directly from the HD Community Repository at the Coriell Institute (www.coriell.org/1/CHDI); see the Table of Materials. - Coomassie stain
- Wash the gel with H2O for 5 min.
- Stain the gel in the Coomassie staining solution (Table 1) by rocking the gel in 30 mL of staining solution for 15 min.
- De-stain by rocking the gel in 50 mL of H2O for 5 min. Repeat twice. Visualize and document the Coomassie-stained gel on an imaging station.
- Silver stain using a commercial silver stain kit.
- After SDS-PAGE, fix the gel using Fixing Solution (Table 1) for 1 h to overnight at room temperature.
- Perform the stain, wash, and develop according to the kit's instructions.
- Stop the developing step immediately once the bands reach the desired intensity.
- Document the gel in a gel documentation system equipped with a visible light source.
NOTE: HTT purified at >95% can be detected by Coomassie and silver staining with this protocol. Refer to a previously published protocol32 for details of how to perform quantitative protein analysis.
Results
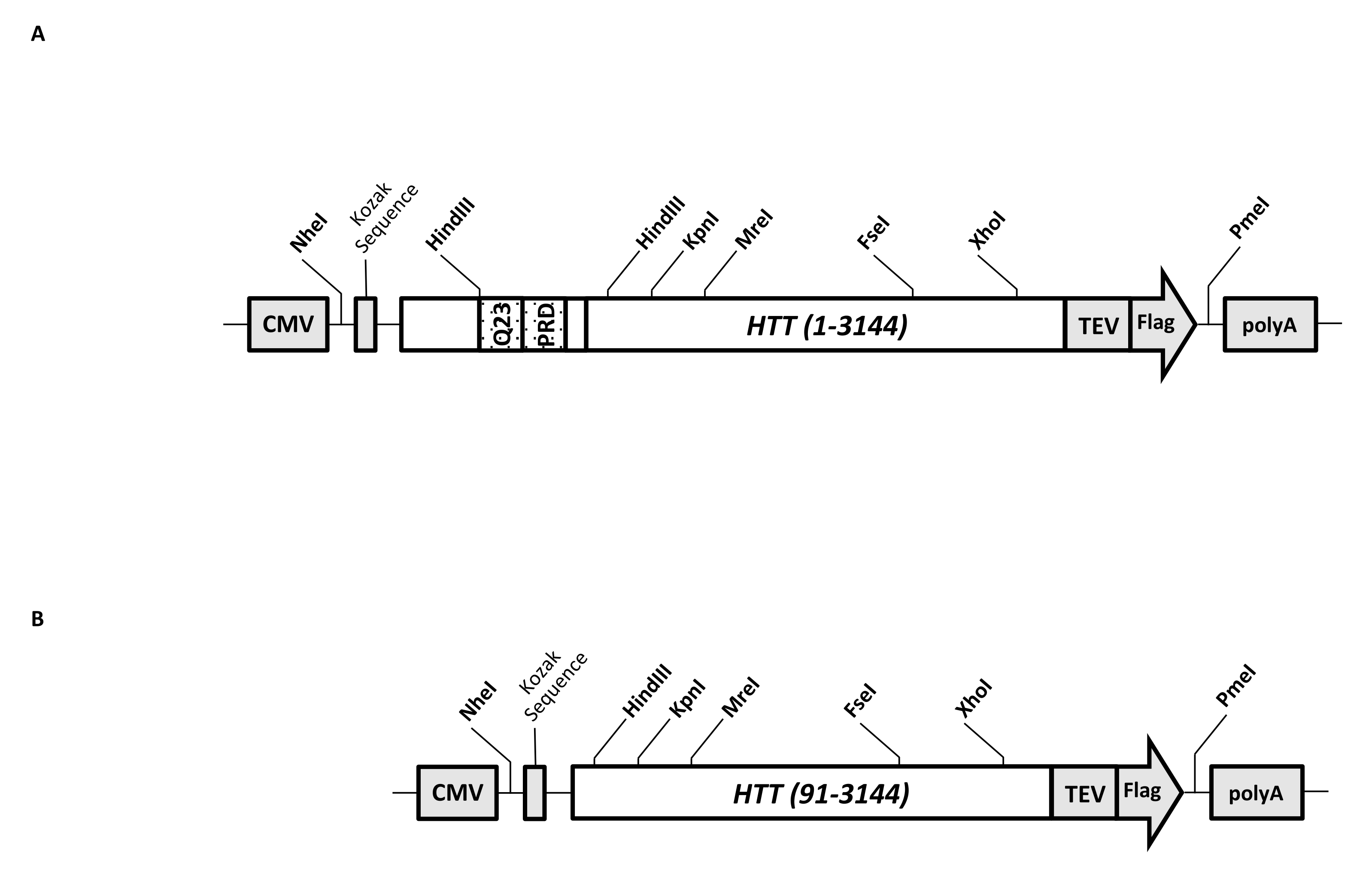
A transient expression vector (pcDNA3.1-Q23-HTT-TEV-FLAG, Figure 1A) is engineered for rapid production in mammalian cells of FL Q23-HTT (aa 1-3,144, based on Q23 numbering). This construct has the features designed to rapidly generate various HTT mutation constructs by cassette cloning, facilitate purification of HTT protein to high quality and homogeneity with minimal chromatographic steps, and have the option to produce untagged FL HTT. The list of the features includes 1. HindIII restriction digestion sites, surrounding the CAG repeat in HTT exon 1, can be used to generate FL HTT mutants with a polyQ stretch of various lengths by restriction enzyme digestion and ligation; 2. the C-terminal end of FL HTT is tagged with a FLAG epitope with a TEV protease recognition site for one-step affinity purification of FL HTT with high purity and optional generation of tag-free FL HTT protein using TEV protease cleavage; 3. Codon-optimized FL HTT sequence for human cell codon usage for high-level expression in HEK293 cells. The pcDNA 3.1 (+) vector is used as the backbone of the construct to take advantage of the high transcriptional activation activity of the CMV promoter in mammalian cell lines.
Using pcDNA3.1-Q23-HTT-TEV-FLAG as the starting template, the Q48 and Q73 FL HTT constructs were produced by synthesizing DNA fragments with proper Q length spanning two HindIII restriction enzyme sites and swapping the same region in the template. The ΔExon1 mutant of FL HTT (aa 91-3,144) (Figure 1B) was produced using primers directed to deleted residues spanning the exon 1 region in the template. HEK293 cells transfected with pcDNA3.1-Q23-HTT-TEV-FLAG using PEI were grown in 5 L shaker flasks under 5% CO2. A typical large-scale purification uses a 2-10 L cell pellet containing 6.0 × 109-3.0 × 1010 cells. Before proceeding to purification, the HTT expression level from each transfection was estimated by quantitative western blotting using purified recombinant FLAG-tagged HTT as a standard and anti-FLAG antibody as the first antibody. Pellets with an estimated HTT expression level at ≥2 pg HTT/cell were used for purification.
Purification of FL HTT consists of a 2-step column process, first with anti-FLAG affinity purification and then with SEC on a gel filtration column with a suitable separation range for HTT (Figure 2A; see Table of Materials for examples). After both steps, HTT was obtained at >95% sample purity, as determined by SDS-PAGE with Coomassie blue and >65% monomer content based on analytical SEC-MALS. Because both prolonged purification time and temperature have a negative impact on the final HTT monomer content, FPLC was used in both purification steps to minimize handling and obtain consistent sample quality. The major contaminant during the anti-FLAG purification was the chaperone Hsp70 as determined by mass spectrometry (Figure 2B, lane 2). This is consistent with the finding that Hsp70 is co-purified with FL HTT stably expressed in human cell lines24, suggesting that Hsp70 may be a common stabilizer for FL HTT in vivo.
Hsp70 contamination can be eliminated by extensively washing with magnesium chloride and ATP during the anti-FLAG affinity purification step (Figure 2B, lane 1). Upon removal of Hsp70, FL HTT is prone to form higher-ordered oligomers24 and has to be maintained at a concentration ≤ 1 mg/mL. The concentration step before SEC can often result in significant aggregation. Therefore, the best practice is to directly load peak fractions from anti-FLAG purification onto the size exclusion column without concentrating. After SEC, the sample was concentrated to ≤1 mg/mL for maximum recovery of monomeric FL HTT. The amount of HTT recovered from each purification step was estimated by either Coomassie blue or quantitative western blotting using purified FL HTT as a quantification standard (Table 2). The typical yield of purified FL HTT proteins produced by the described method is approximately 1 mg/L of cell culture but may fall well below that (Table 3) due to batch-to-batch variability, or if the anti-FLAG purification resin is reused multiple times.
Overexpression of FL HTT can result in fragmentation of the protein22. FL Q23-HTT produced by the method described here resolved as a single band with the correct MW of 350 kDa by SDS PAGE, stained either by Coomassie G250 or by silver staining (Figure 2C). By western blotting, FL Q23-HTT reacted with antibodies raised against epitopes at the N-terminal, C-terminal, and several intermediate domains, with no additional fragment-related bands observed, indicating that the protein was isolated without significant detectable truncations (Figure 3A). FL HTT polyQ length variants Q23, Q48, and Q73 reacted as expected in western blot, showing a progressively stronger signal for polyQ-directed mAb MW1 correlating with increasing Q-length: Q23-HTT < Q48-HTT < Q73-HTT (Figure 3B). No signal was observed for ΔExon1-HTT (aa 91-3,144) when probed with the antibodies MW1 and MAB549, which target the N terminal exon 1 (Figure 3B).
SEC-MALS was employed to analyze the aggregation state and molecular mass of the purified HTT protein. Samples were analyzed by analytical SEC monitored by UV, MALS, and dRI detectors. The absolute molar mass obtained from SEC-MALS does not depend on the shape of molecules33,34; therefore, SEC-MALS provides an unbiased estimation of MW for monomeric and oligomeric fractions when they are well separated. Among the HPLC columns tested, the SEC column (see the Table of Materials) showed sufficient resolution between the HTT monomer and dimer such that molar masses could be distinguished (Figure 4). The protein concentration was determined by dRI detection. Refractive index increments (dn/dc) of FL HTT are 0.1853 mL/g as calculated by the SEDFIT software35. Similar analytical SEC elution patterns were observed for ΔExon1 HTT (91-3,144), FL Q23, Q48, and Q73 HTT (1-3,144), each consisting of a major monomer peak with minor dimeric and oligomeric peaks (Table 4). The calculated MW for the monomeric form is greater than the theoretical MW. This is probably caused by overlapping species from higher-ordered oligomeric peaks and errors resulting from weak dRI signals as HTT proteins are maintained in low concentration to avoid forming higher-ordered oligomers. By integrating the UV peaks of several batches of purified FL HTT variants, no clear correlation between polyQ length and aggregate profile was observed (Table 4).
In addition to analytical SEC, traditional native PAGE was performed to determine whether it can be used as a complementary method to characterize the FL HTT oligomeric state. Higher-ordered oligomers were resolved through 3-8% Tris-acetate gels using native buffer without detergent. Purified FL HTT from SEC showed multiple bands corresponding to the oligomerization states (Figure 5A). The lowest band was located between native marker 480 kDa and 720 kDa, similar to previous results reported for FL HTT purified from insect cells22. However, the HTT monomer was not the most abundant band when using traditional native PAGE, and the results do not correlate with the aggregate profile determined by analytical SEC-MALS. Several hydrophobic patches present in FL HTT36,37,38, especially the hydrophobic interface between HAP40 and FL HTT25, are likely to contribute to the formation of higher-ordered oligomers during migration within the gel. This is because hydrophobic regions are known to interact with each other in the absence of detergent or stabilizing protein-protein interactions. Consistent with the hydrophobic properties of HTT, FL HTT forms increasing amounts of higher-ordered oligomeric fractions in the absence of CHAPS during the SEC purification step.
Blue Native PAGE, which is widely used to investigate membrane proteins and large protein complexes containing hydrophobic patches39, was compared to traditional native PAGE. Purified HTT showed three major bands on Blue Native PAGE with estimated MW of 643, 927, and 1070 kDa (Figure 5B) that likely represent the monomeric, dimeric, and trimeric species of HTT, respectively. The monomer band remained the most abundant band in the Blue Native PAGE, corresponding well to the analytical SEC profile of the same samples. The overestimation of MW of the HTT monomer by Blue Native PAGE may result from the unique hollow spherical structure or hydrophobic regions of HTT that cause slower migration relative to corresponding molecular weight markers11,23,25. Overall, FL Q23-HTT, FL Q48-HTT, FL Q73-HTT, and ΔExon1-HTT have similar Blue Native PAGE profiles with only slight differences in the protein band migration due to their molecular weight differences.
As an additional check of the quality of the purified proteins, the C-terminal FLAG tag can be removed from FL HTT by treatment with TEV protease. After proteolytic cleavage, samples were analyzed by western blot using four antibodies to confirm FLAG tag removal and detect HTT degradation. Immunoreactivity to anti-FLAG M2 and three huntingtin-specific antibodies with epitopes to the N-terminus, intermediate domains, and C-terminus of HTT showed successful FLAG tag removal and no HTT-specific degradation products (Supplemental Figure S1).

Figure 1: Construct for full-length HTT expression. (A) Full-length Q23 HTT was codon-optimized and cloned into pcDNA3.1 (+) plasmid. The 3' end of HTT was tagged with Flag epitope and TEV protease cleavage site to produce tag-free HTT protein. The polyglutamine stretch and proline-rich domain were engineered with flanked HindIII restriction endonuclease sites to insert additional CAG repeats using cassette cloning, i.e., Q48 and Q73, to produce HTT variants with different polyQ lengths. (B) ΔExon1 construct was made PCR mutagenesis using pcDNA3.1-Q23-HTT as the template. Residues 91-3,144 of HTT remained in the ΔExon1 construct for expression. Abbreviations: HTT = huntingtin; CMV = cytomegalovirus; Q23 = polyglutamine stretch; PRD = proline-rich domain; TEV = tobacco etch virus cleavage site. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Large-scale purification of HTT. (A) SEC profile of anti-Flag-purified full-length Q23-HTT on an FPLC column. High-ordered oligomers, dimer, and monomer peaks of Q23-HTT are labeled. Fractions containing monomer were collected as the final HTT sample. (B) SDS-PAGE of purified Q23-HTT with ATP/magnesium washing step (lane 1) or without ATP/magnesium washing results in Hsp70 co-elution (lane 2). (C) Final purified full-length HTT variants on SDS-PAGE stained with Coomassie blue G-250 or silver stain. Abbreviations: FL = full length; HTT = huntingtin; SEC = size exclusion chromatography; FPLC = fast protein liquid chromatography; O = oligomer; D = dimer; M = monomer; SDS-PAGE = sodium dodecylsulfate polyacrylamide gel electrophoresis; Hsp70 = heat shock protein 70. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Western blot analysis of purified HTT Variants. (A) Purified FL Q23-HTT was run on SDS-PAGE and transferred to PVDF membrane.The primary antibodies and interacting epitopes are Lane 1, α-FLAG M2, FLAG tag; Lane 2, MAB5492, HTT aa. 1-82; Lane 3, MAB5490, HTT aa 115-129; Lane 4, MAB2166, HTT aa 181-810; Lane 5, MAB3E10, HTT aa 1,171-1,177; Lane 6, MAB4E10, HTT aa 1,844-2,131; Lane 7, MAB2168, HTT aa 2,146-2,541; Lane 8, MAB8A4, HTT aa 2,703-2,911. (B) 1 µg of purified FL HTT variants were run on SDS-PAGE and transferred to PVDF (left), and a duplicate SDS gel was run and stained with Coomassie Blue (right). The primary antibodies and interacting epitopes are Row 1, MW1, expanded PolyQ repeats; Row 2, MAB2166, HTT aa 181-810; Row 3, MAB5492, HTT aa 1-82. Abbreviations: FLL Q23-HTT = full-length huntingtin protein containing 23 glutamine residues; SDS-PAGE = sodium dodecylsulfate polyacrylamide gel electrophoresis; WB = western blot; M = marker; PVDF = polyvinylidene fluoride. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: SEC-MALS analysis of full-length HTT. Purified full-length Q23-HTT was eluted on a UPLC column. Peak positions of predicted monomer, dimer, and oligomer are indicated. Molecular weights were calculated for monomer, dimer, and trimer peaks and listed in Table 5. Similar elution profiles are observed for Q48, Q73, and ΔExon1 HTT, with variable monomer, dimer, and oligomer contents in each purification. Abbreviations: SEC-MALS = Size exclusion chromatography with multi-angle light scattering; UV = Ultra Violet; LS = Light Scattering; MW = Molecular Weight; Q23-HTT = huntingtin protein containing 23 glutamine residues; M = monomer; D = dimer; O = oligomer. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Characterization of purified HTT using clear Native PAGE or Blue Native PAGE gel. Native Marker and apparent monomeric Q23-HTT from SEC were resolved on 3-8% Tris-acetate gels in a non-denaturing PAGE system (A) and a Blue Native PAGE system (B). Abbreviations: FL = full-length; Q23-HTT = huntingtin protein containing 23 glutamine residues; PAGE = polyacrylamide gel electrophoresis; M = marker. Please click here to view a larger version of this figure.
{kind=link}
| Step | Name | Composition | ||
| 6.1.1 | Buffer A | 50 mM Tris, 500 mM NaCl, 5% v/v glycerol, 5 mM EDTA, 0.01% v/v Tween-20, pH 8.0. | ||
| 6.1.2 | Lysis Buffer | 50 mM Tris, 500 mM NaCl , 5% v/v glycerol, 5 mM EDTA, and 1x protease inhibitor cocktail | ||
| 6.1.4.2 | Buffer A | 50 mM Tris, 500 mM NaCl, 5% v/v glycerol, 5 mM EDTA, 0.01% v/v Tween-20, pH 8.0. | ||
| 6.1.4.3 | Buffer B | 50 mM Tris; 500 mM KCl; 5 mM MgCl2; 5% v/v glycerol; 0.01% v/v Tween-20, pH 8.0 | ||
| 6.1.4.4 | Buffer C | 20 mM Tris; 200 mM KCl; 5 mM MgCl2; 5 mM ATP; 0.01% v/v Tween-20; 5% v/v glycerol, pH 8.0 | ||
| 6.1.4.5 | Buffer D | 50 mM Tris; 500 mM NaCl; 5% v/v glycerol; 5 mM EDTA; 0.5% w/v CHAPS, pH 8.0 | ||
| 6.1.4.6 | Elution Buffer | 50 mM Tris; 500 mM NaCl; 5% v/v glycerol; 0.5% w/v CHAPS; 0.2 mg/mL DYKDDDDK peptide, pH 8.0 | ||
| 6.1.6 | Regeneration Buffer | 0.1 M glycine HCl, pH 3.5; 0.01% v/v Tween-20 | ||
| 6.2.1 | SEC Buffer | 50 mM Tris, 500 mM NaCl, 5% v/v glycerol, 0.5% w/v CHAPS, 1 mM TCEP | ||
| 7.3 | SEC-MALS Buffer | 50 mM HEPES, pH 7.2, 500 mM NaCl, 5% v/v glycerol, 0.5% w/v CHAPS | ||
| 8.7 | Destaining Solution | 40% v/v methanol and 7% v/v acetic acid | ||
| 9.4.2 | Coomassie Staining Solution | 0.01% w /v Coomassie G250, 50% v/v/ methanol, 10% v/v acetic acid | ||
| 9.5.1 | Fixing Solution | 50% v/v methanol , 10% v/v acetic acid, 50 μL of formaldehyde/100 mL of solution | ||
Table 1: Composition of buffers and solutions
| Steps | HTT concentration (mg/ml) | Total Volume (mL) | HTT content (mg) | HTT yield per cell (pg/cell) | % Yield |
| Supernatant | 0.1792 | 220 | 39.4 | 4.4 | 100 |
| Anti-Flag | 1.524 | 8.6 | 13.1 | 1.47 | 33.4 |
| SEC | 0.91 | 3.9 | 3.54 | 0.4 | 9.1 |
Table 2: HTT yield from a 2 L HEK293 pellet transfected with pcDNA3.1-Q23-HTT-TEV-Flag. Abbreviations: FL Q23-HTT = full-length huntingtin protein containing 23 glutamine residues; TEV = tobacco etch virus cleavage site; SEC = size exclusion chromatography.
| HTT Sample | HTT Yield (mg/L) | Avg Purity (%) | ||
| BCA | A280 | |||
| 1 | FL DEx1-HTT (N=3) | 0.67-1.30 | 0.69-1.18 | 99.3 |
| 2 | FL Q23-HTT (N=3) | 0.25-0.92 | 0.28-0.98 | 96.9 |
| 3 | FL Q48-HTT (N=3) | 0.28-1.15 | 0.38-1.16 | 97.4 |
| 4 | FL Q73-HTT (N=3) | 0.58-1.05 | 0.57-0.97 | 98.8 |
Table 3: Summary of the protein yield of four FL HTT variant purifications and their final purity. Abbreviation: FLL HTT = full-length huntingtin protein.
| HTT Sample | A | D | M |
| FL Q23-HTT | 4.2-6.9% | 18.7-29.3% | 66.5-76.0% |
| FL Q48-HTT | 4.0-9.4% | 10.6-17.8% | 73.6-85.4% |
| FL Q73-HTT | 2.0-14.0% | 16.9-24.6% | 65.1-81.1% |
Table 4: Summary of the representative aggregate, dimer, and monomer content of FL HTT variants from the purification. Abbreviations: FL HTT = full-length huntingtin protein; A = aggregate; D = dimer; M = monomer; SEC = size exclusion chromatography.
Supplemental Figure S1: Western blot analysis following TEV protease digestion. Purified FL Q23-HTT and FL Q48-HTT were run on SDS-PAGE, transferred to PVDF membranes, and analyzed by western blotting following TEV digest. Primary antibodies used were anti-Flag M2 (Flag tag), MAB5492 (HTT aa 1-82), MAB3E10 (HTT aa 997-1,276), and MAB2168 (HTT aa 2,146-2,541). Lane 1, Protein Standard; Lane 2, Q23-HTT-TEV-Flag; Lane 3, Q48-HTT-TEV-Flag; Lane 4, Q23-HTT-TEV-Flag treated with TEV protease at 1:5, overnight at 4 °C; Lane 5, Q48-HTT-TEV-Flag treated with TEV protease at 1:5, overnight at 4 °C. Abbreviations: FL HTT = full-length huntingtin protein; SDS-PAGE = sodium dodecylsulfate polyacrylamide gel electrophoresis; TEV = tobacco etch virus; PVDF = polyvinylidene fluoride. Please click here to download this File.
Supplemental Figure S2: SEC-MALS analysis of FL HTT variants subjected to freeze-thaw cycles. Purified Q23-HTT (A) and Q48-HTT (B) were frozen at -80 °C and thawed at room temperature for up to 6 times. Q23-HTT and Q48-HTT after the first freeze-thaw and the sixth freeze-thaw cycles were then analyzed by SEC-MALS. A slight decrease in monomer fraction and increase in dimer and high-order oligomer fractions were observed by light scattering after repeated freeze-thaw cycles. Peak positions of predicted monomer, dimer, and high-order oligomer are indicated. Abbreviations: FL HTT = full-length huntingtin protein; O = oligomer; D = dimer; M = monomer; SEC-MALS = Size exclusion chromatography with multi-angle light scattering. Please click here to download this File.
Supplemental Figure S3: SDS PAGE of FL HTT variants subjected to freeze-thaw cycles. Purified Q23-HTT (lanes 2-7) and Q48-HTT (lanes 9-14) were frozen at -80 °C and thawed at room temperature for up to 6 times. Aliquots of Q23-HTT and Q48-HTT were stored after each freeze-thaw cycle and then analyzed by SDS PAGE. No increase in aggregate or degradation products was observed; the samples were considered stable and >95% pure by band densitometry. Abbreviations: FL HTT = full-length huntingtin protein; SDS-PAGE = sodium dodecylsulfate polyacrylamide gel electrophoresis. Please click here to download this File.
Supplemental File 1: FPLC 15 mL anti-FLAG HTT script. Abbreviations = FPLC = fast protein liquid chromatography; HTT = huntingtin protein. Please click here to download this File.
Supplemental File 2: FPLC SEC_MALS HTT script. Abbreviations: SEC-MALS = Size exclusion chromatography with multi-angle light scattering; FPLC = fast protein liquid chromatography; HTT = huntingtin protein. Please click here to download this File.
Discussion
We describe here a transient transfection, expression, and purification method to generate multiple FL HTT protein constructs with suitable purity and homogeneity for use as standards for immunoassay and MS assay development, controls for western blot analysis, and for structure-function studies. This transient expression method is scalable and versatile and enables the user to generate low-milligram quantities of FL HTT variants more efficiently than using stable cell lines or virus-based methods described previously21,22,23,24. Routinely, 2-5 mg of highly purified FL HTT can be generated from a 2 L scale protein production run in less than a week using the transient expression method once the plasmid is constructed, with a typical yield of 1-2.5 mg of FL HTT per liter of cell culture.
The transient expression method described here overcomes many obstacles in stable cell line expression, such as the long time needed to establish cell lines and difficulties in storage and maintaining stable cell lines. PEI is also relatively inexpensive compared to other transfection reagents in the market, making large-scale transfection economically viable. There are also limitations in the protocol: transfection efficiency largely depends on the quality of the plasmids, optimal cell growth, and how well PEI is stored and prepared. Operators need to take special care and perform quality controls in those critical steps to avoid a drastic drop in protein yields. Anti-FLAG resin used in the protocol is also relatively expensive and shows reduced capture of FL HTT after several purifications and regenerations. Some researchers may find it more practical to switch to a different tag to allow more robust regeneration of the affinity resin.
Various cell lines and expression conditions were tested to optimize FL HTT expression levels. HEK293 cells were chosen for the expression of FL HTT because of the high expression of protein and the ease of handling in a suspension culture format, making the method suitable for large-scale expression in either shakers or bioreactors. A higher FL HTT protein expression level can be achieved at lower culturing temperatures such as 32 °C rather than using the customary temperature of 37 °C. It is possible that the lower temperature may slow protein synthesis and promote correct folding of FL HTT40. However, this phenomenon is not specific to FL HTT or the cell lines tested. The reduced posttransfection temperature has been widely used in pharmaceutical protein expression in CHO cells. Although the mechanism is not fully understood, it is thought that low temperatures arrest the cell cycle in the G1 phase and divert cellular energy to protein production41.
Full-length HTT purified from mammalian cells co-elutes with the chaperone Hsp7024, and Mg-ATP washing steps can remove the Hsp70 protein. Interestingly, co-eluted Hsp70 is not observed in FL HTT purified from an insect cell expression system21,22,23. This may reflect a difference in the PTMs of FL HTT or heat shock protein responses to the overexpression of FL HTT in mammalian and insect cells. Once the recombinant protein has been stripped of Hsp70, non-ionic detergents such as CHAPS or DDM are required to stabilize the monomeric form of FL HTT.
The oligomerization states of FL HTT variants were analyzed using Blue Native PAGE and SEC-MALS. A small fraction of dimeric and higher-order oligomeric HTT was present when analyzed by either Blue Native PAGE or SEC-MALS. Of note, higher-ordered oligomers formed by FL HTT do not appear to correlate with polyQ length, and even the Exon1 deletion mutant displays a similar oligomer-dimer-monomer ratio. The actual variations in oligomer content among these constructs are likely due to minor differences in the production and handling of each batch. In contrast to aggregates and fibrils formed by HTT Exon140,41, the higher-ordered oligomers of FL HTT remained soluble and could be analyzed by SEC and Native PAGE.
Purified monomeric FL HTT is only relatively stable. Prolonged storage at 4 °C, short incubations at room temperature, or concentrations > 1 mg/mL will all convert monomeric FL HTT to dimeric and higher-ordered oligomeric forms even though there is no visible precipitation observed under those conditions. Purified monomeric FL HTT maintained at ≤1 mg/mL remained relatively stable at -80 °C in storage buffer (50 mM Tris, pH 8.0, 500 mM NaCl, 5% v/v glycerol, 0.5% w/v CHAPS, and 5 mM DTT) as previously described24. Up to 6 freeze-thaw cycles of FL HTT prepared and stored in this manner did not cause visible precipitation of the protein, although a slight shift to a higher oligomeric state was observed by SEC-MALS (Supplemental Figure S2). Samples were also analyzed by SDS PAGE following repeated freeze-thaw cycles. No visible precipitates were observed; no aggregates or additional degradation products were seen by SDS-PAGE (Supplemental Figure S3). The long-term stability of purified FL HTT is still under investigation. In the absence of conclusive long-term data, we recommend storing purified FL HTT at -80 °C for no longer than 6 months.
High-quality, recombinant FL HTT protein variants and the methods to produce them are in high demand by the HD research community. These proteins are in use as immunoassay and MS analytical standards, in structural studies, and for the development of novel FL HTT-specific assays. The large-scale transient expression methods described here have consistently produced milligram quantities of FL HTT variants with >95% purity, providing essential tools for HTT studies. Production of tens of milligrams of highly purified FL HTT polyQ variants and other mutants in support of HD research has become routine.
Disclosures
The authors declare that they have no conflicts of interest with the contents of this article.
Acknowledgements
We thank the Department of Pharmaceutical Sciences, The State University of New York at Buffalo, for performing MS analysis of HTT. This work was a collaborative effort with the CHDI Foundation. We specifically thank Elizabeth M. Doherty; Ignacio Munoz-Sanjuan; Douglas Macdonald, CHDI Foundation; and Rory Curtis, Curia, for their invaluable input during the preparation of this manuscript. We are also grateful to Michele Luche, Mithra Mahmoudi, and Stephanie Fox for their support of this research effort.
Materials
| Name | Company | Catalog Number | Comments |
| 100 kDa concentrator-Amicon | Millipore | UFC910096 | Protocol Section Number-6.2.4 |
| 20x blue native PAGE running buffer | Invitrogen | BN2001 | Protocol Section Number-8.1 |
| 20x TBS | Thermo Fisher | PI28358 | Protocol Section Number-5.1 |
| 4x blue native PAGE sample buffer | Invitrogen | BN2003 | Protocol Section Number-8.3 |
| 4x LDS loading buffer | Invitrogen | NP0007 | Protocol Section Number-5.3 |
| 5 L Erlenmeyer flasks | Corning | 431685 | Protocol Section Number-4.2 |
| Agarose gel extraction kit | Qiagen | 28704 | Protocol Section Number-2.2 |
| Anti-clumping agent | Thermo Fisher | 0010057AE | Protocol Section Number-4.8 |
| anti-FLAG M2 affinity gel | Sigma | A2220 | Protocol Section Number-6.1.1 |
| anti-FLAG M2 | Sigma | F3165 | Protocol Section Number-5.7 |
| Anti foam-Excell anti foam | Sigma | 59920C-1B | Protocol Section Number-4.8 |
| ATP | Sigma | A6419 | Protocol Section Number-6.1.4.4 |
| BEH 450 SEC | Waters | 186006851 | 2.5 µm x 4.6 mm x 150 mm Protocol Section Number-7.3 |
| blue native PAGE 5% G-250 sample additive | Invitrogen | BN2004 | Protocol Section Number-8.3 |
| carbenicillin | Thermo Fisher | 10177012 | Protocol Section Number-2.5 |
| centrifuge - Sorvall Lynx 6000 | Thermo Fisher | 75006590 | Protocol Section Number-6.1.3 |
| Cell Counter - ViCELL | BECKMAN COULTER | Protocol Section Number-4.3 | |
| CHAPS | Anatrace | C316S | Protocol Section Number-6.1.4.6 |
| Competent E. coli cells-TOP10 | Invitrogen | C404010 | Protocol Section Number-2.4 |
| digitonin | Sigma | D141 | Protocol Section Number-5.1 |
| differential refractive index detector | Wyatt | Protocol Section Number-7.1 | |
| DYKDDDDK peptide | Genscript | Peptide synthesis service Protocol Section Number-6.1.4.6 | |
| EDTA | Sigma | EDS | Protocol Section Number-5.1 |
| EndoFree Plasmid Giga Kit | Qiagen | 12391 | Protocol Section Number-3.3 |
| Endotoxin free water | Cytiva | SH30529.03 | Protocol Section Number-4.1 |
| endotoxin quantification kit-CRL Endosafe Nexgen-PTS detection system | Charles River | PTS150K | Protocol Section Number-3.4 |
| fixed angle rotor A23-6x100 rotor | Thermo Fisher | 75003006 | Protocol Section Number-6.1.3 |
| FPLC software- Unicorn 6.2 | Cytiva | Protocol Section Number-6.1.4 | |
| Gene synthesis | Genscript | Gene synthesis service Protocol Section Number-1.2 | |
| Glycerol | Fisher Scientific Glycerol (Certified ACS) | G33-4 | Protocol Section Number-5.6 |
| Growth Medium-Expi293 expression medium | Thermo Fisher | A1435102 | Protocol Section Number-4.2 |
| HEK293 cells | Thermo Fisher | R79007 | Protocol Section Number-4 |
| high shear homogenizer-Microfluidizer | MicroFluidics | LM10 | Protocol Section Number-6.1.3 |
| HPLC - 1260 infinity II Bio-Insert HPLC | Agilent | Protocol Section Number-7.1 | |
| Image Studio | LiCor | Image analysis software Protocol Section Number-5.1 | |
| MAB2166 | Sigma | MAB2166 | Protocol Section Number-5.7 |
| MAB2168 | EMD | MAB2168 | Protocol Section Number-5.7 |
| MAB3E10 | Santa Cruz | SC-47757 | Protocol Section Number-5.7 |
| MAB4E10 | Santa Cruz | SC-7757 | Protocol Section Number-5.7 |
| MAB5490 | Sigma | MAB5490 | Protocol Section Number-5.7 |
| MAB5492 | Sigma | MAB5492 | Protocol Section Number-5.7 |
| MAB8A4 | Santa Cruz | SC-47759 | Protocol Section Number-5.7 |
| multi-angle light scattering detector | Wyatt | Protocol Section Number-7.1 | |
| NativeMark Unstained Protein Standard | Invitrogen | LC0725 | Protocol Section Number-8.4 |
| NaCl | Sigma | S9888 | Protocol Section Number-5.6 |
| NheI | New England Biolab | R0131S | Hi-Fi version available Protocol Section Number-2.2 |
| NuPAGE 3–8% Tris acetate gels | Invitrogen | EA0375PK2 | Protocol Section Number-5.4 |
| NuPAGE Tris-Acetate SDS Running buffer | Invitrogen | LA0041 | Protocol Section Number-5.4 |
| PEI 25K | Polysciences | 23966-1 | Protocol Section Number-4.1 |
| Penicillin-Streptomycin | Thermo Fisher | 15070063 | Protocol Section Number-4.2 |
| Phosphate Buffered Saline (PBS) | Cytiva | SH30256.02 | Protocol Section Number-4.5 |
| plasmid miniprep kit | Qiagen | 27104 | Protocol Section Number-2.6 |
| PmeI | New England Biolab | R0560S | Protocol Section Number-2.2 |
| precast Bis-tris gel- 3-12% NativePAGE Novex Bis-Tris Gel | Invitrogen | BN1003BOX | Protocol Section Number-8.4 |
| protease inhibitor cocktail | GoldBio | GB-331-1 | Protocol Section Number-5.1 |
| SEC-MALS analysis software - Astra 7 | Wyatt Technology | Protocol Section Number-7.6 | |
| secondary antibody -IRdye 800 CW goat anti-mouse IgG | LiCor | 926-32210 | Protocol Section Number-5.9 |
| Superose 6 pg XK 16/70 | Cytiva | 90100042 | Protocol Section Number-6.2 |
| Tris base | Fisher | BP152 | Protocol Section Number-5.6 |
| Tween-20 | Thermo Fisher | AAJ20605AP | Protocol Section Number-6.1.1 |
| UV spectrometer - Nanodrop 8000 | Thermo Fisher | ND-8000-GL | Protocol Section Number-2.2 |
| XK26/100 | Cytiva | 28988951 | Protocol Section Number-6.1.1 |
References
- Walker, F. O. Huntington's disease. Lancet. 369 (9557), 218-228 (2007).
- McColgan, P., Tabrizi, S. J. Huntington's disease: a clinical review. European Journal of Neurology. 25 (1), 24-34 (2018).
- Duyao, M., et al. Trinucleotide repeat length instability and age of onset in Huntington's disease. Nature Genetics. 4 (4), 387-392 (1993).
- MacDonald, M. E., et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 72 (6), 971-983 (1993).
- Nasir, J., et al. Targeted disruption of the Huntington's disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 81 (5), 811-823 (1995).
- Dragatsis, I., Levine, M. S., Zeitlin, S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nature Genetics. 26 (3), 300-306 (2000).
- Anne, S. L., Saudou, F., Humbert, S. Phosphorylation of huntingtin by cyclin-dependent kinase 5 is induced by DNA damage and regulates wild-type and mutant huntingtin toxicity in neurons. Journal of Neuroscience. 27 (27), 7318-7328 (2007).
- Dietrich, P., Johnson, I. M., Alli, S., Dragatsis, I. Elimination of huntingtin in the adult mouse leads to progressive behavioral deficits, bilateral thalamic calcification, and altered brain iron homeostasis. PLoS Genetics. 13 (7), 1006846(2017).
- Dragatsis, I., et al. Effect of early embryonic deletion of huntingtin from pyramidal neurons on the development and long-term survival of neurons in cerebral cortex and striatum. Neurobiology of Disease. 111, 102-117 (2018).
- Benn, C. L., et al. Huntingtin modulates transcription, occupies gene promoters in vivo, and binds directly to DNA in a polyglutamine-dependent manner. Journal of Neuroscience. 28 (42), 10720-10733 (2008).
- Saudou, F., Humbert, S. The biology of huntingtin. Neuron. 89 (5), 910-926 (2016).
- Davies, S. W., et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 90 (3), 537-548 (1997).
- DiFiglia, M., et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 277 (5334), 1990-1993 (1997).
- Gutekunst, C. A., et al. Nuclear and neuropil aggregates in Huntington's disease: Relationship to neuropathology. Journal of Neuroscience. 19 (7), 2522-2534 (1999).
- Hodgson, J. G., et al. A YAC mouse model for Huntington's disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 23 (1), 181-192 (1999).
- Hoffner, G., Djian, P. Polyglutamine aggregation in Huntington disease: does structure determine toxicity. Molecular Neurobiology. 52 (3), 1297-1314 (2015).
- Waldvogel, H. J., Kim, E. H., Tippett, L. J., Vonsattel, J. P. G., Faull, R. L. M. The neuropathology of Huntington's disease. Current Topics in Behavioral Neurosciences. 22, 33-80 (2014).
- Kim, M. Beta conformation of polyglutamine track revealed by a crystal structure of huntingtin N-terminal region with insertion of three histidine residues. Prion. 7 (3), 221-228 (2013).
- Hoop, C. L., et al. Huntingtin exon 1 fibrils feature an interdigitated β-hairpin-based polyglutamine core. Proceedings of the National Academy of Sciences of the United States of America. 113 (6), 1546-1551 (2016).
- Vieweg, S., Ansaloni, A., Wang, Z. M., Warner, J. B., Lashuel, H. A. An intein-based strategy for the production of tag-free huntingtin exon 1 proteins enables new insights into the polyglutamine dependence of Httex1 aggregation and fibril formation. Journal of Biological Chemistry. 291 (23), 12074-12086 (2016).
- Seong, I. S., et al. Huntingtin facilitates polycomb repressive complex 2. Human Molecular Genetics. 19 (4), 573-583 (2009).
- Li, W., Serpell, L. C., Carter, W. J., Rubinsztein, D. C., Huntington, J. A. Expression and characterization of full-length human huntingtin, an elongated HEAT repeat protein. Journal of Biological Chemistry. 281 (23), 15916-15922 (2006).
- Vijayvargia, R., et al. Huntingtin's spherical solenoid structure enables polyglutamine tract-dependent modulation of its structure and function. eLife. 5, 11184(2016).
- Huang, B., et al. Scalable production in human cells and biochemical characterization of full-length normal and mutant huntingtin. PLoS ONE. 10 (3), 0121055(2015).
- Guo, Q., et al. The cryo-electron microscopy structure of huntingtin. Nature. 555 (7694), 117-120 (2018).
- Harding, R. J., et al. Design and characterization of mutant and wildtype huntingtin proteins produced from a toolkit of scalable eukaryotic expression systems. Journal of Biological Chemistry. 294 (17), 6986-7001 (2019).
- Harding, R. J., et al. HAP40 orchestrates huntingtin structure for 1 differential interaction with polyglutamine 2 expanded exon 1. bioRxiv. , (2021).
- Huang, B., et al. Pathological polyQ expansion does not alter the conformation of the Huntingtin-HAP40 complex. Structure. 29 (8), 804-809 (2021).
- Colin, E., et al. Huntingtin phosphorylation acts as a molecular switch for anterograde/retrograde transport in neurons. EMBO Journal. 27 (15), 2124-2134 (2008).
- Thompson, L. M., et al. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. Journal of Cell Biology. 187 (7), 1083-1099 (2009).
- Ratovitski, T., et al. Post-translational modifications (PTMs), identified on endogenous Huntingtin, cluster within proteolytic domains between HEAT repeats. Journal of Proteome Research. 16 (8), 2692-2708 (2017).
- Taylor, S. C., Berkelman, T., Yadav, G., Hammond, M. A defined methodology for reliable quantification of western blot data. Molecular Biotechnology. 55 (3), 217-226 (2013).
- Tarazona, M. P., Saiz, E. Combination of SEC/MALS experimental procedures and theoretical analysis for studying the solution properties of macromolecules. Journal of Biochemical and Biophysical Methods. 56 (1-3), 95-116 (2003).
- Folta-Stogniew, E. Oligomeric states of proteins determined by size-exclusion chromatography coupled with light scattering, absorbance, and refractive index detectors. Methods in Molecular Biology. 328, Clifton, N.J. 97-112 (2006).
- McMeekin, T. L., Wilensky, M., Groves, M. L. Refractive indices of proteins in relation to amino acid composition and specific volume. Biochemical and Biophysical Research Communications. 7 (2), 151-156 (1962).
- Atwal, R. S., et al. Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity. Human Molecular Genetics. 16 (21), 2600-2615 (2007).
- Kegel-Gleason, K. B. Huntingtin interactions with membrane phospholipids: Strategic targets for therapeutic intervention. Journal of Huntington's Disease. 2 (3), 239-250 (2013).
- Michalek, M., Salnikov, E. S., Werten, S., Bechinger, B. Membrane interactions of the amphipathic amino terminus of huntingtin. Biochemistry. 52 (5), 847-858 (2013).
- Wittig, I., Braun, H. P., Schägger, H. Blue native PAGE. Nature Protocols. 1 (1), 418-428 (2006).
- Nissley, D. A., O'Brien, E. P. Altered co-translational processing plays a role in huntington's pathogenesis-A hypothesis. Frontiers in Molecular Neuroscience. 9, 54(2016).
- Kumar, N., Gammell, P., Clynes, M. Proliferation control strategies to improve productivity and survival during CHO based production culture: A summary of recent methods employed and the effects of proliferation control in product secreting CHO cell lines. Cytotechnology. 53 (1-3), 33-46 (2007).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved