
Method Article
A Gravity-Fed Transcardial Perfusion Method for Histologic Analysis of the Mouse Central Nervous System
In This Article
Summary
A convenient gravity-fed perfusion method for histological analysis of the mouse central nervous system is presented. Immunofluorescent detection of phosphorylated α-synuclein is demonstrated in a mouse model of Parkinson's disease. This work also comprehensively describes the transcardial perfusion, dissection, tissue freezing/embedding, and frozen sectioning steps.
Abstract
The histologic analysis of brain and spinal cord specimens isolated from mice is common practice for the assessment of pathology in this model system. To maintain the morphology of these delicate tissues, it is routine to administer a chemical fixative such as paraformaldehyde via cannulation of the heart in anesthetized animals (transcardial perfusion). Transcardial perfusion of the mouse heart has traditionally relied on the use of peristaltic pumps or air pressure to deliver both the saline and fixative solutions necessary for this process. As an easily accessible alternative to these methods, this work demonstrates the use of a gravity-fed method of perfusate delivery that uses materials available in most hardware stores.
To validate this new perfusion method, this work demonstrates all the subsequent steps necessary for the sensitive detection of phosphorylated α-synuclein in both the brain and spinal cord. Included in these steps are the dissection of the fixed brain and spinal cord tissues, rapid freezing/embedding and cryosectioning of the tissues, and immunofluorescent staining. As this method results in whole-body delivery of the fixative, it may also be used to prepare other non-neuronal tissues for histologic analysis.
Introduction
Histologic characterization of pathology in the mouse central nervous system (CNS) is a routine technique used in studies of neurodegeneration. As neuronal tissues rapidly degrade after death, it is common practice to deliver a chemical fixative such as paraformaldehyde to the CNS tissues to preserve their morphology1,2. Chemical fixation can be performed either through whole-body perfusion with a fixative solution or through the isolation of tissues and their immersion in a fixative solution (termed "drop fixation"). Perfusion is the preferred method of fixative delivery, as drop fixation may not allow rapid penetration of the fixative solution into deep CNS structures3,4,5. Furthermore, as it is difficult to remove the unfixed spinal cord from the vertebral column, delivery of the fixative solution via perfusion allows for the in situ preservation of spinal cord microscopic and gross anatomy and stiffens the tissue to minimize damage during removal.
Delivery of the buffer and fixative solutions necessary for fixation is commonly performed using commercially available pumps or air pressure. Gravity delivery of perfusate may serve as an alternative to pump delivery for the following reasons: (1) Pump or air pressure delivery may in some cases require a user to manually maintain pressure in the system throughout the perfusion. Gravity delivery of perfusate can be maintained without user intervention. (2) A gravity-delivered perfusate apparatus can be constructed at low cost to the user by obtaining materials available from standard scientific vendors. This work describes how to construct a simple gravity perfusion device using wash bottles and vinyl tubing. Using a mouse model of Parkinson's disease, this work demonstrates the efficacy of this system in perfusing the brain and spinal cord tissues prior to their isolation for frozen sectioning. This work comprehensively describes all steps, techniques, and materials needed to dissect the fixed tissue out of the animal, rapidly freeze/embed and cryosection the tissue, and detect the presence of phosphorylated α-synuclein in both the brain and spinal cord via indirect immunofluorescence microscopy.
Protocol
The data and experimental steps presented in this protocol were generated using C57BL/6J mice. All methods involving animals were approved by the State University of New York Upstate Medical University Institutional Animal Care and Use Committee.
1. Construction of perfusion apparatus and dissection platform
- As shown in Figure 1, trim the inner straws from two 500 mL wash bottles by cutting these flush to the inner side of the screw-on cap with a sharp razor blade. Cut a 4 cm x 4 cm square hole in the bottom of each buffer bottle. Cut a small hole in the bottom of each bottle to allow the passage of a bent, stainless steel microspatula.
- Create an "S" shaped bend in the microspatulas so that they may be used as hooks for hanging the buffer bottles. Insert the bent microspatulas into the appropriate openings in the buffer bottles.
- Cut two 25 cm lengths of tubing and tightly connect these with the outer bottle straw outlets. Join the ends of these tubes together with the Y connector. Cut 2 m of tubing and join this to the free end of the Y connector.
- Remove the plunger from the 1 mL syringe and cut the syringe approximately 6 cm away from the syringe tip. Cut the syringe by first scoring the plastic with a razor blade and then sharply snapping the syringe plastic.
- Insert the cut face of the syringe into the free end of the plastic tubing. Insert to a sufficient depth to ensure a tight seal.
NOTE: It is important that all connections within the perfusion apparatus are sufficiently tight to avoid leakage. In the case of loose connections, small stainless steel hose clamps can be placed and tightened at junctions as desired to ensure a tight seal if necessary. - Label one buffer bottle as PFA and one bottle as PBS (buffer). Measure approximately 1/3rd of the length away from the bottle opening and draw a line around the bottle circumference at this point to denote the appropriate filling level of the perfusate solutions.
- Straighten and then create a loop in a large paper clip that can fit around the circumference of the tubing. Place this loop around the tubing just proximal to the syringe.
- Place an appropriately sized Styrofoam block in the glass tray. Place the ends of the paperclip into the Styrofoam block to affix the tubing.
- Using paper towels, raise the front end of the glass tray by approximately 2 cm.
NOTE: This will place the mouse in roughly 20° of the Trendelenburg position (backward tilt) to allow for easier visualization of the diaphragm during dissection and encourage the drainage of fluids during perfusion.

Figure 1: Diagram depicting the assembled perfusion apparatus. Abbreviations: PBS = phosphate-buffered saline; PFA = paraformaldehyde. Please click here to view a larger version of this figure.
{kind=link}
2. Preparation of paraformaldehyde (PFA) solution
NOTE: PFA solution must be prepared fresh on the day of perfusion and discarded at the end of the perfusion in a designated waste container prior to disposal by trained personnel. This protocol makes 1 L of 4% PFA solution, which is sufficient to perfuse approximately 4 mice. PFA is highly toxic, and care must be taken to avoid inhalation or direct skin contact in either the powdered or liquid form. Most preparation steps are therefore performed while wearing gloves, protectove goggles, and a lab coat under a fume hood.
- Rinse a 1 L beaker using distilled water and fill with approximately 800 mL of 18 mΩ molecular-biology-grade H2O.
- Heat the beaker of H2O in a microwave for 3 min or until the water temperature reaches 65 °C. Place on a heating/stirring plate kept in a fume hood.
- Rinse a stirring rod with distilled water and place it in the hot water. Start the stirrer and turn the hot plate up to medium heat. Ensure that the water temperature does not go above 70 °C.
- Wear a surgical mask, gloves, protective goggles, and lab coat and measure 40 g of PFA powder. Pour this powder into the heated water.
- Using a transfer pipette, add a few drops of 5 M NaOH to the solution. Allow the PFA powder to dissolve fully. If the powder has not fully dissolved after a few minutes, add drops of 5 M NaOH as needed.
- Once nearly all the PFA has dissolved (will appear slightly turbid), stop the stirring/ heating and immediately add 100 mL of 10x phosphate-buffered saline (PBS). Finally, top up the water to the 1 L mark on the beaker using 18 mΩ molecular-biology-grade H2O. Cover the beaker with plastic wrap and place it in a -20 °C freezer until the solution reaches room temperature (approximately 45 min).
- Calibrate a pH meter using the appropriate standards. While the beaker is on a stirring plate, measure the pH of the solution and add HCl until the pH reaches 7.4. If necessary, add 5 M NaOH to increase the pH if it is too low.
- Connect a vacuum flask to vacuum and place a clean ceramic Büchner funnel with filter paper in the flask. Turn on the vacuum and wet the filter paper using a transfer pipette filled with the 4% PFA solution.
- Slowly pour the 4% PFA solution onto the filter paper until all the solution has been filtered.
- Transfer the filtered solution to a clean, light-protected container and store it at 4 °C until use (store no longer than 24 h).
3. Transcardial "pump-free" perfusion
- In the fume hood, place a Styrofoam dissecting block in a glass tray. Ensure that the dissecting block has 5-6 short needles for restraining the mouse during surgery and 2 long needles for supporting the perfusion tubing.
- Rinse the perfusion apparatus using distilled water. Allow all the water to drain prior to hanging the perfusion apparatus. Hang the perfusion bottles 1 m above the perfused animal (for a 20-30 g mouse).
- Prepare a non-sterile solution of 1x PBS using 10x PBS diluted with 18 mΩ molecular-biology-grade H2O.
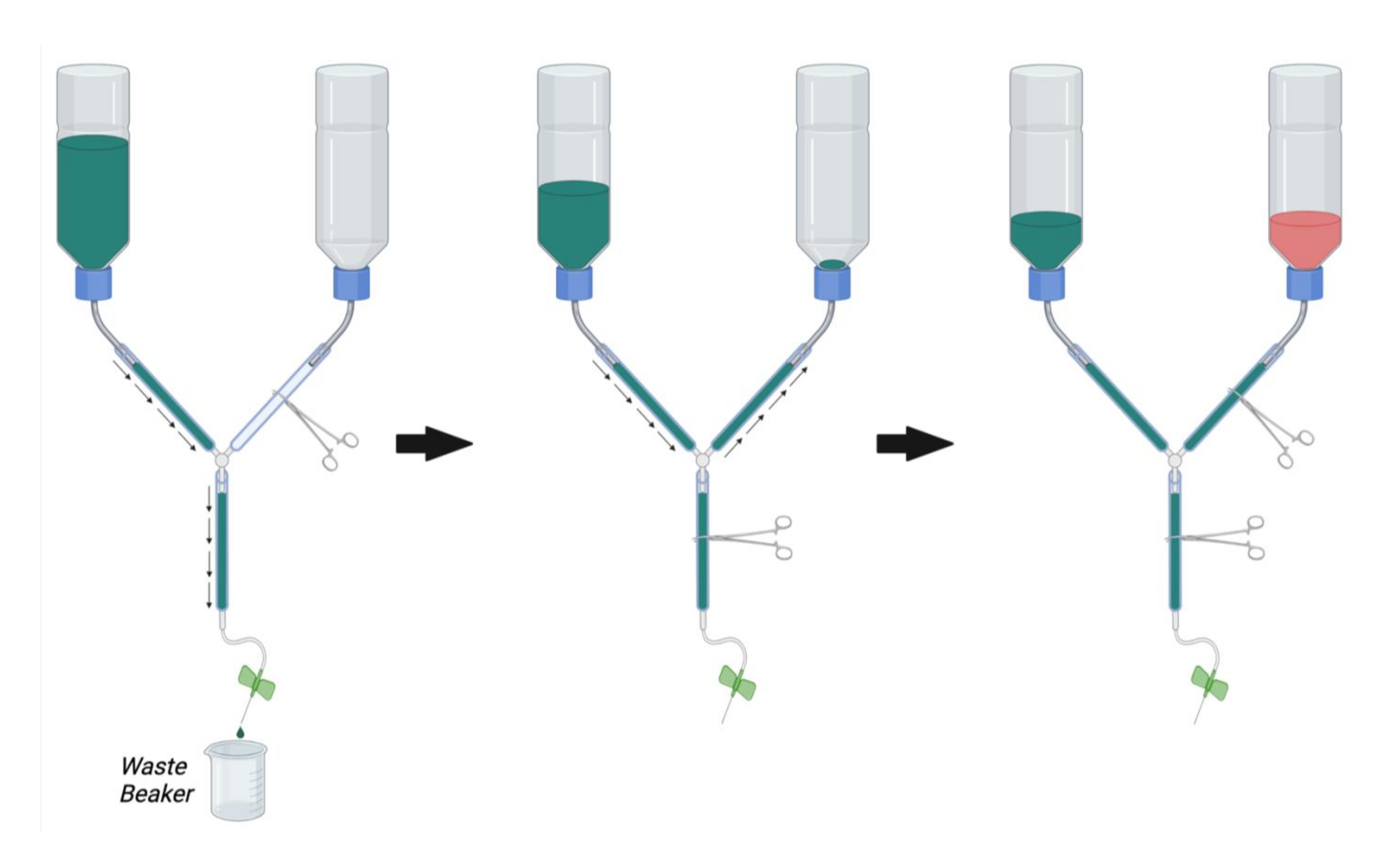
- As shown in Figure 2, clamp the main line of the perfusion apparatus using a hemostat. Clamp the PFA line with another hemostat.
NOTE: Occlusion with multiple hemostats may be necessary per line to completely prevent flow. - Fill the buffer container with 1x PBS at room temperature.
- Place the syringe end of the perfusion apparatus in a beaker to collect waste buffer and remove the hemostat occluding the main line (Figure 2, left).
- Allow PBS to flow through the line while removing trapped air by vigorously tapping on the walls of the tube.
- Once all the air has been removed from the buffer and main lines, occlude the flow by placing a hemostat on the main line.
- Remove the hemostat from the PFA line and allow the PBS to flow in a retrograde manner up to the PFA bottle while tapping out any bubbles in the PFA line (Figure 2, middle). Continue to allow the PBS into the PFA line until PBS can be seen just above the opening of the bottle. Occlude the PFA line with a hemostat to stop the flow into the PFA bottle.
- Connect the butterfly infusion needle to the perfusion syringe and flush PBS through the line (by opening the main line hemostat) to remove bubbles from the perfusion syringe. Close the main-line hemostat.
- Ensure that the PBS bottle is now approximately 1/3rd full of PBS. If necessary, flush PBS through the main line or fill more PBS into the buffer bottle until 1/3rd of its full capacity.
- Once all bubbles have been removed from the PBS, PFA, and main lines, fill the PFA bottle with 4% PFA solution at room temperature up to the black mark on the bottle (approximately 1/3rd full) (Figure 2, right).

Figure 2: Preparation of the perfusion apparatus for perfusion surgery. First, close the PFA line hemostat and open the hemostat on the PBS line and the main line. Fill PBS and remove bubbles from the PBS line and main line. Next, fill the PFA line with PBS by opening the hemostat on the PFA line and closing the hemostat on the main line. Remove bubbles in the PFA line. Finally, close the hemostat on the PFA line when the PBS reaches the opening of the PFA bottle. Fill the PFA bottle 1/3rd full of PFA. Ensure that the level of PBS in the PBS bottle is 1/3rd full and either fill with PBS or drain PBS by opening the main line hemostat if necessary. Abbreviations: PBS = phosphate-buffered saline; PFA = paraformaldehyde. Please click here to view a larger version of this figure.
{kind=link}
- Prepare for non-survival surgery by cleaning the following instruments with water followed by 70% ethanol: large scissors, fine dissecting scissors, skin forceps, fine fenestrated curved forceps.
- Prepare the anesthesia by placing dust-free wipes in a 50 mL conical tube. In a fume hood, soak the dust-free wipes thoroughly with isoflurane and place the opened tube upside down in a 500 mL beaker. Ensure that no liquid isoflurane is present at the bottom of the beaker and discard any liquid isoflurane prior to placing a mouse in the beaker.
- Place the mouse in the beaker and place plastic wrap over the opening to begin anesthesia.
- Administer anesthesia until the mouse stops breathing (approximately 1 min and 30 s).
- When breathing has stopped, immediately remove the mouse from the beaker and replace the cap on the 50 mL conical tube.
- Check that the mouse is sufficiently anesthetized by using the toe pinch reflex. If the animal is not sufficiently anesthetized, administer more isoflurane as described in step 3.15.
- Working quickly, place the mouse on the Styrofoam block and pin the paws outwards using four short needles (e.g., 22 G syringe needles).
- Lifting the abdominal skin with skin forceps, use the large scissors to cut through the abdominal wall. Ensure that no abdominal organs are cut!
- Continue the abdominal cut superiorly towards the liver. Detach the liver from the anterior abdominal wall and continue the initial incision superiorly towards the diaphragm. Stop this incision approximately 1 cm inferior to the diaphragm. Take care to ensure that the liver is not cut!
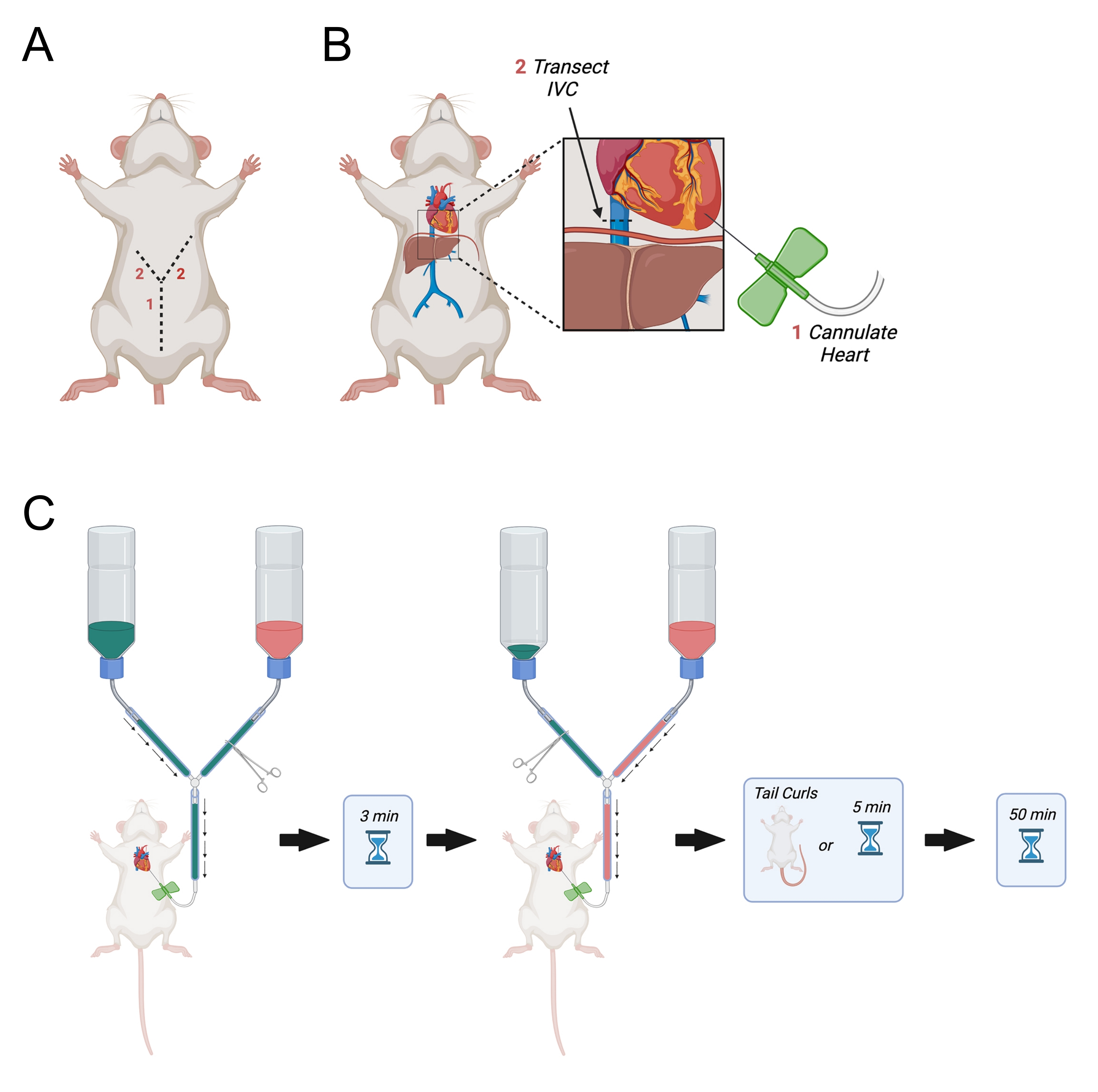
- Continue the initial incision laterally up towards the right and left sides of the diaphragm to make a "Y" shaped incision (Figure 3A).
- When the incision reaches the diaphragm, make a hole in the diaphragm using the fine dissecting scissors and cut through the ribs on the animal's right side. Cut the ribs approximately halfway to the right axilla.
- Make a similar but longer incision through the left side of the diaphragm almost all the way to the left axilla.
- Reflect the chest wall and pin it to the Styrofoam block.
- If necessary, dissect away the fat from around the heart to expose the left ventricle. Identify the left ventricle based on the relatively lighter color when compared with the right ventricle.
- After identifying the left ventricle, hold the heart steady using light pressure with the fine curved forceps. Open the main-line hemostat to allow the PBS to flow through the needle. Immediately pierce the left ventricle and assure that the needle is inserted no more than 0.5 cm into the ventricle. Withdraw the needle slightly if necessary.
NOTE: Accurate placement of the needle in the left ventricle is indicated by retrograde flow of blood into the needle tubing. It is important to ensure that the needle is not too deeply inserted into the ventricle. This can result in retrograde flow through the pulmonary veins or piercing the interventricular septum. - Rest the butterfly tubing on an X made in the Styrofoam using two large 18 G needles.
NOTE: This is critical as it will avoid movement of the butterfly needle within the heart during perfusion. - Identify the inferior vena cava as it exits the liver and transect it using the fine dissecting scissors (Figure 3B). Alternatively, open the right atrium with scissors.
- Immediately open the main line to allow PBS perfusion to start (Figure 3C).
- Assure that the PBS is draining off the Styrofoam block and into the glass tray below. If this is not occurring, readjust the angle of the tray or the Styrofoak block to allow draining of fluids into the glass tray.
- Continue PBS perfusion until the liquid flowing out of the inferior vena cava is blood-free (approximately 3 min).
NOTE: Proper placement of the needle in the heart will result in the visual clearing of blood from the liver, which will turn from red to straw-colored. If this does not occur, it may be remedied by repositioning the infusion needle within the left ventricle. A small incision can be made in the ventral tail base to assess the quality of perfusion. Proper perfusion will result in clear PBS flowing from the tail base incision. - Working quickly, occlude the PBS line with a hemostat and open the PFA line.
- Allow PFA to perfuse for a few minutes until the tail starts to curl. Once the tail starts to curl, begin a 50 min timer. If the tail does not curl after 5 min of PFA perfusion, begin a 50 min timer for the PFA perfusion.
- Monitor the PFA level in the bottle continuously throughout the perfusion to ensure that the level does not drop too low. Fill more PFA in the bottle if the level drops below 2 cm above the bottle lid.

Figure 3: Diagram depicting transcardial perfusion. (A) The abdominal wall is first cut, followed by two laterally pointing incisions towards the axilla forming a "Y." (B) After entering the thoracic cavity and exposing the heart, the needle is passed into the left ventricle. Next, the IVC or right atrium is transected to allow for drainage of perfusates after they have circulated through the body. The IVC is cut superior to the diaphragm. (C) Procedure for administration of perfusates. Abbreviation = IVC = inferior vena cava. Please click here to view a larger version of this figure.
{kind=link}
- After 50 min have elapsed, occlude the main line with a hemostat and withdraw the needle from the left ventricle.
NOTE: The entire mouse is stiff at this point and can now be placed in a labeled container of 4% PFA overnight at 4 °C. At this stage, it is possible to dissect out the CNS tissues and place those individually in fixative overnight. In this work, the entire mouse is placed in fixative as this shortens the interval between fixation and placement at 4 °C. This also avoids any possible mechanical distortion of nervous tissue that may occur if animals are pre-dissected before overnight fixation is complete.
4. CNS dissection
- Prepare the following instruments for dissection by washing with water followed by 70% ethanol: scissors, skin forceps, curved fenestrated forceps, curved fine forceps, straight fine forceps, fine scissors.
- Label four tubes per mouse as follows and fill with sterile 20% sucrose: Mouse ID, Brain 1, Mouse ID, Brain 2, Mouse ID, Lumbar, Mouse ID, Cervical.
- Remove the mouse from the PFA solution and blot it dry with a paper towel to remove excess PFA.
- Using the large scissors, remove the head of the mouse by cutting at the neck.
- Place the body in the PFA solution. Retain the head to dissect the brain from the skull.
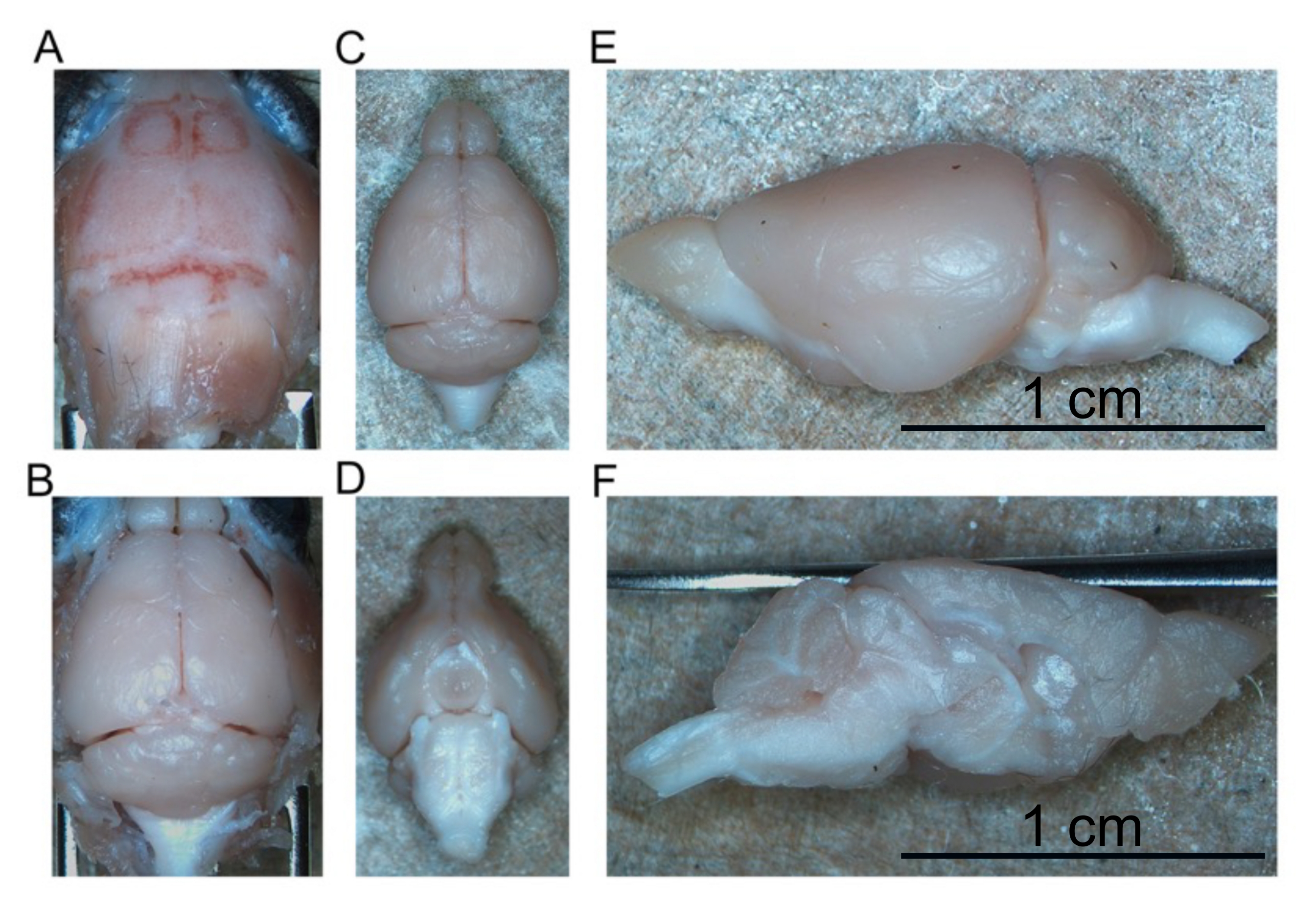
- To dissect the brain, begin by reflecting the cranial skin forward to expose the entire skull (Figure 4A).
- Make a shallow cut into the auditory canal using the fine dissecting scissors to gain entry into the skull
- Continue this cut along the transverse sinus until the skull is cut all the way between both auditory canals.
- Make a cut perpendicular to the previous cut along the longitudinal fissure all the way to the most rostral portion of the skull (approximately between the eyes).
- Using the fenestrated curved forceps, reflect the skull laterally to expose the forebrain. Continue removing the skull until the entire forebrain, including the olfactory bulbs, are exposed.
- Begin dissecting the caudal region of the skull by making a cut through the occipital bone and slowly continuing this cut down towards the foramen magnum.
- Reflect the two pieces of skull laterally to expose the cerebellum and brainstem caudally (Figure 4B).
- Use the ultrafine curved forceps to separate the olfactory bulbs from the anterior skull and begin peeling the brain away from the skull base starting at the olfactory bulbs.
- As the brain is peeled away from the skull base, use ultrafine forceps to cut the cranial nerves and allow removal of the brain.
- Continue to peel away the brain from the skull base until the entire intact brain is removed (Figure 4C,D).
- Cut the brain in half along the longitudinal fissure to separate the right and left hemispheres from one another (Figure 4E,F).
- Place one hemisphere in the brain 1 tube and the second hemisphere in the brain 2 tube. Store these tubes at 4 °C until the brain hemispheres sink (approximately overnight).

Figure 4: Removal of fixed brain. (A) Top of the skull. (B) Exposed brain within the skull. (C) Isolated brain (dorsal aspect). (D) Isolated brain (ventral aspect). (E) Left hemisphere (lateral aspect). (F) Left hemisphere (medial aspect). Scale bars = 1 cm (E and F). Please click here to view a larger version of this figure.
{kind=link}
- To dissect the spinal cord, remove the mouse body from the PFA and blot it dry.
- Place the mouse on a Styrofoam dissecting block in the prone position and pin the paws into the block using four needles.
- Begin the dissection by cutting the skin down the midline of the mouse from neck to tail to expose the entire backside.
- Reflect the muscle and fascia on the back to expose the posterior part of the vertebral column.
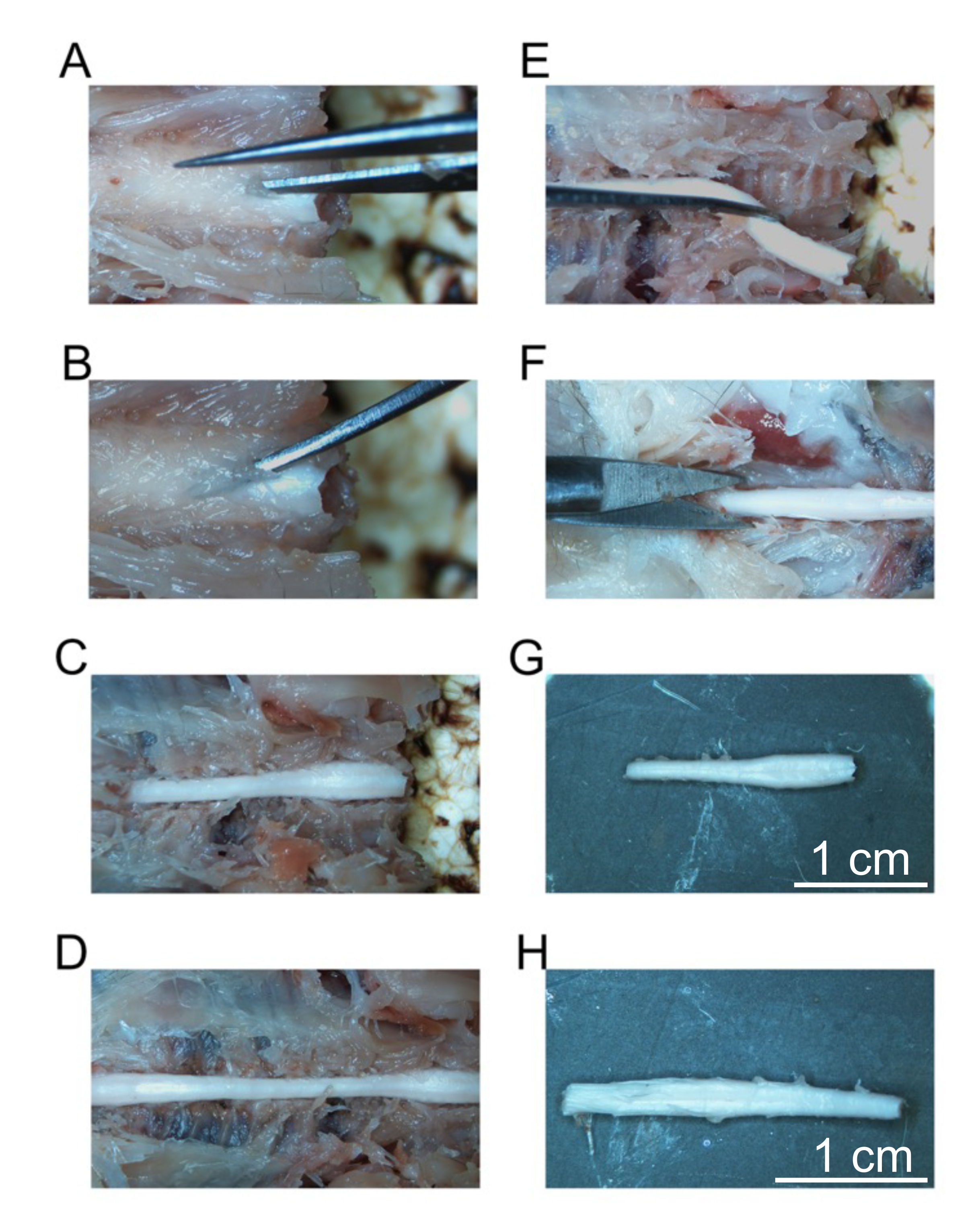
- Beginning at the most cranial vertebrae, use the fine dissecting scissors to cut through the lamina while avoiding the spinal cord (Figure 5A).
- Place the ultrafine curved forceps under the lamina and pull upwards to fracture the vertebrae to expose the spinal cord (Figure 5B).
- Use the curved forceps to reflect the fractured vertebrae and expose the most lateral regions of the spinal cord.
- Continue this process for the next vertebrae by placing the ultrafine curved forceps below the lamina and fracturing the vertebrae. Continue to fracture vertebrae until the entire cervical spine and thoracic spine are exposed (Figure 5C).
- Continue to expose the spinal cord until the end of the lumbar spinal cord (Figure 5D).
- Using a sharp razor blade, cut all the way through the midthoracic spinal cord.
- Using the ultrafine curved forceps, transect the spinal nerves laterally from the spine and the fascia anterior to the spinal cord to separate the cervical spinal cord slowly from the vertebral column.
- Continue to dissect out the cervical spinal cord until it is free from the vertebrae (Figure 5E). Place the cervical-midthoracic spinal cord in the tube labeled cervical. Store this tube at 4 °C until the cervical spinal cord sinks (approximately overnight).
- Continue to fracture the thoracic vertebrae all the way to the cauda equina as detailed in steps 4.22 through 4.25. When the cauda equina is exposed, use a sharp razor blade to cut the spinal cord 1 cm below the lumbar spinal cord (Figure 5F).
- Dissect the lumbar spinal cord away from the vertebrae as outlined in steps 4.26-4.27. Place the lumbar-mid cauda equina spinal cord into the tube labeled lumbar. Store this tube at 4 °C until the lumbar spinal cord sinks (approximately overnight).
- When all the tissues have sunk in 20% sucrose, transfer them to labeled tubes of 30% sucrose until they have sunk or for 3 days.
NOTE: Spinal tissues often do not sink at 30% sucrose.

Figure 5: Removal of fixed spinal cord segments. (A) Initial cut into the lamina of the cervical vertebrae. (B) Placement of curved forceps to fracture individual vertebrae. (C) The exposed cervical spine. (D) Exposed cervical, thoracic, and lumbar spine. (E) Removal of the cervical spine after cutting spinal nerves. (F) Cutting the sacral spine. (G) Isolated cervical spine. (H) Isolated lumbar spine. Scale bars = 1 cm (G and H). Please click here to view a larger version of this figure.
{kind=link}
5. OCT embedding and tissue storage
- For each mouse, label four rectangular-shaped cryomolds: Mouse ID, Right Hemisphere, Date of embedding; Mouse ID, Left Hemisphere, Date of embedding; Mouse ID, Cervical, Date of embedding; Mouse ID, Lumbar, Date of embedding.
- Set up a Styrofoam container and hang a metal cup above the container.
- Fill the Styrofoam container with liquid nitrogen approximately halfway. Fill the metal cup with fresh 2-methylbutane approximately halfway.
NOTE: 2-Methylbutane is toxic, highly volatile, and flammable. Care must be taken to avoid inhalation by performing subsequent steps in a fume hood and away from combustion sources. - Slowly lower the metal cup into the liquid nitrogen and avoid any splashing of the liquid nitrogen into the metal cup.
- Allow the 2-methylbutane to begin freezing. While the 2-Methylbutane is freezing, take the desired tissue out of the 30% sucrose solution and blot it dry on a dust-free wipe.
- Place the tissue in a cryomold with the rostral region pointed towards the top (unlabeled) part of the mold.
- Sparingly pour optimal cutting temperature embedding medium (OCT) over the tissue in the cryomold, using only enough to cover the tissue completely. Avoid using excessive OCT as this can cause cracking. Remove any bubbles from the OCT.
- When frozen 2-methylbutane has fully covered the inner surface of the metal cup, fully immerse the cryomold in the overlying liquid phase 2-methylbutane for 12 s. After 12 s, place the cryomold to drain in a labeled aluminum foil square and wrap the entire cryomold. Immediately place the wrapped cryomold on dry ice and avoid thawing. Ensure that the frozen OCT/cryomold are always covered with dry ice.
- Repeat steps 5.6 through 5.8 for all tissues. Place the tissue for one mouse in a sealed small plastic bag and then into a cryo-safe, sealed container. Store this container in the -80 °C deep freezer until sections are cut.
6. Cryosectioning
- Prior to cryosectioning, ensure that the cryostat chamber temperature and specimen head are set to the appropriate settings.
NOTE: A chamber temperature of -20 °C, a specimen head temperature of -20 °C, and a section thickness of 30 µm are used for cutting brain sections. For cutting spinal cord sections, a chamber temperature of -23 °C, a specimen head temperature of -30 °C, and a section thickness of 30 µm are used. It is important to note that the cryostat settings (especially the specimen head temperature) will need to be adjusted during sectioning to address problems with tissue quality. Generally, the specimen head temperature is lowered to correct for tissue smearing. Conversely, the specimen head temperature is increased to correct for excessive tissue curling. - Remove the desired OCT block from the -80 °C freezer and place the unwrapped cryomold/OCT block in the cryostat chamber.
- Allow the OCT block to acclimate to the chamber temperature for 30 min.
- Clean a chuck with 70% ethanol and wipe it dry with a dust-free wipe. Place the cleaned chuck in the cryostat chamber and make a coin-sized circle of OCT on the chuck. Allow the OCT to freeze (approximately 1-2 min).
- When the OCT has frozen, place a pea-sized dot of fresh OCT on the chuck and immediately place the tissue OCT block on the chuck. Ensure that the tissue is perfectly perpendicular to the chuck before the OCT completely freezes.
NOTE: Generally, the most rostral region of the brain is placed towards the chuck so that sectioning begins from the caudal end. For the spinal cord, generally, the most caudal region is placed on the chuck so that sectioning begins from the rostral end. - When the OCT has hardened, place more OCT around the base of the OCT block to act as structural support during sectioning. When this support has slightly hardened, place the chuck on the specimen head and allow the OCT block to reach the temperature of the specimen head for 30 min.
- Place a microtome blade in the blade holder and clean the antiroll plate. Adjust the antiroll plate distance to ensure that the tissue is passing just under the plate during sectioning. For more information on the correct positioning of the antiroll plate, consult the cryostat manual.
- After the OCT block has acclimated to the specimen head temperature, begin trimming the OCT block until the tissue is reached. When tissue is visible, switch from trimming to sectioning and begin cutting 30 μm sections. Move aside the antiroll plate and, using a microscope slide, pick up the section.
- Continue to cut and collect sections until satisfied with the anatomical location of the sections or all the tissue has been cut. Place the slides to dry at room temperature for 1-3 days. After drying, place the slides in a slide box and place this box in a sealed plastic bag. Label the bag with the mouse ID and tissue information prior to placing the slides in the -80 °C deep freezer.
7. Immunofluorescent staining
- Thaw the slides for 1 h at room temperature.
- Place the slides in a horizontal slide jar and wash the sections 3 times in PBS for 10 min each wash at room temperature on a shaker.
- Prepare blocking buffer (2% BSA + 0.3% Triton-X-100 in 1x PBS). Use approximately 1 mL per slide.
- Create a humidified chamber by adding water to the bottom. Lay the slides face up horizontally and do not allow them to dry out. Add 1 mL of blocking buffer to each slide and incubate for at least 1 h at room temperature.
- Prepare primary antibody solution by diluting the primary antibody in antibody buffer (0.7% BSA + 0.3% Triton-X-100 in 1x PBS).
NOTE: For the phosphorylated α-synuclein antibody, a dilution factor of 1:500 was used. - Add 200-300 µL of primary antibody solution per slide. Cover with a coverslip to disperse the antibody and incubate at 4 °C overnight.
- The next day, remove the slides from the humidified chamber and place them in a vertical Coplin jar filled with 1x PBS to allow the coverslip to fall off. Do this for each slide individually and take care not to disturb the tissue section when removing the coverslip.
- Place the slides in a horizontal slide jar and wash the sections 3 times in PBS for 10 min each wash at room temperature on a shaker.
NOTE: All subsequent steps must be performed with the lights off to avoid photobleaching the fluorophore! - Prepare the secondary antibody solution by diluting the secondary antibody in antibody buffer.
NOTE: For the detection of phosphorylated α-synuclein, anti-rabbit secondary conjugated to Alexa 488 with a dilution factor of 1:500 in antibody buffer (0.7% BSA + 0.3% Triton-X-100 in 1x PBS) was used. - Lay the slides horizontally in the humidified chamber and add 200-300 µL of secondary antibody solution per slide. Cover with a coverslip to disperse the antibody. Incubate at room temperature for at least 2 h.
- Remove the slides from the humidified chamber and place them in a vertical Coplin jar filled with 1x PBS to allow the coverslip to fall off. Do this for each slide individually and take care not to disturb the tissue section when removing the coverslip.
- Place the slides in a horizontal slide jar and wash the sections 3 times in PBS for 10 min each wash at room temperature on a shaker.
- Blot-dry the side of the slides on a towel and shake off excess PBS.
- Add 3 drops of mounting medium to each slide. Add the coverslip and gently push out bubbles with a pair of forceps by pressing the coverslip before imaging by confocal microscopy.
Results
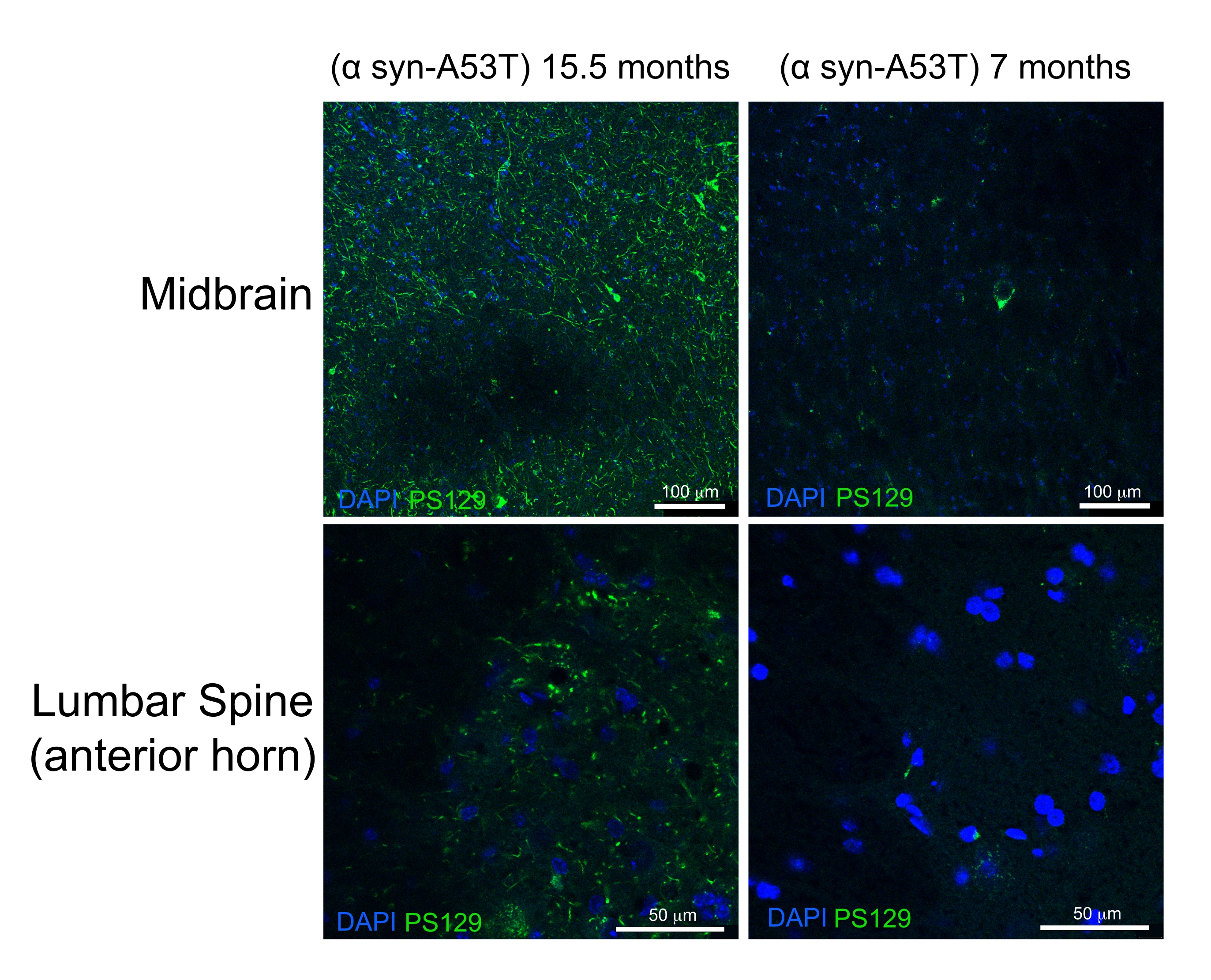
High-quality perfusion is indicated by the absence of blood in the liver, spinal cord, and deep CNS structures (Figure 4C and Figure 5G,H). Retained blood below the dura mater (for example, within the venous sinuses) or between the dura mater and the skull has not been problematic as this blood is not within the brain parenchyma. The blood seen in Figure 4A is located between the skull and the dura matter and is therefore not problematic or suggestive of poor-quality perfusion. The fresh brain and spinal cord are quite soft and easily damaged during handling. Adequately fixed tissues, by comparison, are firm. To assess the quality of tissue and the preservation of morphology of tissues by this perfusion method, this work demonstrates the detection of phosphorylated α-synuclein in the midbrain and lumbar spinal cord of a 15-month-old mouse and a 7 month-old-mouse expressing human A53T α-synuclein (Figure 6).
The A53T mutation is overrepresented in patients with autosomal dominant Parkinson's disease (PD). Furthermore, human α-synuclein with the A53T mutation can recapitulate many of the features of human PD when expressed in mice6,7. Phosphorylation of α-synuclein at the Serine 129 residue has been shown in vivo and in vitro to induce α-synuclein aggregation8. Lewey bodies are the classical histologic finding present in patients with PD or Lewey body dementia9. A majority of the α-synuclein present in Lewey bodies is phosphorylated at Serine 12910,11. As a result, the accumulation of phosphorylated α-synuclein is used as a marker of the histologic severity of PD pathology. The present study finds that phosphorylated α-synuclein accumulates at a significantly higher level in 15.5-month-old symptomatic mice relative to 7-month-old asymptomatic mice expressing human A53T α-synuclein. This is consistent with reports describing an enrichment of cytopathology in the anterior horns of the spinal cord and the midbrain of these mice.6 From this, it is concluded that the simplified perfusion method described here provides high-quality fixation of CNS tissues for downstream histologic characterization.

Figure 6: Label for phosphorylated α-synuclein in midbrain and lumbar spinal cord tissues from a mouse model of Parkinson's Disease. The aged (15.5-month-old) end-stage paralyzed mouse is compared with the 7-month-old healthy mouse. Both mice express a misfolding prone mutant variant (A53T) of human α-synuclein that induces Parkinson's-like pathology. Scale bars = 100 µm (upper panels) and 50 µm (lower panels). Abbreviation: α syn-A53T = A53T mutant of α-synuclein; DAPI = 4',6-diamidino-2-phenylindole; PS129 = phosphorylated Serine 129 of α-synuclein. Please click here to view a larger version of this figure.
{kind=link}
Discussion
This work describes the critical steps to performing transcardial perfusion. When constructing the perfusion apparatus (protocol section 1), it is important to use tubing that is flexible enough to be completely occluded by a hemostat. Some stiff tubing may not be sufficiently occluded by a hemostat and may still allow PFA to leak into the main line during the initial PBS perfusion. When preparing the 4% PFA solution, it is important to ensure that the pH is physiologic (7.4). As preparation of the PFA solution involves heating it to 65 °C, the solution must be cooled back down to 25 °C prior to measuring the pH as this is the temperature at which pH is calibrated on the meter.
When making the initial incision into the abdomen, care must be taken to avoid laceration of the abdominal organs (protocol section 2). When dissecting superiorly towards the diaphragm, it is important to avoid laceration of the liver as it is common for the liver to be adherent with the anterior abdominal wall. To overcome this, the liver is carefully and bluntly dissected away from the anterior wall before continuing an incision towards the diaphragm. When entering the thoracic cavity through the diaphragm, it is important to avoid laceration of the heart, great vessels, and the lung. To avoid this, the scissor tip is kept superficially and at an acute angle with the ribcage.
The initial dissection to expose the heart takes approximately 2 min from the initial incision. It is expected that during this time, some air has entered the tip of the butterfly needle. The introduction of this air into the circulation of the mouse will yield poor-quality perfusion. Therefore, it is critical that the main line is opened and PBS is flushed through the needle immediately prior to cannulation of the heart to remove air bubbles. Ideally, the heart is cannulated while a small PBS trickle flows through the needle tip to ensure the complete absence of air when puncturing the LV.
When the needle enters the LV, it must not go so deep as to introduce the needle tip into the right ventricle (RV). Placement of the needle in either the RV or beyond the mitral valve will result in immediate "inflation" of the lungs when perfusion is started. This is undesirable, and the needle must be withdrawn slightly to ensure LV placement. If the needle is placed properly, the lungs will remain flat throughout perfusion. When perfusion is initiated, it is sometimes observed that a clear liquid is emerging from the open mouth of the animal. This is usually due to a perfusion pressure that is too high or due to misplacement of the needle within the heart. The authors speculate that elevated perfusion pressures result in extravasation of the perfusate from the arteriolar capillary bed and retrograde flow of PBS via the bronchial tree into the esophagus and oral cavity.
The perfusion pressure must be lowered by either decreasing the level of PBS in the PBS bottle or by lowering the height of the PBS bottle. Alternatively, if the needle is placed too deeply into the left ventricle, it may travel through the mitral valve and deliver perfusate to the left atrium. This may result in retrograde flow through the pulmonary veins and extravasation of perfusate into the arterioles, as described above. Thorough clearance of blood from the circulatory system with PBS is especially important to avoid fixative-induced cross-linking of blood components resulting in vessel occlusion upon subsequent fixative perfusion. Clearance is effectively assessed by a color change of the liver and PBS flow from an incision in the ventral tail base. Blood clearance is generally complete by 3 min of perfusion with PBS; however, if visual signs of clearance occur at shorter times, then fixative is introduced sooner than 3 min. Longer clearance times are not recommended as delayed fixative perfusion leads to artifacts in CNS fine structure1.
When PFA is being administered, it is important to monitor the level of PFA solution in the PFA bottle. Fill up the PFA bottle if the level of PFA drops to less than 4 cm above the mouth of the PFA bottle. After perfusion has been completed, the perfusion apparatus must be thoroughly rinsed with distilled water. This is important as residual PFA in the main line will contaminate the initial PBS perfusion with PFA and result in poor-quality perfusion. Finally, 25 G butterfly needles are generally recommended for average-sized adult mice in the 20-30 g range. However, larger or smaller mice may require slightly larger or smaller gauge needles in addition to the adjustment of the fixative bottles to provide optimal flow rates.
For CNS dissection and OCT embedding (protocol section 3), it is common for spinal cord tissues to not completely sink in 30% sucrose. These tissues are therefore left in sucrose for 2 days and then embedded in OCT, regardless of whether they sink or not. When freezing down tissues in OCT, it is possible that certain tissues may crack when placed in cooled 2-methylbutane. This is more common with the brain and usually occurs when too much OCT is placed on the tissue. To avoid this, place only enough OCT to cover the tissue surfaces prior to immediate freezing. In some protocols, cracking is less common despite complete immersion in OCT. This is usually due to a slower freezing method such as when using dry-ice-cooled 2-methylbutane or placing the cryomold on a block of dry ice directly. Liquid-nitrogen-cooled 2-methylbutane is preferred in this work as the rate of freezing is substantially more rapid and may better preserve tissue morphology than slower freezing methods.
When cryosectioning the tissue (protocol section 4), it is important to avoid multiple freeze-thaw cycles. Therefore, it is optimal to cut all sections from a single OCT block to obtain a specific brain region for analysis instead of thawing and refreezing selected areas. If this is not viable, after obtaining a few sections, users may refreeze and store the OCT blocks in the -80 °C deep freezer 1-2 more times for future use.
The major benefits of this method over more traditional pump or air pressure delivery of perfusate are as follows: (1) low cost and accessibility of the perfusion apparatus. (2) Users do not need to manually maintain pressure in the perfusion apparatus throughout the perfusion. (3) Lower and more consistent perfusion pressure than other low-cost alternatives for perfusion such as via syringe delivery. Using Bernoulli's equation, it is calculated that the gravity-fed perfusion apparatus constructed here will maintain a perfusion pressure of approximately 73 mm Hg when the perfusate bottles are placed at 1 m of elevation relative to the needle. Given that this is significantly below the systolic blood pressure of these animals, this perfusion pressure is likely sufficiently low to avoid vascular rupture12.
The authors have thus far successfully used this perfusion system to detect the presence of phosphorylated α-synuclein in a mouse model of Parkinson's disease. During this time, significant limitations with this perfusion method have not been encountered that are not present with a pump perfusion delivery method. The major limitation of this technique is the time-consuming nature of perfusion versus drop fixation. This technique is preferable to drop fixation as perfusion results in the deeper penetration of fixative to the CNS structures. A second limitation of this technique is that it requires some surgical skill to perform, as the heart must be cannulated quickly following entrance into the thoracic cavity. However, with experience, trained users can routinely canulate the heart within 1 min of the initial incision into the abdomen.
Disclosures
The authors declare no competing financial interests.
Acknowledgements
The authors thank Xiaowen Wang, Liam Coyne, and Jason Grullon for their assistance in developing this protocol. This work was supported by the NIH grants AG061204 and AG063499.
Materials
| Name | Company | Catalog Number | Comments |
| 2-Methylbutane (Certified ACS), Fisher Chemical | Fisher Scientific | 03551-4 | |
| Andwin Scientific Tissue-Tek O.C.T Compound | Fisher Scientific | NC1029572 | |
| Artman Instruments 4.5" Straight Castroveijo Spring Action Scissors | Amazon | B0752XHK2X | "fine scissors" |
| BD General Use and PrecisionGlide Hypodermic Needles, 18 G | Fisher Scientific | 14-826-5D | |
| BD General Use and PrecisionGlide Hypodermic Needles, 22 G | Fisher Scientific | 14-826B | |
| Corning PES Syringe Filters | Fisher Scientific | 09-754-29 | |
| Cytiva HyClone Phosphate Buffered Saline (PBS), 10x | Fisher Scientific | SH30258 | |
| Falcon 50 mL Conical Centrifuge Tubes | Fisher Scientific | 14-432-22 | |
| Fisher BioReagents Bovine Serum Albumin (BSA) Protease-free Powder | Fisher Scientific | BP9703100 | |
| Fisherbrand Curved Medium Point General Purpose Forceps | Fisher Scientific | 16-100-110 | "curved fenestrated forceps" |
| Fisherbrand Curved Very Fine Precision Tip Forceps | Fisher Scientific | 16-100-123 | "curved fine forceps" |
| Fisherbrand Disposable Graduated Transfer Pipettes | Fisher Scientific | 13-711-9AM | |
| Fisherbrand High Precision Straight Tapered Ultra Fine Point Tweezers/Forceps | Fisher Scientific | 12-000-122 | "straight fine forceps" |
| Fisherbrand Micro Spatulas with Rounded Ends | Fisher Scientific | 21-401-5 | |
| Fisherbrand Porcelain Buchner Funnels with Fixed Perforated Plates | Fisher Scientific | FB966J | |
| Fisherbrand Premium Cover Glass, 24 x 50 | Fisher Scientific | 12-548-5M | |
| Fisherbrand Premium Tissue Forceps 1X2 Teeth 5 in. German Steel | Fisher Scientific | 13-820-074 | "skin forceps" |
| Fisherbrand Reusable Heavy-Wall Filter Flasks | Fisher Scientific | FB3002000 | |
| Fisherbrand Standard Dissecting Scissors | Fisher Scientific | 08-951-20 | "dissecting scissors" |
| Fisherbrand Sterile Syringes for Single Use | Fisher Scientific | 14-955-464 | |
| Fisherbrand Straight Locking Hemostats | Fisher Scientific | 16-100-115 | |
| Fisherbrand Superfrost Plus Microscope Slides | Fisher Scientific | 12-550-15 | |
| Fisherbrand Vinyl Tubing and Connector Kits, 1/4 in. | Fisher Scientific | 14-174-1C | |
| Fisherbrand Wet-Strengthened Qualitative Filter Paper Circles | Fisher Scientific | 09-790-12F | |
| Fisherbrand Y Connector with 1/4 in. ID - Polypropylene - QC | Fisher Scientific | 01-000-686 | |
| Garvey Economy Single Edge Cutter Blade | Amazon | B001GXFAEQ | |
| Goat anti-Rabbit IgG (H+L) Secondary Antibody, DyLight 488 | ThermoFisher Scientific | 35552 | |
| Ideal Clamp Stant High-Pressure Clamps | Fisher Scientific | 14-198-5A | "hose clamps" |
| IMEB, Inc Sakura Accu-Edge Low Profile Microtome Blades, Dispoisable | Fisher Scientific | NC9822467 | |
| Kawasumi 25 Gauge Standard Winged Blood Collection Set | Fisher Scientific | 22-010-137 | "butterfly needle" |
| Kimberly-Clark Professional Kimtech Science Kimwipes Delicate Task Wipers, 1-Ply | Fisher Scientific | 06-666 | |
| Molecular Probes ProLong Diamond Antifade Mountant with DAPI | Fisher Scientific | P36962 | |
| Nalgene Narrow-Mouth Right-to-Know LDPE Wash Bottles | ThermoFisher Scientific | 2425-0506 | "buffer bottles" |
| PAP pen | abcam | ab2601 | |
| Paraformaldehyde Granular | Electron Microscopy Systems | 19210 | |
| Pyrex Glass Drying Dishes, 34.9 x 24.9 x 6 cm | Fisher Scientific | 15-242D | |
| Recombinant Anti-Alpha-synuclein (phospho S129) antibody [EP1536Y] | abcam | ab51253 | |
| Sodium Hydroxide | Sigma-Aldrich | 221465-2.5kg | |
| Stainless Steel Drinking Cup 18-oz | amazon | B0039PPO9U | |
| Sucrose for Molecular Biology CAS: 57-50-1 | Us Biological | S8010 | |
| Triton X-100 | Us Biological | T8655 | |
| Vetone Fluriso Isoflurane USP | MWI Animal Health | 502017 |
References
- Tao-Cheng, J. H., Gallant, P. E., Brightman, M. W., Dosemeci, A., Reese, T. S. Structural changes at synapses after delayed perfusion fixation in different regions of the mouse brain. Journal of Comparative Neurology. 501 (5), 731-740 (2007).
- Gage, G. J., Kipke, D. R., Shain, W. Whole animal perfusion fixation for rodents. Journal of Visualized Experiments: JoVE. (65), e3564(2012).
- McFadden, W. C., et al. Perfusion fixation in brain banking: a systematic review. Acta Neuropathologica Communications. 7 (1), 146(2019).
- Lamberts, R., Goldsmith, P. C. Fixation, fine structure, and immunostaining for neuropeptides: perfusion versus immersion of the neuroendocrine hypothalamus. Journal of Histochemistry and Cytochemistry. 34 (3), 389-398 (1986).
- Adickes, E. D., Folkerth, R. D., Sims, K. L. Use of perfusion fixation for improved neuropathologic examination. Archives of Pathology and Laboratory Medicine. 121 (11), 1199-1206 (1997).
- Giasson, B. I., et al. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron. 34 (4), 521-533 (2002).
- Martin, L. J., et al. Parkinson's disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. Journal of Neuroscience. 26 (1), 41-50 (2006).
- Fujiwara, H., et al. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nature Cell Biology. 4 (2), 160-164 (2002).
- Kalia, L. V., Lang, A. E. Parkinson's disease. Lancet. 386 (9996), 896-912 (2015).
- Anderson, J. P., et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. Journal of Biological Chemistry. 281 (40), 29739-29752 (2006).
- Kahle, P. J., et al. Hyperphosphorylation and insolubility of alpha-synuclein in transgenic mouse oligodendrocytes. EMBO Reports. 3 (6), 583-588 (2002).
- Mattson, D. L. Comparison of arterial blood pressure in different strains of mice. American Journal of Hypertension. 14, 405-408 (2001).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved