È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Caratterizzazione tridimensionale dei siti di contatto interorganelle negli epatociti mediante microscopia elettronica a sezione seriale
In questo articolo
Riepilogo
Un protocollo semplice e completo per acquisire dettagli tridimensionali dei siti di contatto della membrana tra organelli negli epatociti dal fegato o cellule in altri tessuti.
Abstract
La microscopia elettronica a trasmissione è stata a lungo considerata il gold standard per la visualizzazione dell'ultrastruttura cellulare. Tuttavia, l'analisi è spesso limitata a due dimensioni, ostacolando la capacità di descrivere completamente l'ultrastruttura tridimensionale (3D) e la relazione funzionale tra gli organelli. La microscopia elettronica volumetrica (vEM) descrive una raccolta di tecniche che consentono l'interrogazione dell'ultrastruttura cellulare in 3D a mesoscala, microscala e risoluzioni su scala nanometrica.
Questo protocollo fornisce un metodo accessibile e robusto per acquisire dati vEM utilizzando la trasmissione a sezione seriale EM (TEM) e copre gli aspetti tecnici dell'elaborazione dei campioni fino alla ricostruzione 3D digitale in un unico flusso di lavoro semplice. Per dimostrare l'utilità di questa tecnica, viene presentata la relazione ultrastrutturale 3D tra il reticolo endoplasmatico e i mitocondri e i loro siti di contatto negli epatociti epatici. I contatti interorganelle svolgono ruoli vitali nel trasferimento di ioni, lipidi, sostanze nutritive e altre piccole molecole tra gli organelli. Tuttavia, nonostante la loro scoperta iniziale negli epatociti, c'è ancora molto da imparare sulle loro caratteristiche fisiche, dinamiche e funzioni.
I contatti interorganelle possono mostrare una gamma di morfologie, che variano nella vicinanza dei due organelli l'uno all'altro (in genere ~ 10-30 nm) e l'estensione del sito di contatto (dai contatti puntati ai contatti 3D più grandi simili a cisternali). L'esame dei contatti stretti richiede l'imaging ad alta risoluzione e la sezione seriale TEM è adatta per visualizzare l'ultrastrutturale 3D dei contatti interorganelle durante la differenziazione degli epatociti, nonché le alterazioni dell'architettura degli epatociti associate a malattie metaboliche.
Introduzione
Dalla loro invenzione nel 1930, i microscopi elettronici hanno permesso ai ricercatori di visualizzare i componenti strutturali di cellule e tessuti 1,2. La maggior parte delle indagini ha fornito informazioni 2D, poiché la costruzione di modelli 3D richiede una scrupolosa raccolta di sezioni seriali, fotografia manuale, elaborazione negativa, tracciamento manuale e la creazione e l'assemblaggio di modelli 3D da lastre di vetro, plastica o polistirolo 3,4. Quasi 70 anni dopo, ci sono stati notevoli progressi in numerosi aspetti del processo, dalle prestazioni del microscopio, alla raccolta di sezioni seriali, all'imaging digitale automatizzato, al sofisticato software e hardware per la ricostruzione, la visualizzazione e l'analisi 3D ad approcci alternativi per quello che ora è collettivamente chiamato volume EM (vEM). Queste tecniche vEM sono generalmente considerate per fornire informazioni ultrastrutturali 3D a risoluzioni nanometriche su scale micron e comprendono la microscopia elettronica a trasmissione (TEM) e le più recenti tecniche di microscopia elettronica a scansione (SEM); vedi recensioni 5,6,7,8.
Ad esempio, il fascio ionico focalizzato SEM (FIB-SEM) utilizza un fascio ionico focalizzato all'interno di un SEM per fresare la superficie del blocco tra scansioni sequenziali seM della superficie del blocco, consentendo la fresatura / imaging automatizzato ripetuto di un campione e la creazione di un set di dati 3D per la ricostruzione 9,10. Al contrario, il blocco seriale FACCIA SEM (SBF-SEM) utilizza un ultramicrotomo all'interno del SEM per rimuovere materiale dalla faccia del blocco prima dell'imaging 11,12, mentre la tomografia array è un processo non distruttivo che richiede la raccolta di sezioni seriali, su coverslip, wafer o nastro, prima di impostare un flusso di lavoro automatizzato di imaging della regione di interesse nelle sezioni sequenziali nel SEM per generare il set di dati 3D13 . Simile alla tomografia ad array, la sezione seriale TEM (ssTEM) richiede che le sezioni fisiche siano raccolte prima dell'imaging; tuttavia, queste sezioni sono raccolte su griglie TEM e fotografate in un TEM 14,15,16. ssTEM può essere esteso eseguendo la tomografia tilt 17,18,19. La tomografia inclinabile seriale fornisce la migliore risoluzione in x, y e z e, sebbene sia stata utilizzata per ricostruire intere celle20, è ragionevolmente impegnativa. Questo protocollo si concentra sugli aspetti pratici di ssTEM come la tecnica vEM più accessibile disponibile per molti laboratori EM che potrebbero non avere attualmente accesso a strumenti specializzati di sezionamento o vEM, ma trarrebbero vantaggio dalla generazione di dati vEM 3D.
L'ultramicrotomia seriale per la ricostruzione 3D è stata precedentemente considerata impegnativa. Era difficile tagliare nastri dritti di spessore di sezione uniforme, essere in grado di disporre e raccogliere nastri delle dimensioni corrette, nell'ordine corretto, su griglie con supporto sufficiente, ma senza barre della griglia che oscuravano le regioni di interesse e, soprattutto, senza perdere sezioni, poiché una serie incompleta potrebbe impedire la ricostruzione 3D completa21. Tuttavia, i miglioramenti agli ultramicrotomi commerciali, i coltelli da taglio e rifilatura diamantati 22,23, le pellicole di supporto elettrone lucente sulle griglie 21,24 e gli adesivi per aiutare l'adesione della sezione e la conservazione del nastro13,21 sono solo alcuni dei progressi incrementali nel corso degli anni che hanno reso la tecnica più di routine in molti laboratori. Una volta raccolte le sezioni seriali, l'imaging seriale in TEM è semplice e può fornire immagini EM con dimensioni px subnanometriche in x e y, consentendo l'interrogazione ad alta risoluzione delle strutture subcellulari, un potenziale requisito per molte domande di ricerca. Il caso di studio qui presentato dimostra l'uso di ssTEM e ricostruzione 3D nello studio dei contatti reticolo endoplasmatico (ER)-organello negli epatociti epatici, dove i contatti ER-organello sono stati osservati per la prima volta25,26.
Pur essendo contiguo con l'involucro nucleare, l'ER ha anche stretti contatti con numerosi altri organelli cellulari, tra cui lisosomi, mitocondri, goccioline lipidiche e la membrana plasmatica27. I contatti ER-organello sono stati implicati nel metabolismo lipidico28, nella segnalazione fosfoinositide e calcio29, nella regolazione dell'autofagia e nella risposta allo stress30,31. I contatti ER-organello e altri contatti interorganelle sono strutture altamente dinamiche che rispondono alle esigenze metaboliche cellulari e ai segnali extracellulari. È stato dimostrato che variano morfologicamente nelle loro dimensioni e forma e nelle distanze tra le membrane degli organelli32,33. Si ritiene che queste differenze ultrastrutturali riflettano probabilmente le loro diverse composizioni proteiche / lipidiche e la funzione34,35. Tuttavia, è ancora un compito impegnativo definire i contatti interorganelle e analizzarli36. Pertanto, è necessario un protocollo affidabile ma semplice per esaminare e caratterizzare i contatti interorganelle per ulteriori indagini.
Poiché i contatti ER-organello possono variare da 10 a 30 nm nella separazione membrana-membrana, il gold standard per l'identificazione è stato storicamente TEM. Il TEM a sezione sottile ha rivelato la localizzazione di sottodomini specifici per le proteine ER residenti a contatti di membrana distinti37. Tradizionalmente, questo ha rivelato contatti ER-organello con risoluzione nm, ma spesso ha presentato solo una visione 2D di queste interazioni. Tuttavia, gli approcci vEM rivelano la presentazione ultrastrutturale e il contesto di questi siti di contatto in 3D, consentendo la ricostruzione completa dei contatti e una classificazione più accurata dei contatti (punto vs. tubolare vs. simile a cisternale) e la quantificazione38,39. Oltre ad essere il primo tipo di cellula in cui sono stati osservati contatti ER-organello25,26, gli epatociti hanno un ampio sistema di altri contatti interorganelli che svolgono ruoli vitali nella loro architettura e fisiologia28,40. Tuttavia, manca ancora un'accurata caratterizzazione morfologica dell'ER-organello e di altri contatti interorganelli negli epatociti. Di conseguenza, il modo in cui i contatti interorganelle si formano e rimodellano durante la rigenerazione e la riparazione è di particolare rilevanza per la biologia degli epatociti e la funzionalità epatica.
Protocollo
Tutti gli animali sono stati alloggiati in conformità con le linee guida del Ministero degli Interni del Regno Unito e la raccolta dei tessuti è stata effettuata in conformità con l'Animal (Scientific Procedures) Act del Regno Unito del 1986.
1. Fissazione e preparazione del campione
- Sezionare il tessuto epatico in pezzi di dimensioni appropriate, circa 8 mm x 8 mm x 3 mm, e posizionare i pezzi in soluzione salina calda tamponata con fosfato (PBS, 37 °C).
- Iniettare la temperatura ambiente (20-25 °C) fissativo (1,5% glutaraldeide in 1% di saccarosio, 0,1 M di cacodilato di sodio) nei pezzi di fegato e trasferirli da PBS a fissativo per un massimo di 20 minuti a temperatura ambiente. Tenere sempre il tessuto immerso in soluzioni per evitare l'essiccazione.
NOTA: Le aldeidi sono sostanze irritanti corrosive e potenzialmente cancerogene. Il cacodilato di sodio è tossico se ingerito o inalato. Tutti i fissativi devono essere maneggiati indossando adeguati dispositivi di protezione individuale e l'esperimento deve essere eseguito in una cappa aspirante. Una buona fissazione si tradurrà in un tessuto più solido. - Impostare il microtomo vibrante con una lama, un bagno di ghiaccio e un vassoio tampone freddo riempito di PBS. Montare il primo pezzo di tessuto epatico fisso su un supporto per campioni con colla cianoacrilata e trasferire il blocco al microtomo vibrante.
- Seguendo le raccomandazioni del produttore, avvicinarsi al tessuto e tagliare il fegato fisso in fette spesse 100 μm.
- Raccogliere le fette con una spatola o un pennello naturale per capelli e trasferirle in un piatto a 12 o 24 pozzetti contenente una dose ghiacciata (1,5% glutaraldeide, 0,1 M di cacodilato di sodio) su ghiaccio. Lasciare le fette sul ghiaccio fino a quando tutti i campioni sono stati affettati e sono pronti per essere ulteriormente lavorati.
- Selezionare le fette contenenti le regioni di interesse per un'ulteriore lavorazione e lavare con delicata agitazione. Eseguire tre lavaggi di 5 minuti con cacodilato di sodio a temperatura ambiente 0,1 M in un piatto da 12 o 24 pozzetti, assicurandosi che le fette abbiano un buffer sufficiente per muoversi liberamente.
NOTA: In generale, le regioni di interesse sono selezionate in base alle caratteristiche anatomiche relative alla questione biologica dello studio e guidate da regioni del tessuto che possono essere presenti nell'intera serie, ad esempio non sul bordo della sezione, e che sono ben conservate. - In una cappa aspirante, sostituire il cacodilato di sodio 0,1 M con tetrossido di osmio all'1% appena preparato/ ferricianuro di potassio all'1,5%. Posizionare la parabola da 12 o 24 pozzetti in un contenitore sigillato e trasferire il contenitore in un frigorifero per sostanze chimiche pericolose per 1 ora.
NOTA: l'osmio è estremamente pericoloso in caso di ingestione, inalazione e contatto con la pelle. Il ferricianuro di potassio è irritante ed è dannoso per inalazione e contatto con la pelle. Maneggiare sempre utilizzando adeguati dispositivi di protezione individuale ed eseguire l'esperimento in una cappa aspirante. - In una cappa aspirante, rimuovere il tetrossido di osmio / ferricianuro di potassio in una bottiglia di scarico di osmio dedicata e lavare i campioni per 5 minuti con cacodilato di sodio 0,1 M tre volte. Lasciare i campioni in un contenitore sigillato per una notte a 4 °C.
NOTA: potenziale punto di pausa. I campioni possono essere conservati in cacodilato di sodio 0,1 M in un contenitore sigillato a 4 °C per settimane con pochi danni alla conservazione. Assicurarsi che vi sia un tampone sufficiente per evitare l'essiccazione. - Incubare i campioni con acido tannico all'1% appena preparato in cacodilato di sodio 0,05 M per 45 minuti al buio a temperatura ambiente.
NOTA: L'acido tannico è irritante e può causare danni agli occhi. Indossare adeguati dispositivi di protezione individuale ed eseguire l'esperimento in una cappa aspirante. - Eseguire tre lavaggi di 5 minuti con ddH2O prima della disidratazione e dell'incorporamento.
2. Disidratazione del campione, incorporamento della resina Epon e montaggio
- Preparare la resina Epon secondo il seguente rapporto in peso (vedere punto 2.2). Tarare una bilancia con un becher di plastica monouso da 100 ml contenente una barra di agitazione. Tagliare le estremità da 5 pipette Pasteur in plastica monouso e utilizzarle per trasferire componenti in resina viscosa nel becher.
- Aggiungere in sequenza 19,2 g di resina-812, 7,6 g di DDSA, 13,2 g di MNA e 0,8 g di acceleratore DMP-30 nel becher. Utilizzando la quinta pipetta Pasteur in plastica pulita, mescolare accuratamente i componenti in resina a mano.
NOTA: Evitare l'introduzione di bolle ma garantire una miscelazione sufficiente della resina inferiore con la parte superiore per ottenere un cambiamento di colore e una miscelazione approssimativa degli strati componenti. Tutti i componenti della resina sono irritanti e sono dannosi per inalazione e contatto con la pelle. DMP-30 è corrosivo e può causare corrosione cutanea. Indossare adeguati dispositivi di protezione individuale. - Posizionare il becher su un agitatore magnetico e lasciare mescolare delicatamente, mescolando periodicamente la resina manualmente.
- Lavare i campioni con agitazione delicata per 5 minuti con etanolo al 70%; ripetere una volta.
- Lavare i campioni con agitazione delicata per 5 minuti con etanolo al 90%; ripetere una volta.
- Lavare i campioni con agitazione delicata per 5 minuti con etanolo al 100%; ripetere una volta.
- Mentre i campioni sono in etanolo al 100% lavati in una cappa aspirante, preparare una miscela di ossido di propilene (PO) 50:50 (v/v):Epon in una fiala di vetro con un coperchio di plastica resistente all'ossido di propilene (PO). Agganciare con cautela ma saldamente il coperchio del flaconcino di vetro e, tenendo premuti sia il coperchio che il flaconcino, agitare o vorticare per mescolare.
NOTA: L'ossido di propilene è un irritante acutamente tossico e infiammabile che scioglie alcune materie plastiche. Indossare adeguati dispositivi di protezione individuale ed eseguire l'esperimento in una cappa aspirante. - Dopo il passaggio 2.6, incubare i campioni con PO:Epon per 1 ora in un contenitore resistente alla PO (ad esempio, vassoi di alluminio o fiale di vetro), con un leggero dondolo / agitazione nella cappa aspirante.
- Nella cappa aspirante, trasferire i campioni a 100% Epon. Incubare per 2 ore a temperatura ambiente nella cappa aspirante con dondolo/rotazione/agitazione. Trasferire la miscela PO:Epon in una bottiglia di vetro Epon dedicata.
- Ripetere il passaggio 2.9 una volta.
- Montare i campioni per l'incorporamento. A seconda delle dimensioni delle fette e della regione di interesse, montare direttamente le fette su mozziconi di resina prepolimerizzati o incorporarle piatte per la dissezione e reimposizionarle in un secondo momento.
NOTA: per l'incorporamento piatto, è possibile utilizzare una "diapositiva cast-a" per incorporare più sezioni contemporaneamente. La resina rimanente può essere utilizzata per riempire capsule di travi e cotta per realizzare mozziconi prepolimerizzati o congelata per un uso successivo. - Una volta montati e ricoperti di resina sufficiente per riempire la cavità "cast-a slide", cuocere i campioni durante la notte in un forno a 60 °C.
NOTA: potenziale punto di pausa. I campioni possono essere conservati a temperatura ambiente per anni. - Per il reincorporazione, identificare la regione di interesse nelle fette di tessuto piatte incorporate. Utilizzando una sega da gioielliere, ritagliare il pezzo di tessuto di dimensioni appropriate (da 1 mm2 a 4 mm2) e reincorporare utilizzando resina preparata, come al punto 2.2, sulla parte superiore di un blocco prepolimerizzato e cuocere per una notte in un forno a 60 °C.
NOTA: In alternativa, il pezzo di tessuto può essere incollato a uno stub o un perno con resina epossidica in due parti. Lasciare impostare durante la notte. Potenziale punto di pausa.
3. Taglio e sezionamento seriale di campioni incorporati
NOTA: il sezionamento è un'abilità appresa; gli utenti dovrebbero essere abili nel sezionamento ultrasottile prima di tentare il sezionamento seriale. Poiché i controlli esatti dei microtomi variano da produttore a produttore, seguire le istruzioni e le linee guida del produttore.
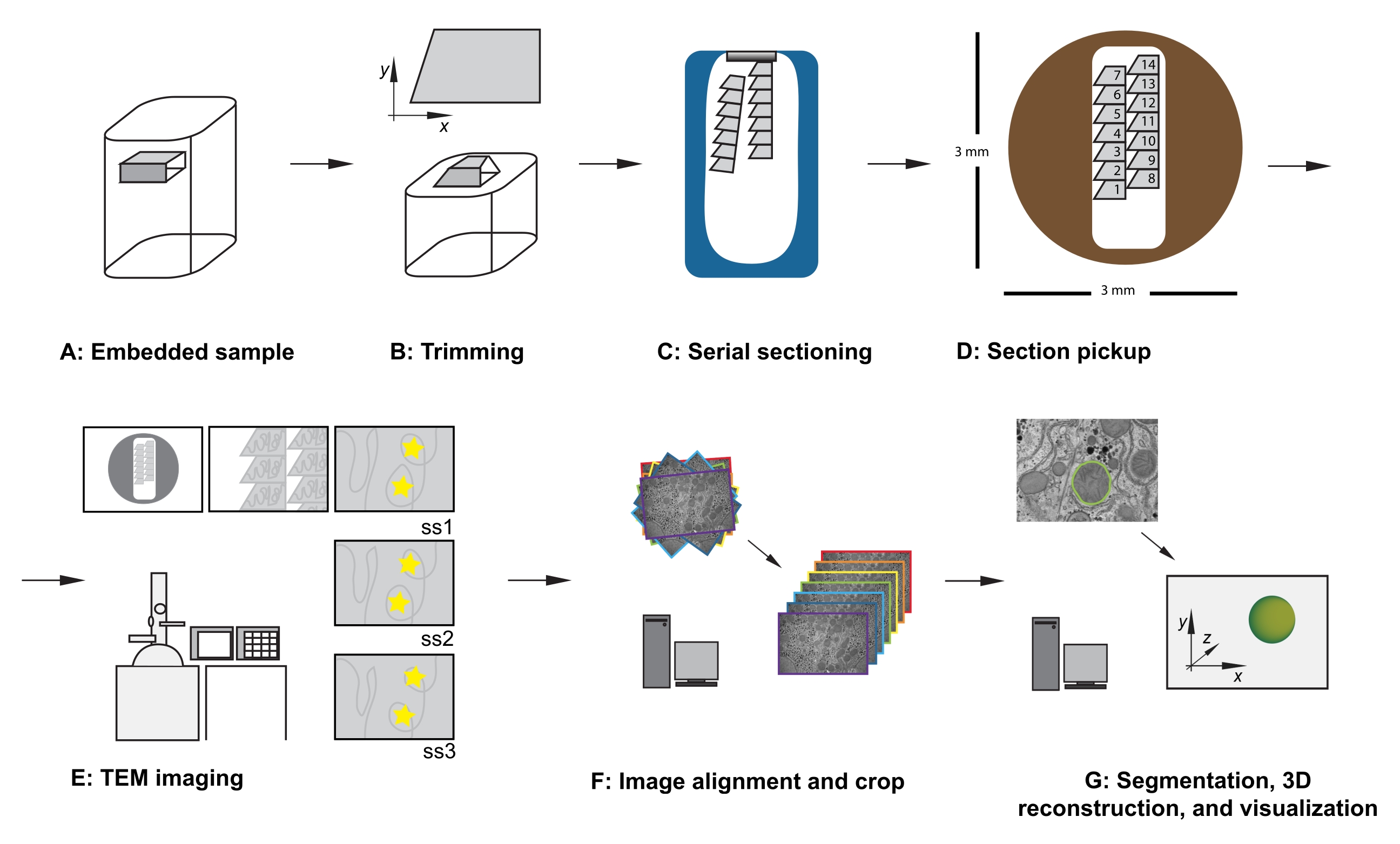
- Con il campione bloccato nell'adattatore di rifilatura, utilizzare una lama di rasoio per tagliare con cura il tessuto incorporato in resina per soddisfare i seguenti criteri (vedere figura 1A,B):
- Assicurarsi che vi sia una superficie superiore piatta che esponga il tessuto intorno alla regione di interesse.
- Garantire una forma trapezoidale con i bordi superiore e inferiore puliti e paralleli.
- Garantire dimensioni complessive di 200-500 μm in x, 100-500 μm in y.
- Garantire una faccia asimmetrica del blocco, ad esempio angoli laterali a destra di ~ 90 °, angolo in alto a sinistra ottuso e angolo inferiore sinistro acuto.
NOTA: un cryoknife di taglio può essere uno strumento alternativo per una lama di rasoio. Gli altri consigli sono per la comodità dell'utente per ordinare le sezioni durante l'imaging. Facoltativo: se le sezioni non riescono a formare nastri stabili, è possibile applicare un cemento di contatto sul bordo anteriore della faccia del blocco per favorire la formazione del nastro. Le lame di rasoio sono affilate; fare attenzione a tenere la lama del rasoio in modo tale che sia improbabile che scivolamenti accidentali provochino danni personali.

Figura 1: Flusso di lavoro TEM della sezione seriale. (A) Diagramma del campione nel blocco di resina. (B) Tagliare il blocco per generare una forma trapezoidale con bordi adatti per il sezionamento seriale e la faccia asimmetrica del blocco per garantire l'orientamento noto. (C) Diagramma che mostra nastri di sezioni seriali, che galleggiano sulla superficie dell'acqua nella barca del coltello diamantato. (D) Diagramma che mostra l'organizzazione della sezione e del nastro, dettando l'ordine delle sezioni, su una griglia di fessure TEM di 3 mm di diametro. (E) Imaging e navigazione TEM. Visualizzazione dell'ordine del nastro e delle sezioni e utilizzo di "adesivi a stella gialla" sul monitor per il riferimento allo schermo per garantire la reimaging della stessa regione di interesse nelle sezioni successive. (F) Allineamento e ritaglio dell'immagine. (G) Segmentazione, ricostruzione 3D e visualizzazione. Abbreviazione: TEM = microscopia elettronica a trasmissione. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
- Una volta tagliato, trasferire l'arco del campione, insieme al mandrino e al campione, al braccio del campione del microtomo, posizionando l'arco del campione in modo che l'intervallo di corsa dell'arco vada dall'alto verso il basso; fissare l'arco del campione in posizione.
- Posizionare e bloccare il coltello diamantato nel portacoltelli, assicurandosi che l'angolo di taglio sia impostato in modo appropriato sul coltello. Blocca saldamente il portacoltelli nel palco.
- Con il palco acceso, usa l'avanzamento del coltello mentre controlli costantemente la relazione tra la faccia del blocco e il bordo del coltello. Avanzare con cautela il coltello verso il campione, regolando continuamente l'angolo laterale del coltello, l'inclinazione del campione e la rotazione del campione regolando le manopole pertinenti fino a quando il blocco non è allineato al bordo del coltello.
- Spegnere l'uplighting del palco; accendere il downlighting del palco; impostare la parte superiore e inferiore della finestra di taglio del braccio del campione; e lasciare il campione appena sotto il bordo del coltello.
- Riempire la barca del coltello con ddH2O pulito e assicurarsi che la superficie dell'acqua sia a livello del bordo del coltello e leggermente concava.
- Opzionale: immergere una ciglia nello 0,1% Triton X-100 e poi nell'acqua della barca del coltello per ridurre la tensione superficiale dell'acqua per aiutare il cloroformaggio e il prelievo del nastro.
- Preparare la postazione di lavoro con ciglia (ciglia incollate a un bastoncino da cocktail), griglie a fessura rivestite di formvar, pinze crossover etichettate, cloroformio, soluzione Triton X-100 allo 0,1%, acqua distillata, carta da filtro e griglia con note a griglia.
- Impostare la velocità di taglio a 1 mm/s e lo spessore di taglio iniziale a 100 nm e avviare il ciclo di taglio.
- Dopo aver tagliato la prima sezione, modificare i parametri di taglio a una velocità di taglio a 0,8 mm/s e lo spessore di taglio a 70 nm e continuare il taglio, consentendo alle sezioni di formare un nastro che si muove lungo la superficie della barca coltello riempita d'acqua (Figura 1C).
NOTA: È importante essere consapevoli del colore delle sezioni prodotte in quanto questa è spesso una guida più accurata allo spessore delle sezioni in resina. Le sezioni d'argento hanno di solito uno spessore di circa 70 nm, mentre le sezioni grigie sono più sottili e le sezioni dorate sono più spesse. - Lasciare che il microtomo continui a tagliare le sezioni e il nastro si allunghi.
NOTA: È importante evitare grandi vibrazioni e disturbi fisici nella stanza. Le correnti d'aria possono far sì che le sezioni si muovano sulla superficie dell'acqua nella barca del coltello e le vibrazioni fisiche possono causare il taglio non uniforme del microtomo. - Una volta raccolte abbastanza sezioni e prima che il nastro arrivi alla fine della barca, interrompere il taglio (subito dopo che il campione ha superato il bordo del coltello).
NOTA: il numero di sezioni necessarie dipende dalle dimensioni della faccia del blocco e dalla dimensione del set di dati da raccogliere. Pertanto, è utile essere consapevoli della relazione tra la dimensione del blocco e la griglia della fessura mentre le sezioni di taglio si staccano. - Usando una ciglia in ogni mano, rompere delicatamente il nastro in nastri più piccoli che possono adattarsi alla lunghezza della griglia della fessura, avendo cura di prendere nota delle loro posizioni relative dall'interno del campione.
NOTA: se la loro larghezza combinata si adatta, più nastri possono essere posizionati delicatamente uno accanto all'altro e raccolti insieme in un'unica griglia di fessure. Se si raccolgono più nastri su una singola griglia di slot, prestare attenzione all'ordine e alla posizione relativa dei nastri. Ad esempio, posizionare sempre i nastri più avanti nell'esempio a destra di un nastro già presente nell'esempio (Figura 1D). - Opzionale: utilizzando un'asta di applicazione in vetro, passare una goccia di cloroformio sulle sezioni per appiattirle.
NOTA: Il cloroformio è tossico e irritante. Non lasciare che il cloroformio tocchi la superficie o le sezioni dell'acqua. Se lo fa accidentalmente, l'acqua deve essere rimossa e il coltello lavato prima di tornare al sezionamento. Il cloroformio può danneggiare le sezioni e degradare la colla che fissa il diamante nella barca del coltello. - Usando la prima pinza numerata, prendi la prima griglia di slot vuota (sul lato destro della fessura, lato formvar verso il basso), immergi delicatamente nel Triton X-100 e poi due volte nell'acqua distillata prima di rimuovere l'acqua in eccesso dal bordo della pinza usando un pezzo di carta da filtro.
- Con una ciglia in una mano e la pinza nell'altra, abbassare delicatamente circa 2/3 della griglia a fessura rivestita di formvar nell'acqua della barca del coltello (lontano dalle sezioni), in modo che il lato formvar sia rivolto verso il basso e il bordo lungo destro della fessura sia sulla superficie dell'acqua e parallelo al bordo dell'acqua.
- Muovere delicatamente la griglia nell'acqua verso i nastri in modo che al momento del tratto di ritorno, le sezioni si spostino verso la griglia. Continuate a farlo in waft sempre più piccoli fino a quando il bordo destro del nastro si allinea con il bordo destro dello slot. Quindi, con l'ultimo waft, porta delicatamente la griglia verso l'alto per raccogliere le sezioni nella griglia della fessura.
- Lasciare asciugare la griglia nella pinza prima di riporla nella casella della griglia, opportunamente annotata sul foglio di riferimento della casella della griglia.
- Ripetere il passaggio 3.16 fino a quando tutti i nastri non vengono raccolti, assicurando che l'ordine dei nastri venga mantenuto.
- Se sono necessarie ulteriori sezioni, ritrarre il coltello di circa 150 nm, controllare il livello dell'acqua nella barca e aggiungerne altre se necessario. Avviare nuovamente il processo di taglio, seguendo i passaggi 3.11-3.18.
- Una volta raccolte tutte le sezioni, assicurarsi che il bordo del coltello sia privo di detriti di sezione, ritrarre il coltello lontano dalla faccia del blocco e rimuovere e pulire il coltello.
4. Colorazione della griglia
- Una volta asciutte, macchiate le sezioni con il citrato di piombo di Reynolds su parafilm sulla panca o in una capsula di Petri. Posizionare diversi pellet di idrossido di sodio sotto il coperchio di una capsula di Petri per fornire un ambiente privo di anidride carbonica. Quindi, con attenzione, lontano dai pellet, pipettare 40 μL di gocce di citrato di piombo di Reynolds sul parafilm, una per ogni griglia.
NOTA: non macchiare troppe griglie contemporaneamente; ad esempio, il massimo dovrebbe essere 6. Cerca di non respirare direttamente sul piatto colorante. L'anidride carbonica può reagire con il citrato di piombo e causare precipitati indesiderati sulle griglie. - Capovolgere ogni griglia (sezione rivolta verso il basso) sulla goccia di citrato di piombo e lasciare protetto dal coperchio della capsula di Petri per 7-10 minuti. Mentre le griglie si colorano, preparare un secondo pezzo più grande di parafilm sul banco con cinque gocce da 300 μL di acqua distillata per ogni griglia.
- Alla fine dell'incubazione del citrato di piombo, trasferire ogni griglia in una goccia di acqua distillata per lavare per 1 minuto senza respirare direttamente sulle griglie.
- Ripetere il passaggio 4.3 per un totale di cinque volte.
- Usando una pinza incrociata numerata, prendi la prima griglia, tocca il bordo della griglia per filtrare la carta per allontanare la maggior parte dell'acqua e lascia asciugare nella pinza (per almeno 20 minuti). Ripetere l'operazione per ogni griglia.
5. Acquisizione di immagini da parte di TEM
NOTA: poiché i controlli TEM esatti variano da produttore a produttore, seguire le istruzioni e le linee guida del produttore. I seguenti passaggi devono essere eseguiti da utenti che sono già esperti nell'uso di TEM.
- Prima dell'imaging, eseguire i controlli usuali, ad esempio l'allineamento del fascio, i riferimenti di guadagno e l'eucentrismo del campione.
- Caricare con cura la prima griglia di sezioni seriali nel portacampioni, avendo cura di allineare la fessura (e quindi le sezioni) all'asse verticale dello stadio del microscopio.
NOTA: questa precisione non è essenziale, ma consente di risparmiare tempo durante le fasi di acquisizione e gestione dei dati future. Quando si inserisce la griglia (sezione rivolta verso il basso o sezione rivolta verso l'alto), fare attenzione a visualizzare tutte le griglie con lo stesso orientamento. - A basso ingrandimento, osservare l'ordine, la posizione e la posizione delle sezioni seriali (Figura 1E). Passare alla sezione centrale della serie sulla griglia.
NOTA: a seconda dell'obiettivo esatto della ricerca, gli approcci per l'imaging possono variare; tuttavia, quanto segue è un utile punto di partenza. La forma delle sezioni e la relazione dei nastri (come rilevato nel passaggio 3.14) determinano quale sezione era la prima e quale sezione era l'ultima sulla griglia. - Sfoglia l'esempio e identifica una regione di interesse. Osserva il campione all'ingrandimento desiderato e considera la possibilità di raccogliere la serie con un ingrandimento leggermente inferiore, poiché le sezioni spesso non sono perfettamente allineate e le immagini potrebbero dover essere ritagliate in seguito.
- Scatta immagini di riferimento a ingrandimenti inferiori per apprezzare il contesto della regione di interesse, la sua posizione approssimativa a diversi ingrandimenti in relazione ai confini della sezione e le caratteristiche di riferimento all'interno del campione. Utilizzali per firmare la regione di interesse in altre sezioni.
- Facoltativo: per i riferimenti allo schermo, utilizzare stucco adesivo riutilizzabile, adesivi o un pezzo di carta per lavagna luminosa (OHP), fissato sullo schermo, per posizionare marcatori temporanei sullo schermo per consentire la reimaging di routine delle stesse caratteristiche della regione di interesse al centro dell'immagine, in tutto il set di dati (vedere stelle gialle nella Figura 1E).
- Utilizzando le immagini di riferimento, passare alla regione di interesse nella prima sezione della griglia e acquisire un'immagine all'ingrandimento desiderato.
NOTA: quando si salvano le immagini, annotare il primo nome file della prima immagine della serie e utilizzare la nomenclatura dei nomi sequenziali in modo che tutti i nomi delle immagini seguano l'ordine sequenziale delle sezioni seriali. - Passare alla sezione successiva e ripetere il passaggio 5.7 fino a quando tutte le sezioni non sono state visualizzate per l'area di interesse.
6. Esportazione di immagini e registrazione dell'allineamento della sezione seriale
- Esporta i file di immagine appartenenti allo stesso stack in un'unica cartella. Assicurati che la cartella sia ordinata in base al nome del file.
NOTA: le immagini dovrebbero idealmente avere lo stesso nome radice e seguire l'ordine in cui sono state acquisite. - Apri Fiji e fai clic su File | Importare | Sequenza di immagini.
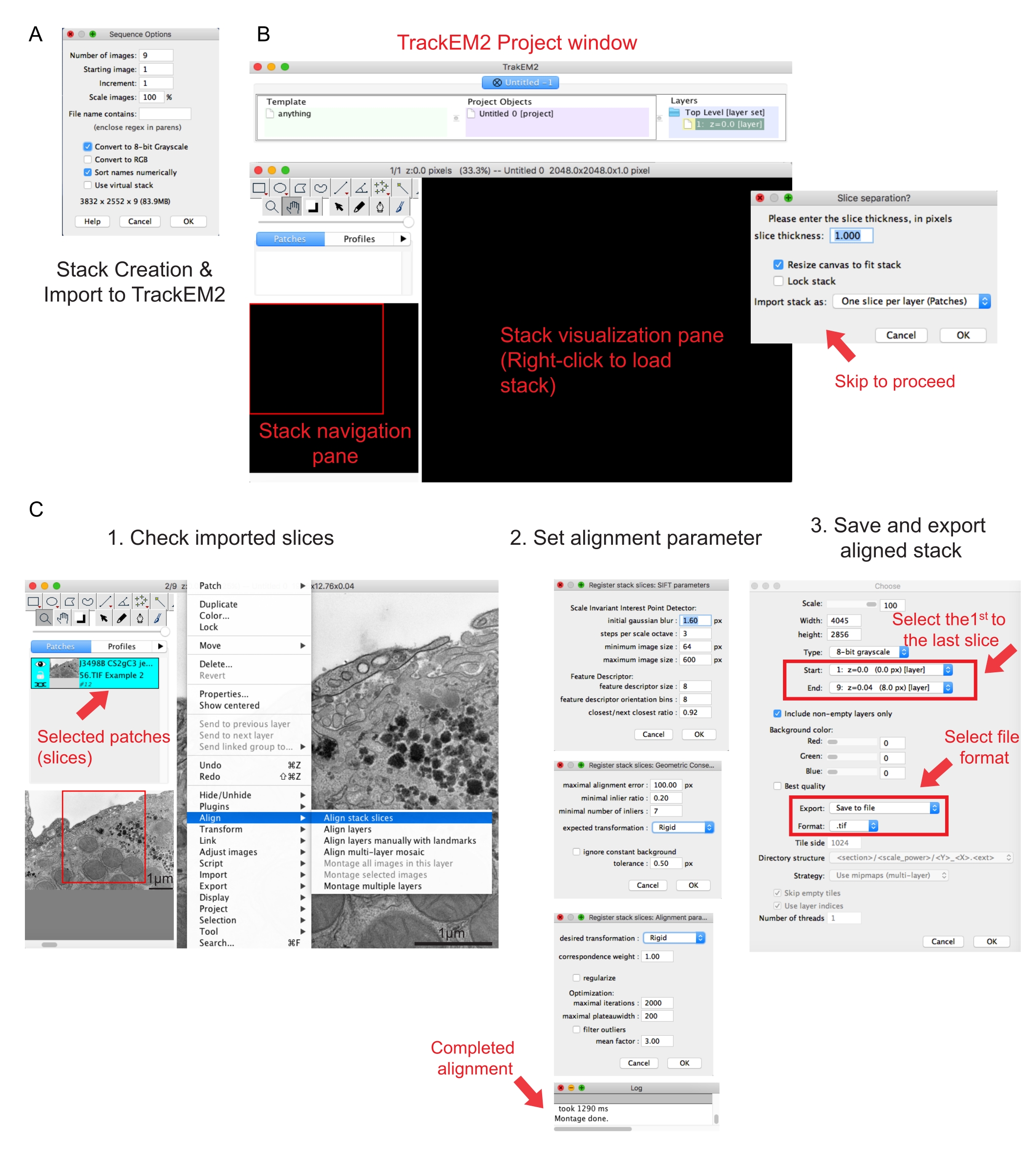
- Fare clic sulla prima immagine della cartella e fare clic su Apri. Attendere la visualizzazione di una finestra popup di Opzioni sequenza (Figura 2A).

Figura 2: Creazione di uno stack seriale e allineamento della sezione seriale utilizzando Fiji. (A) Screenshot che mostra le opzioni di sequenza durante il caricamento delle immagini per la creazione di uno stack seriale. (B) Screenshot del plugin TrackEM2 e delle finestre chiave del plugin. Premete OK nella separazione slice per procedere con l'allineamento. (C) Screenshot dopo aver caricato correttamente lo stack seriale nel riquadro di visualizzazione. Una volta selezionate allinea le sezioni dello stack , verranno visualizzate tre finestre sequenziali dei parametri di allineamento. Esportare lo stack allineato una volta completato l'allineamento. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
- Fare clic su Ordina nomi numericamente | Converti in opzione a 8 bit . Premere OK.
NOTA: la conversione a 8 bit aiuta l'importazione dei dati in Amira e riduce le dimensioni del file, consentendo velocità di elaborazione più rapide nelle fasi successive. - Controllare la completezza, la sequenza e l'ingrandimento della pila creata. Salvare lo stack creato come file .tif.
NOTA: le immagini dovrebbero essere state acquisite con lo stesso ingrandimento. - Eseguire il plugin TrakEM241. Vai a File | Nuovi | TrackEM2 (vuoto).
NOTA: Il plugin chiederà all'utente di salvare i file TrackEM2. Se necessario, salvate i file TrackEM2 nella cartella delle immagini. Dovrebbero essere visualizzate tre finestre: una finestra di progetto, una finestra di navigazione dello stack (a sinistra) e un riquadro di visualizzazione dello stack (Figura 2B). - Fare clic con il pulsante destro del mouse sul riquadro di visualizzazione nero. Fai clic su Importa | Importa stack e seleziona lo stack creato in precedenza.
- Fare clic su OK per caricare lo stack nella finestra di navigazione dello stack.
NOTA: verrà visualizzata una finestra di separazione delle sezioni per chiedere la relazione tra pixel e dimensione. Per il solo allineamento dello stack, fare clic su OK per ignorare questo passaggio. - Utilizzare il dispositivo di scorrimento per controllare tutte le sezioni dello stack. Cerca la sezione caricata, che verrà visualizzata come patch nel piano di navigazione. Selezionate le patch che verranno incluse nel seguente allineamento.
NOTA: le patch selezionate diventeranno blu. - Passare il puntatore del mouse sul riquadro di visualizzazione. Fare clic con il pulsante destro del mouse sull'immagine, selezionare Allinea | Allineare le sezioni dello stack (Figura 2C-1).
- Specificate i parametri di allineamento tramite un insieme di tre finestre sequenziali.
NOTA: per la maggior parte dei dati, iniziare con un allineamento rigido (consente la rotazione e la traslazione ma non la trasformazione) e mantenere gli altri parametri come predefiniti (Figura 2C-2). - Consenti l'esecuzione dell'allineamento fino a quando il registro di lettura non dice Montaggio completato.
NOTA: il runtime dipende dal numero di voxel e dalla velocità del computer. - Controllare lo stack allineato nel riquadro di visualizzazione. Premi Alt e - (nel PC) o i tasti Ctrl e - (in Mac) per una visualizzazione zoom-out dello stack allineato.
- Se si è soddisfatti dello stack allineato, fare clic con il pulsante destro del mouse su Esporta | Crea un'immagine piatta per salvare la pila allineata.
- Selezionare la prima immagine come inizio dello stack e l'ultima immagine come fine dello stack, fare clic su OK (Figura 2C-3). Salvare lo stack allineato come .tif.
NOTA: per ridurre le dimensioni del file, ritagliare i dati in modo che contengano solo l'area di interesse necessaria. - Se necessario, eseguire un allineamento affine sullo stack allineato. Apri lo stack allineato nelle Figi, seleziona Plugin | | di registrazione StackReg.
- Scegliete l'opzione affine e premete OK. Attendere il completamento del programma.
- Salvare lo stack allineato affine con un nome di file diverso.
7. Segmentazione e ricostruzione 3D
- Aperto Amira42. Fare clic su File | Open Data per caricare lo stack allineato.
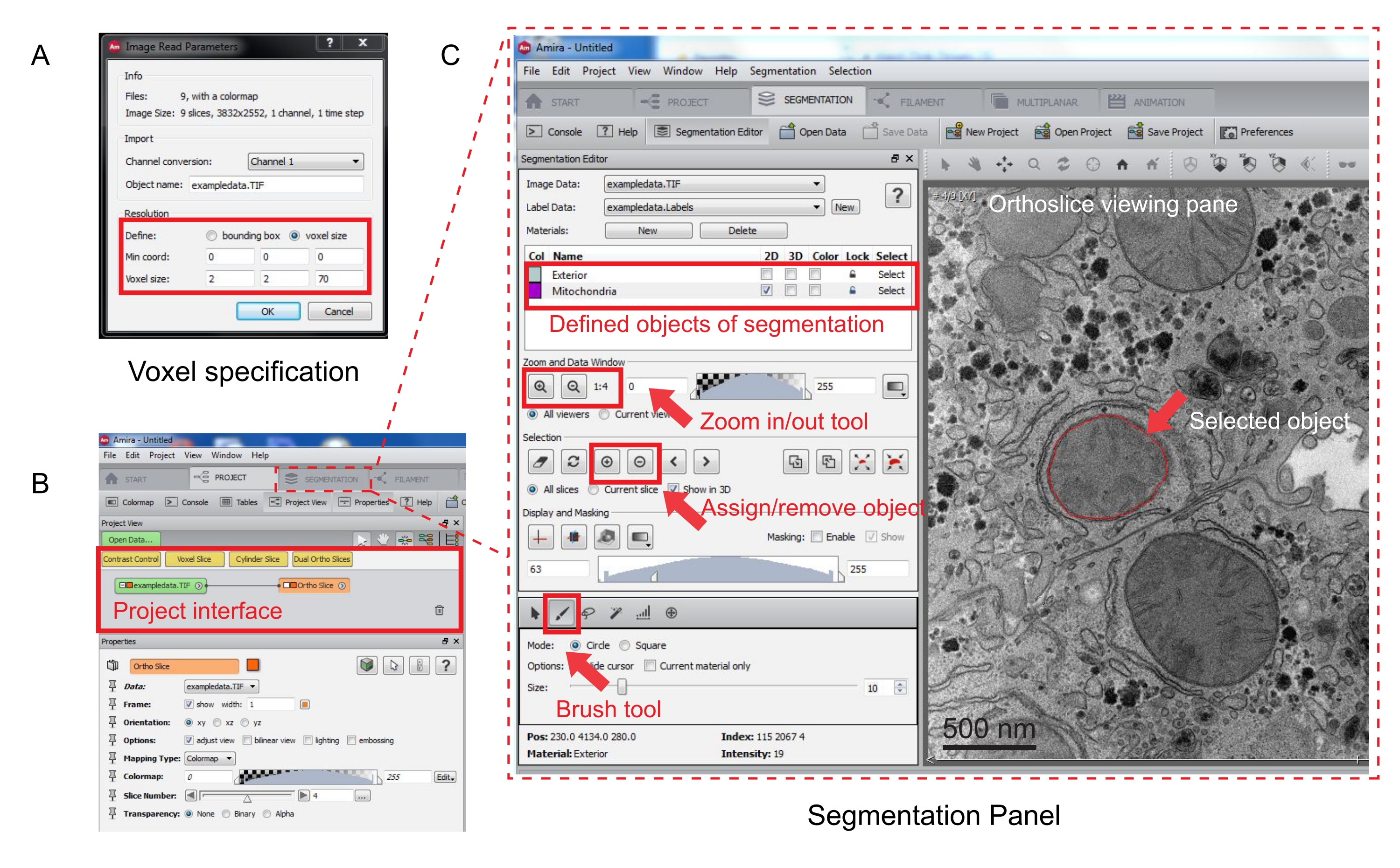
- Specificare le misure del voxel nella nuova finestra popup (Figura 3A).

Figura 3: Segmentazione dello stack seriale utilizzando Amira. (A) La finestra popup di definizione voxel prima di caricare uno stack allineato. (B) Screenshot dell'interfaccia del progetto dopo l'importazione di uno stack. Selezionate la scheda Segmentazione per avviare la traccia degli oggetti nel pannello Editor segmentazione. (C) Caratteristiche principali della scheda segmentazione. Definire gli oggetti per la segmentazione nella sezione Editor segmentazione della scheda Segmentazione. Utilizzare la funzione di zoom per facilitare l'identificazione degli oggetti. Selezionate lo strumento pennello e tracciate il contorno dell'oggetto. Fare clic sul simbolo + in Selezione per assegnare la traccia. Un oggetto assegnato apparirà con un limite rosso nel riquadro di visualizzazione dell'ortoslice. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
NOTA: nell'interfaccia del progetto verrà visualizzato un nodo dello stack di immagini e un'ortoslice nel riquadro di visualizzazione a destra (Figura 3B).
- Per avviare la segmentazione, selezionare la scheda Segmentazione (Figura 3B).
NOTA: si consiglia di salvare l'avanzamento della segmentazione prima e durante la segmentazione. Vai a Model | Salva modello come qualsiasi file .am adatto. - Fate clic su Nuovo nel pannello dell'editor di segmentazione per definire nuovi oggetti nell'elenco dei materiali. Fare clic con il pulsante destro del mouse per modificare il colore dell'oggetto e fare doppio clic per rinominare l'oggetto.
- Per la segmentazione manuale, scegli lo strumento di segmentazione sotto l'elenco dei materiali. Selezionate lo strumento Pennello di default per evidenziare i pixel (Figura 3C).
NOTA: in alternativa, utilizzate lo strumento pennello per tracciare il contorno dell'oggetto e premete Maiusc + F per riempire l'oggetto. - Per convertire lo strumento pennello in una gomma, premete continuamente Ctrl mentre selezionate i pixel da correggere. Annotare ogni fetta della pila.
- Una volta confermata, assegna la selezione a un'etichetta facendo clic sul segno + . Fare clic sul segno - per rimuovere la selezione.
- Torna all'interfaccia del progetto una volta completata la segmentazione. Cerca un nodo con un'estensione ".label" connessa allo stack di immagini.
- Fare clic con il pulsante destro del mouse sull'estensione ".label" e selezionare Genera superficie | Applicare per creare un file .surf.
- Per eseguire il rendering del modello 3D di un oggetto segmentato, fate clic con il pulsante destro del mouse sul file .surf e selezionate Vista superficie (Surface view ) per generare un modello 3D nel riquadro di visualizzazione.
- Salvare il modello 3D per la visualizzazione o ulteriori analisi quantitative.
Risultati
Per questa tecnica, le regioni di interesse vengono selezionate in base all'obiettivo della ricerca biologica e identificate prima del taglio e del sezionamento del tessuto incorporato. Allo stesso modo, la dimensione della faccia del blocco può essere dettata dalla domanda di ricerca; in questo caso, il campione è stato tagliato per lasciare una faccia di blocco di circa 0,3 mm x 0,15 mm (Figura 4A). Ciò ha permesso di creare due griglie di 9 sezioni seriali per griglia, fornendo 18 sezi...
Discussione
In questo protocollo è descritta una tecnica vEM accessibile per visualizzare la struttura e le interazioni degli organelli in 3D. La morfologia dei contatti interorganelle negli epatociti è presentata come un caso di studio qui. Tuttavia, questo approccio è stato applicato anche per studiare una varietà di altri campioni e aree di ricerca, tra cui le interazioni cellula-endotelio di Schwann nei nervi periferici45, la biogenesi del corpo di Weibel Palade nelle cellule endoteliali
Divulgazioni
Gli autori non hanno conflitti di interesse da divulgare.
Riconoscimenti
Ringraziamo Joanna Hanley, Rebecca Fiadeiro e Ania Straatman-Iwanowska per l'assistenza tecnica esperta. Ringraziamo anche i membri del laboratorio Stefan e Ian J. White per le discussioni utili. J.J.B. è supportato da finanziamenti MRC al MrC Laboratory of Molecular Cell Biology presso uCL, codice di assegnazione MC_U12266B. C.J.S. è supportato dal finanziamento MRC al MRC Laboratory of Molecular Cell Biology University Unit presso uCL, codice di assegnazione MC_UU_00012/6. P.G. è finanziato dal Consiglio europeo della ricerca, codice di sovvenzione ERC-2013-StG-337057.
Materiali
| Name | Company | Catalog Number | Comments |
| 0.22 µm syringe filter | Sarstedt | 83.1826.001 | |
| Aluminum trays | Agar Scientific | AGG3912 | |
| Amira v6 | ThermoFisher | https://www.thermofisher.com | |
| Chloroform | Fisher | C/4960/PB08 | |
| DDSA/Dodecenyl Succinic Anhydride | TAAB | T027 | Epon ingredient |
| Diamond knife | DiaTOME | ultra 45° | |
| DMP-30/2,4,6-tri (Dimethylaminomethyl) phenol | TAAB | D032 | Epon ingredient |
| Dumont Tweezers N5 | Agar Scientific | AGT5293 | |
| Fiji | https://imagej.net/ | ||
| Fiji TrakEM2 plugin | https://imagej.net/ | ||
| Formaldehyde 36% solution | TAAB | F003 | |
| Formvar coated slot grid | Homemade | Alternative: EMS diasum (FF2010-Cu) | |
| Glass bottle with applicator rod | Medisca | 6258 | |
| Glass vials | Fisher Scientific | 15364769 | |
| Gluteraldehyde 25% solution | TAAB | G011 | |
| MNA/Methyl Nadic Anhydride | TAAB | M011 | Epon ingredient |
| Osmium Tetroxide 2% solution | TAAB | O005 | |
| Potassium Ferricyanide | Sigma-Aldrich | P-8131 | |
| Propylene oxide | Fisher Scientific | E/0050/PB08 | |
| Reuseable adhesive | Blue Tack | ||
| Reynolds Lead Citrate | TAAB | L037 | Section stain |
| Sodium Cacodylate | Sigma-Aldrich | C-0250 | to make 0.1 M Caco buffer |
| Super Glue | RS Components | 918-6872 | Cyanoacrylate glue, Step 1.3 |
| TAAB 812 Resin | TAAB | T023 | Epon ingredient |
| Tannic acid | TAAB | T046 | |
| Triton X-100 | Sigma-Aldrich | T9284 | |
| Two part Epoxy Resin | RS Components | 132-605 | Alternative: Step 2.13 |
| Ultramicrotome | Leica | UC7 | |
| Vibrating microtome | Leica | 100 µm thick slices, 0.16 mm/s cutting at 1 mm amplitude . | |
| Weldwood Original Contact cement | DAP | 107 | Contact adhesive: Step 3.1.4 |
Riferimenti
- Knoll, M., Ruska, E. Das elektronenmikroskop. Zeitschrift für Physik. 78 (5), 318-339 (1932).
- von Ardenne, M. Daselektronen-rastermikroskop. Zeitschrift für Physik. 109 (9), 553-572 (1938).
- Bang, B. H., Bang, F. B. Graphic reconstruction of the third dimension from serial electron microphotographs. Journal of Ultrastructure Research. 1 (2), 138-139 (1957).
- Birch-Andersen, A. Reconstruction of the nuclear sites of Salmonella typhimurium from electron micrographs of serial sections. Journal of General Microbiology. 13 (2), 327-329 (1955).
- Denk, W., Horstmann, H. Serial block-face scanning electron microscopy to reconstruct three-dimensional tissue nanostructure. PLoS Biology. 2 (11), 329 (2004).
- Peddie, C. J., Collinson, L. M. Exploring the third dimension: volume electron microscopy comes of age. Micron. 61, 9-19 (2014).
- Titze, B., Genoud, C. Volume scanning electron microscopy for imaging biological ultrastructure. Biology of the Cell. 108 (11), 307-323 (2016).
- Kornfeld, J., Denk, W. Progress and remaining challenges in high-throughput volume electron microscopy. Current Opinion in Neurobiology. 50, 261-267 (2018).
- Heymann, J. A., et al. Site-specific 3D imaging of cells and tissues with a dual beam microscope. Journal of Structural Biology. 155 (1), 63-73 (2006).
- Knott, G., Marchman, H., Wall, D., Lich, B. Serial section scanning electron microscopy of adult brain tissue using focused ion beam milling. Journal of Neuroscience. 28 (12), 2959-2964 (2008).
- Leighton, S. B. SEM images of block faces, cut by a miniature microtome within the SEM - a technical note. Scanning Electron Microscopy. , 73-76 (1981).
- Martone, M. E., Deerinck, T. J., Yamada, N., Bushong, E., Ellisman, M. H. Correlated 3D light and electron microscopy: use of high voltage electron microscopy and electron tomography for imaging large biological structures. Journal of Histotechnology. 23 (3), 261-270 (2000).
- Micheva, K. D., Smith, S. J. Array tomography: a new tool for imaging the molecular architecture and ultrastructure of neural circuits. Neuron. 55 (1), 25-36 (2007).
- Sjostrand, F. S. Ultrastructure of retinal rod synapses of the guinea pig eye as revealed by three-dimensional reconstructions from serial sections. Journal of Ultrastructure Research. 2 (1), 122-170 (1958).
- Ware, R. W. Three-dimensional reconstruction from serial sections. International Review of Cytology. 40, 325 (1975).
- Stevens, J. K., Davis, T. L., Friedman, N., Sterling, P. A systematic approach to reconstructing microcircuitry by electron microscopy of serial sections. Cognitive Brain Research. 2 (3), 265-293 (1980).
- Hoppe, W. Three-dimensional electron microscopy. Annual Review of Biophysics. 10, 563-592 (1981).
- Frank, J. . Electron tomography: methods for three-dimensional visualization of structures in the cell. , (2008).
- Baumeister, W. Electron tomography: towards visualizing the molecular organization of the cytoplasm. Current Opinion in Structural Biology. 12 (5), 679-684 (2002).
- Hoog, J. L., Schwartz, C., Noon, A. T., O'Toole, E. T. Organization of interphase microtubules in fission yeast analyzed by electron tomography. Developmental Cell. 12 (3), 349-361 (2007).
- Harris, K. M., Perry, E., Bourne, J., Feinberg, M., Ostroff, L., Hurlburt, J. Uniform serial sectioning for transmission electron microscopy. Journal of Neuroscience. 26 (47), 12101-12103 (2006).
- Jesior, J. C. Use of low-angle diamond knives leads to improved ultrastructural preservation of ultrathin sections. Scanning Microscopy Supplement. 3, 147-152 (1989).
- Studer, D., Gnaegi, H. Minimal compression of ultrathin sections with use of an oscillating diamond knife. Journal of Microscopy. 197, 94-100 (2000).
- Gay, H., Anderson, T. F. Serial sections for electron microscopy. Science. 120 (3130), 1071-1073 (1954).
- Bernhard, W., Rouiller, C. Close topographical relationship between mitochondria and ergastoplasm of liver cells in a definite phase of cellular activity. The Journal of Biophysical and Biochemical Cytology. 2, 73-78 (1956).
- Palade, G. E. An electron microscope study of the mitochondrial structure. The Journal of Histochemistry & Cytochemistry. 1 (4), 188-211 (1953).
- Wu, H., Carvalho, P., Voeltz, G. K. Here, there, and everywhere: The importance of ER membrane contact sites. Science. 361 (6401), (2018).
- Vance, J. E. Inter-organelle membrane contact sites: implications for lipid metabolism. Biology Direct. 15 (1), 24 (2020).
- Stefan, C. J. Endoplasmic reticulum-plasma membrane contacts: Principals of phosphoinositide and calcium signaling. Current Opinion in Cell Biology. 63, 125-134 (2020).
- Zaman, M. F., Nenadic, A., Radojicic, A., Rosado, A., Beh, C. T. Sticking with it: ER-PM membrane contact sites as a coordinating nexus for regulating lipids and proteins at the cell cortex. Frontiers in Cell and Developmental Biology. 8, 675 (2020).
- van Vliet, A. R., Sassano, M. L., Agostinis, P. The unfolded protein response and membrane contact sites: tethering as a matter of life and death. Contact. 1, 1-15 (2018).
- Cohen, S., Valm, A. M., Lippincott-Schwartz, J. Interacting organelles. Current Opinion in Cell Biology. 53, 84-91 (2018).
- Hariri, H., et al. Lipid droplet biogenesis is spatially coordinated at ER-vacuole contacts under nutritional stress. EMBO Reports. 19 (1), 57-72 (2018).
- Stefan, C. J., Trimble, W. S., Grinstein, S., Drin, G. Membrane dynamics and organelle biogenesis-lipid pipelines and vesicular carriers. BMC Biology. 15 (1), 102 (2017).
- Eisenberg-Bord, M., Shai, N., Schuldiner, M., Bohnert, M. A tether is a tether is a tether: tethering at membrane contact sites. Developmental Cell. 39 (4), 395-409 (2016).
- Scorrano, L., De Matteis, M. A., Emr, S., Giordano, F. Coming together to define membrane contact sites. Nature Communications. 10 (1), 1287 (2019).
- Lak, B., Li, S., Belevich, I., Sree, S. Specific subdomain localization of ER resident proteins and membrane contact sites resolved by electron microscopy. European Journal of Cell Biology. 100 (7), 151180 (2021).
- Collado, J., Kalemanov, M., Campelo, F., Bourgoint, C. Tricalbin-mediated contact sites control ER curvature to maintain plasma membrane integrity. Developmental Cell. 51 (4), 476-487 (2019).
- West, M., Zurek, N., Hoenger, A., Voeltz, G. K. A 3D analysis of yeast ER structure reveals how ER domains are organized by membrane curvature. Journal of Cell Biology. 193 (2), 333-346 (2011).
- Ilacqua, N., Anastasia, I., Raimondi, A., Lemieux, P. A three-organelle complex made by wrappER contacts with peroxisomes and mitochondria responds to liver lipid flux changes. Journal of Cell Science. 135 (5), 259091 (2022).
- Cardona, A., Saalfeld, S., Schindelin, J., Arganda-Carreras, I. TrakEM2 software for neural circuit reconstruction. PLoS One. 7 (6), 38011 (2012).
- Stalling, D., Westerhoff, M., Hege, H. -. C. Amira: A highly interactive system for visual data analysis. The Visualization Handbook. 38, 749-767 (2005).
- Hsieh, T. S., Chen, Y. J., Chang, C. L., Lee, W. R., Liou, J. Cortical actin contributes to spatial organization of ER-PM junctions. Molecular Biology of the Cell. 28 (23), 3171-3180 (2017).
- Anastasia, I., Ilacqua, N., Raimondi, A., Lemieux, P. Mitochondria-rough-ER contacts in the liver regulate systemic lipid homeostasis. Cell Reports. 34 (11), 108873 (2021).
- Cattin, A. L., Burden, J. J., Van Emmenis, L., Mackenzie, F. E. Macrophage-Induced Blood Vessels Guide Schwann Cell-Mediated Regeneration of Peripheral Nerves. Cell. 162 (5), 1127-1139 (2015).
- Lopes-da-Silva, M., et al. A GBF1-dependent mechanism for environmentally responsive regulation of ER-Golgi transport. Developmental Cell. 49 (5), 786-801 (2019).
- Banushi, B., Forneris, F., Straatman-Iwanowska, A., Strange, A. Regulation of post-Golgi LH3 trafficking is essential for collagen homeostasis. Nature Communications. 7, 12111 (2016).
- Rey, S. A., et al. Ultrastructural and functional fate of recycled vesicles in hippocampal synapses. Nature Communications. 6, 8043 (2015).
- Belicova, L., Repnik, U., Delpierre, J., Gralinska, E. Anisotropic expansion of hepatocyte lumina enforced by apical bulkheads. Journal of Cell Biology. 220 (10), 202303003 (2021).
- Kizilyaprak, C., Daraspe, J., Humbel, B. M. Focused ion beam scanning electron microscopy in biology. Journal of Microscopy. 254 (3), 109-114 (2014).
- Xu, C. S., Hayworth, K. J., Lu, Z., Grob, P. Enhanced FIB-SEM systems for large-volume 3D imaging. Elife. 6, 1-36 (2017).
- Parlakgül, G., Arruda, A. P., Cagampan, E., Pang, S. High resolution 3D imaging of liver reveals a central role for subcellular architectural organization in metabolism. bioRxiv. , (2020).
- Guerin, C. J., Kremer, A., Borghgraef, P., Lippens, S. Targeted studies using serial block face and focused ion beam scan electron microscopy. The Journal of Visualized Experiments: JoVE. (150), e59480 (2019).
- Kremer, A., et al. A workflow for 3D-CLEM investigating liver tissue. Journal of Microscopy. 281 (3), 231-242 (2021).
- Hayat, M. . Principles and techniques of electron microscopy: biological applications. , (2000).
- Wisse, E., Braet, F., Duimel, H., Vreuls, C. Fixation methods for electron microscopy of human and other liver. World Journal of Gastroenterology. 16 (23), 2851-2866 (2010).
- Hanley, J., Dhar, D. K., Mazzacuva, F., Fiadeiro, R. Vps33b is crucial for structural and functional hepatocyte polarity. Journal of Hepatology. 66 (5), 1001-1011 (2017).
- Deerinck, T. J., Bushong, E. A., Thor, A., Ellisman, M. H. NCMIR methods for 3D EM: a new protocol for preparation of biological specimens for serial block face scanning electron microscopy. Microscopy. 1, 6-8 (2010).
- Miranda, K., Girard-Dias, W., Attias, M., de Souza, W., Ramos, I. Three dimensional reconstruction by electron microscopy in the life sciences: An introduction for cell and tissue biologists. Molecular Reproduction and Development. 82 (7-8), 530-547 (2015).
- Yamaguchi, M., Chibana, H. A method for obtaining serial ultrathin sections of microorganisms in transmission electron microscopy. The Journal of Visualized Experiments: JoVE. (131), e56235 (2018).
- Hall, D. H., Hartwieg, E., Nguyen, K. C. Modern electron microscopy methods for C. elegans. Methods in Cell Biology. 107, 93-149 (2012).
- Hagler, H. K. Ultramicrotomy for biological electron microscopy. Methods in Molecular Biology. 369, 67-96 (2007).
- Arganda-Carreras, I., Beichel, R. R., Sonka, M. Consistent and elastic registration of histological sections using vector-spline regularization. Computer vision approaches to medical image analysis, CVAMIA 2006, Lecture Notes in Computer Science. 4241, 85-95 (2006).
- Belevich, I., Joensuu, M., Kumar, D., Vihinen, H., Jokitalo, E. Microscopy image browser: a platform for segmentation and analysis of multidimensional datasets. PLoS Biology. 14 (1), 1002340 (2016).
- Fiala, J. C. Reconstruct: a free editor for serial section microscopy. Journal of Microscopy. 218, 52-61 (2005).
- Kremer, J. R., Mastronarde, D. N., McIntosh, J. R. Computer visualization of three-dimensional image data using IMOD). Journal of Structural Biology. 116 (1), 71-76 (1996).
- Iudin, A., Korir, P. K., Salavert-Torres, J., Kleywegt, G. J., Patwardhan, A. EMPIAR: a public archive for raw electron microscopy image data. Nature Methods. 13 (5), 387-388 (2016).
- Xu, C. S., Pang, S., Shtengel, G., Muller, A. An open-access volume electron microscopy atlas of whole cells and tissues. Nature. 599 (7883), 147-151 (2021).
- Karabag, C., et al. Semantic segmentation of HeLa cells: An objective comparison between one traditional algorithm and four deep-learning architectures. PLoS One. 15 (10), 0230605 (2020).
- Heinrich, L., Bennett, D., Ackerman, D., Park, W. Whole-cell organelle segmentation in volume electron microscopy. Nature. 599 (7883), 141-146 (2021).
- Kim, J. S., Greene, M. J., Zlateski, A., Lee, K. Space-time wiring specificity supports direction selectivity in the retina. Nature. 509 (7500), 331-336 (2014).
- Spiers, H., Songhurst, H., Nightingale, L., de Folter, J. Deep learning for automatic segmentation of the nuclear envelope in electron microscopy data, trained with volunteer segmentations. Traffic. 22 (7), 240-253 (2021).
- Hasan, N. M., Gupta, A., Polishchuk, E., Yu, C. H. Molecular events initiating exit of a copper-transporting ATPase ATP7B from the trans-Golgi network. The Journal of Biological Chemistry. 287 (43), 36041-36050 (2012).
- Stoeck, I. K., Lee, J. Y., Tabata, K., Romero-Brey, I. Hepatitis C virus replication depends on endosomal cholesterol homeostasis. The Journal of Virology. 92 (1), 01196 (2018).
- Ma, X., Qian, H., Chen, A., Ni, H. M., Ding, W. X. Perspectives on mitochondria-ER and mitochondria-lipid droplet contact in hepatocytes and hepatic lipid metabolism. Cells. 10 (9), 2273 (2021).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati