
Method Article
Differentiation of Functional Osteoclasts from Human Peripheral Blood CD14+ Monocytes
* These authors contributed equally
In This Article
Summary
Osteoclasts are key bone-resorbing cells in the body. This protocol describes a reliable method for the in vitro differentiation of osteoclasts from human peripheral blood monocytes. This method can be used as an important tool to further understand osteoclast biology in homeostasis and in diseases.
Abstract
Osteoclasts (OCs) are bone-resorbing cells that play a pivotal role in skeletal development and adult bone remodeling. Several bone disorders are caused by increased differentiation and activation of OCs, so the inhibition of this pathobiology is a key therapeutic principle.Two key factors drive the differentiation of OCs from myeloid precursors: macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor kappa-B ligand (RANKL). Human circulating CD14+ monocytes have long been known to differentiate into OCs in vitro. However, the exposure time and the concentration of RANKL influence the differentiation efficiency. Indeed, protocols for the generation of human OCs in vitro have been described, but they often result in a poor and lengthy differentiation process. Herein, a robust and standardized protocol for generating functionally active mature human OCs in a timely manner is provided. CD14+ monocytes are enriched from human peripheral blood mononuclear cells (PBMCs) and primed with M-CSF to upregulate RANK. Subsequent exposure to RANKL generates OCs in a dose- and time-dependent manner. OCs are identified and quantified by staining with tartrate acid-resistant phosphatase (TRAP) and light microscopy analysis. Immunofluorescence staining of nuclei and F-actin is used to identify functionally active OCs. In addition, OSCAR+CD14− mature OCs are further enriched via flow cytometry cell sorting, and OC functionality quantified by mineral (or dentine/bone) resorption assays and actin ring formation. Finally, a known OC inhibitor, rotenone, is used on mature OCs, demonstrating that adenosine triphosphate (ATP) production is essential for actin ring integrity and OC function. In conclusion, a robust assay for differentiating high numbers of OCs is established in this work, which in combination with actin ring staining and an ATP assay provides a useful in vitro model to evaluate OC function and to screen for novel therapeutic compounds that can modulate the differentiation process.
Introduction
Osteoclasts (OCs) are multinucleated giant cells of hematopoietic lineage with a unique capacity to resorb bone. They are responsible for the development and continuous remodeling of the skeleton1,2. In the skeletal phases of development, OCs and tissue-resident macrophages are derived from erythro-myeloid progenitors and colonize the bone niche and organ tissues. In physiological conditions, erythro-myeloid progenitors are required for normal bone development and tooth eruption, while the influx of circulating blood monocytes into the bone niche provides postnatal maintenance of the OCs, bone mass, and bone marrow cavity3. Under pathological conditions, monocytes are recruited to sites of active inflammation and can contribute to pathological bone destruction4,5.
Patients with several forms of arthritis experience joint inflammation, leading to progressive joint destruction caused by OCs6. For instance, in rheumatoid arthritis (RA), overactivated OCs are responsible for pathological bone erosion and joint destruction7,8, and current treatments often do not improve or stop the bone damage9,10,11. Alterations in circulating monocytes both in terms of population distribution and transcriptomic and epigenetic signatures have been reported in RA patients12,13,14. Moreover, it has been reported that altered monocyte responses to inflammatory stimulation affect osteoclastogenesis in RA patients with active disease15,16,17.
The differentiation of OCs is a complex multistep process comprising the commitment of myeloid precursor cells to differentiation into OC precursors. During osteoclastogenesis, OCs become giant and multinucleated through cell-cell fusion, incomplete cytokinesis, and a nuclear recycling process described as fission and fusion18,19,20. The ability to differentiate OCs in vitro has allowed for significant advances in the understanding of bone biology21. OCs differentiate from precursors upon exposure to macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor kappa-B ligand (RANKL). The latter is essential for the normal development and function of OCs in vitro and in vivo, even under inflammatory conditions6,22,23. RANKL is presented by osteoblasts and osteocytes, as well as by activated T cells and fibroblasts in the inflamed RA synovium2,24,25. During the OC differentiation process, monocytes exposed to M-CSF upregulate receptor activator of nuclear factor kappa-B (RANK) expression on their cell membrane and, under subsequent stimulation with RANKL, differentiate into tartrate-resistant acid phosphatase (TRAP)-positive mononuclear pre-OCs and then into multinucleated OCs15,26. OCs produce several enzymes, the chief among them is TRAP, which enables the degradation of phosphoproteins within bone27. A regulator and marker of OC differentiation is the OC-associated receptor (OSCAR). It is upregulated early in precursor cells committing to the OC lineage28. Mature giant multinucleated OCs can degrade (resorb) the skeletal matrix by generating a large sealing zone, which is made of an actin ring surrounding a ruffled border21,29,30. The bone resorption capability of OCs requires cytoskeleton reorganization and the consequent polarization and formation of a convoluted membrane, which is the so-called ruffled border. The ruffled border is surrounded by a large circular band of an F-actin-rich structure, which is the actin ring or sealing zone. Actin ring integrity is essential for OCs to resorb bone both in vitro and in vivo, and defective ruffled border formation is associated with lower vacuolar adenosine triphosphatase (V-ATPase) expression31,32,33. Moreover, OCs are mitochondria-rich cells, and adenosine triphosphate (ATP) associates with mitochondrial-like structures in OCs localized at the ruffled border31,32,33. Rotenone acts as a strong inhibitor of the mitochondrial complex I and impacts ATP production. Rotenone has also been shown to inhibit OC differentiation and function34.
This protocol describes an efficient and optimized method of in vitro osteoclastogenesis from human peripheral blood samples. In human peripheral blood, CD14+ monocytes are the main source of OCs15,35,36. In this protocol, the kinetics of exposure and the concentrations of M-CSF and RANKL have been adjusted for optimum osteoclastogenesis. Mononuclear cells are first separated from the erythrocytes and granulocytes present in whole blood by density gradient; they are then enriched for CD14+ monocytes using positive selection by magnetic beads. The isolated CD14+ monocytes are then incubated overnight with M-CSF. This primes the monocytes to upregulate the expression of RANK15,26. The subsequent addition of RANKL induces osteoclastogenesis and multinucleation in a time-dependent manner. Active-resorbing OCs show the characteristic distribution of F-actin rings at the edge of the cell membrane30,32 and staining for TRAP. Mature OCs are analyzed by quantifying TRAP+ multinucleated (more than three nuclei) cells. The functional capacity of mature OCs can be assessed by their resorption, actin ring integrity, and ATP production. Furthermore, differentiated CD14− OSCAR+ OCs can be enriched and used to assess the effects of certain compounds on OC functionality via mineral (or dentine) resorption and F-actin organization. Additionally, in this work, a known OC inhibitor, rotenone, is used as an exemplar of a compound that affects the functionality of OCs. Reduced OC resorption activity under rotenone is associated with reduced ATP production and actin ring fragmentation. In conclusion, this protocol establishes a robust assay that can be used as a reference method to study several biological aspects of OC differentiation and function in vitro.
This methodology can be used to assess (1) the potential of circulating monocytes to differentiate into OCs in health and diseases, as well as (2) the impact of therapeutic candidates on OC differentiation and function. This robust osteoclastogenesis protocol enables the determination of the efficacy and mechanisms of bone-targeted therapies on both OC differentiation from precursor cells and the function of mature OCs.
Protocol
Buffy coats obtained from the Scottish National Blood Transfusion Service (Edinburgh) and leucocyte cones obtained from NHS Blood and Transplant (Newcastle) are provided to University of Glasgow researchers in a fully anonymized (non-identifiable) form from fully consenting NHS blood donors. The buffy coat and leukocyte cone blood components are produced from an NHS standard blood donation given at an NHS blood donor center in Scotland or England. The blood donor gives informed consent at the time of the blood donation for surplus blood not used in standard NHS clinical practice to be used for approved medical research studies. The ethical approval from the NHS Research Ethics Committee and the signed donor consent forms to use these blood donations are held by the NHS blood donation service. Approval to access and use these consented blood donations in approved medical research studies was sought and gained using the standard internal application and review process of the National Blood Transfusion Service (Scotland) and NHS Blood and Transport (England). No further NHS REC approval or internal University of Glasgow ethical committee approval was required to use the blood components for the approved medical research studies.
1. General notes prior to starting
- Proceed with all work with blood with caution. Consider the potential hazards of various infectious agents that may be present in the samples.
- Conduct all the work cautiously in the biosafety laboratory under sterile conditions while wearing gloves and lab coats.
- Perform biosafety disposal according to the local guidelines.
- Obtain appropriate consent and ethical approvals prior to sample collection according to local authority regulations.
- Generally, 1 mL of fresh blood will yield 1 million PBMCs, and CD14+ monocytes account for approximately 10%-30% of PBMCs. In comparison, 10 mL of a leukocyte cone can contain 5 x 108-15 x 108 PBMCs. For further details on PBMC isolation, refer to a previous protocol37.
2. Isolation of peripheral blood mononuclear cells (PBMCs) from whole blood
- Collect the required volume of fresh blood from healthy donors into lithium heparin collection tubes.
NOTE: Other collection tubes with appropriate anticoagulants can also be used (e.g., sodium heparin tubes). For higher cell counts, leukocyte cones or buffy coats can be used. - To isolate the PBMCs, transfer the blood into a new 50 mL tube, and dilute it with sterile 1x phosphate-buffered saline (PBS) in a 1:1 or 1:3 ratio for fresh blood or leukocyte cones/buffy coats, respectively.
- Gently mix the cells several times by inversion.
- Prepare 15 mL tubes containing 3 mL of density gradient medium. Slowly layer 8-10 mL of diluted blood on the top of the density gradient medium, and centrifuge at 400 x g for 30 min at room temperature (RT) with no brake.
NOTE: Apply the blood carefully on top of the density gradient medium to prevent mixing. Mixing might result in the loss of PBMCs. - Carefully discard the top layer (containing the plasma) with a Pasteur pipette, collect the underneath interphase layer containing the PBMCs (white, ring-like structure), and transfer this layer into a new 50 mL tube.
- Suspend the cells with sterile 1x PBS up to 50 mL, and wash off the residual density gradient medium by centrifuging at 300 x g for 10 min at RT with full brake.
- To remove residual platelets, repeat the process with an additional slower spin at 200 x g for 10 min at RT with no brake.
- Optional: To remove the carryover of red blood cells, dilute red blood cell lysis buffer (10x, Table of Materials) 1:10 in distilled water, and apply 3 mL of the diluted buffer onto the pellet. Mix, and incubate for 3 min. Wash the pellet in up to 50 mL of 1x PBS, and centrifuge at 300 x g for 10 min at RT with full brake.
- Resuspend the isolated and purified PBMCs in 20 mL of 1x PBS and count them using a hemacytometer or following other standard methods.
NOTE: The cell dilution and cell counting methods must be adjusted accordingly based on the cell density and counting device used.
3. Enrichment of CD14+ monocytes from PBMCs
- Isolate CD14+ monocytes from the PBMCs with a human CD14+ selection kit according to the manufacturer's protocol (Table of Materials).
NOTE: Classical CD14+CD16− monocytes are the main source of OC precursors35; alternative purification methods can be considered. - Transfer 1 x 107 PBMCs into a suitable polystyrene round-bottom tube (i.e., that fits the selection kit magnet), and pellet the cells at 300 x g for 5 min.
- Discard the supernatant, resuspend the cell pellet in cell separation buffer (PBS, 2% fetal bovine serum [FBS], 1 mM ethylenediaminetetraacetic acid [EDTA]) to a final concentration of 1 x 108 cells/mL, and incubate with 10 µL of antibody cocktail per 100 µL with the lid on for 10 min.
NOTE: The cells are resuspended at 1 x 108 cells/mL; adjust the volumes accordingly. - After incubation, add 10 µL of the magnetic nanoparticle beads per 100 µL, and incubate for 3 min with the lid on.
NOTE: Adjust the volumes of the antibody cocktail and magnetic beads to obtain a concentration of 10 µL/mL. - Top up the volume to 2.5 mL with the cell separation buffer, place the tube into a magnet (without the lid), and incubate for 3 min. Discard the negative cell population by one continuous move by inversion while the tube is still in the magnet.
NOTE: If using >2 x 108 PBMCs and a larger magnet, top up to 5 mL or 10 mL with cell separation buffer following the manufacturer's instructions. - Remove the tube from the magnet, and wash the enriched CD14+ monocytes attached to the magnetic beads by resuspending them in 2.5 mL of the cell separation buffer. Incubate for 3 min inside the magnet as before, discard the negative fraction, and repeat one more time.
- Centrifuge all the collected cells at 300 x g for 5 min, discard the supernatant, and resuspend the cells in 5 mL of alpha minimum essential medium (α-MEM; Table of Materials) supplemented with 1% L-glutamine, 1% penicillin/streptomycin (complete α-MEM), and 10% FBS.
NOTE: A post-enrichment purity check via flow cytometry is recommended, and ≥96% purity should be expected. Additional washing steps (step 3.6) can increase the purity.
4. OC differentiation in vitro
- Count the enriched CD14+ monocytes using a hemacytometer.
- Pellet the cells at 300 x g for 5 min, and resuspend at 1 x 106 cells/mL in complete α-MEM supplemented with 10% FBS.
- To differentiate the OCs, add M-CSF at a final concentration of 25 ng/mL to the cell suspension.
NOTE: For 1 mL of cell suspension, add 0.25 µL of M-CSF from a stock concentration of 100 µg/mL. - Mix by pipetting thoroughly to homogenize the cell suspension and plate 100 µL/well in a flat-bottom 96-well plate to a final cell density of 1 x 105 cells/well.
- Add 200 µL/well of sterile distilled water into the wells around the plated cells to prevent medium evaporation and edge effects in the culture system.
- Incubate the cells overnight, for approximately 18-20 h, at 37 °C with 5% CO2.
- After overnight incubation, carefully remove half of the medium (50 µL/well) by aspiration using a P200 pipette, avoiding touching the bottom of the well, and replace with fresh warm complete α-MEM containing 10% FBS, 25 ng/mL M-CSF, and 50 ng/mL RANKL for a final concentration of 25 ng/mL
NOTE: For 1 mL of medium, add 0.25 µL of M-CSF and 0.5 µL of RANKL from a stock concentration of 100 µg/mL. - Change the media every 3 days and differentiate the cells into OCs for 7-14 days (Figure 1).
NOTE: Keep the M-CSF and RANKL concentrations consistent throughout the culture.

Figure 1: OC differentiation workflow. Schematic overview of CD14+ monocyte isolation from PBMCs and differentiation into mature OCs in the presence of M-CSF and RANKL for 7-14 days. RT = room temperature. Image created with BioRender.com. Please click here to view a larger version of this figure.
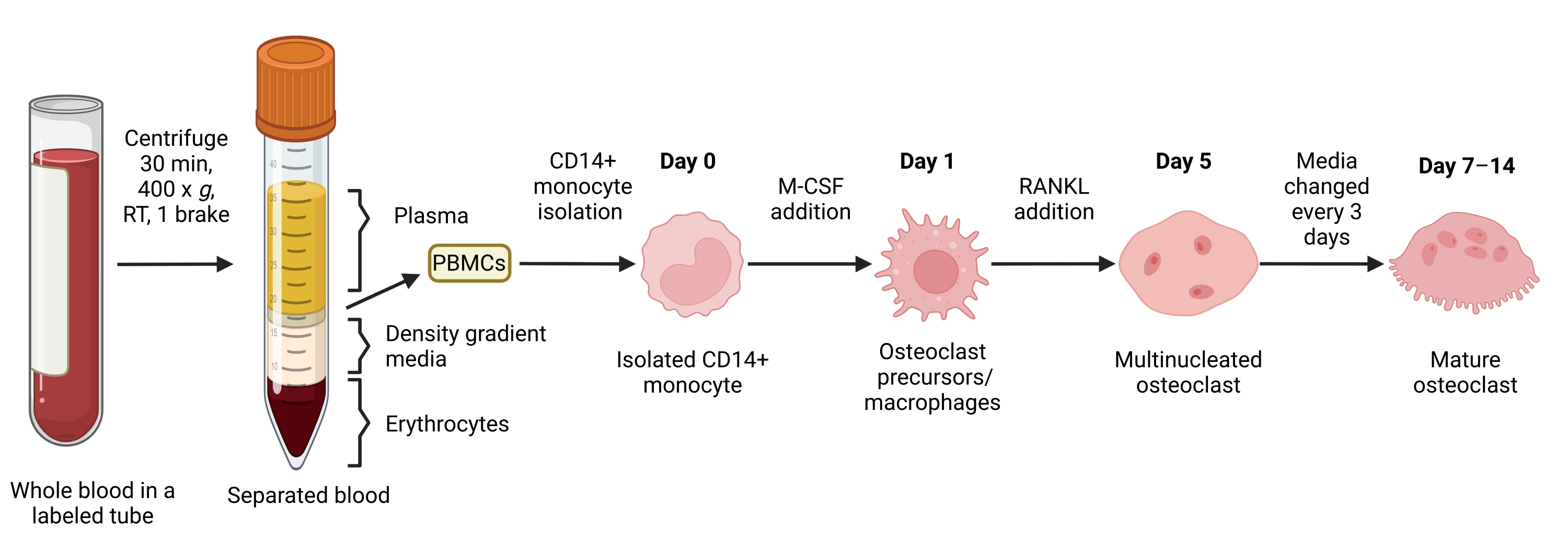{kind=link}
5. TRAP staining for osteoclasts
- Carefully remove the medium, fix the differentiated adherent OCs with 100 µL/well of the previously prepared fixative solution, and incubate for 1 min. Do not touch the bottom of the wells to avoid scratching the adherent cells.
NOTE: The fixative solution is prepared as follows: 12.5 mL of citrate solution (included in the TRAP staining kit), 32.5 mL of acetone, and 5 mL of 37% formaldehyde. - Wash the wells three times with 300 µL of sterile distilled water. Tap the plates dry after washing.
- Prepare a staining solution according to the manufacturer's instructions (Table of Materials); add 5 µL of Fast Garnet and 5 µL of sodium nitrite to make the Fast Garnet solution; mix by inversion, and incubate for 3 min at RT. Prepare 1 mL of staining solution by mixing 900 µL of sterile distilled water, 10 µL of naphthol, 40 µL of acetate solution, 50 µL of tartrate solution, and 10 µL of Fast Garnet solution.
- Add 100 µL/well of freshly prepared staining solution, and incubate the plate at 37 °C in the dark for 20 min.
- After incubation, remove the staining solution by inversion, and wash the plate three times with 300 µL/well of distilled water.
- Remove the excess water by tapping the plates on paper towels. Leave the plates open and protected from the light to air dry overnight.
NOTE: Dry plates can be stored for up to 6 months. Sometimes, residual buffers can promote the development of mold, which is visible under the microscope; this can be removed at any time by washing the affected wells with distilled water and letting them air dry again. - Take images at 10x or 20x using a brightfield microscope with a tile option to capture the entire well surface.
- Manually count the OCs identified as TRAP+ purple stained cells with more than three nuclei using an image analysis software with a cell counter plugin.
NOTE: The numbers of TRAP+ OCs per well are donor dependent and may vary from ~200-1,600 OCs/well, with an average of about 1,000 OCs/well. Moreover, to analyze the data, the OC numbers should be determined in three different wells (technical replicates), and the average should be calculated for each condition and for each biological replicate.
6. Bone resorption assay
- Plate the freshly enriched CD14+ monocytes on 96-well osteo-assay plates coated with calcium phosphate at 1 x 105 cells/well, and differentiate the OCs for 7-14 days, as indicated in steps 4.1-4.8, and changing the medium every 3 days.
NOTE: Dentine/ivory or bovine cortical bone slices can be used in place of osteo-assay plates. If so, the total time of the culture should be prolonged to 14-21 days due to the more complex substrate to be resorbed. - At the end time point, carefully remove the medium, avoiding touching the bottom of the well, and lyse the cells with 10% sodium hypochlorite solution. Wash the wells three times with distilled water.
- Scan the dry plates using a brightfield microscope, and analyze/quantify the acquired images of the resorption pits using an image analysis software.
7. Actin ring fluorescent staining
- Plate 100 µL/well of the isolated CD14+ monocytes in an 18-well chamber slide at a cell density of 1 x 105 cells/well. Differentiate the OCs in the presence of M-CSF and RANKL as described before (steps 4.1-4.8), including changing the medium every 3 days.
- At the end time point, gently remove the medium, and wash each well twice with 200 µL/well of pre-warmed PBS, pH 7.4. Do not let the wells dry in between any of the steps.
- Fix the sample with 100 µL/well of 4% formaldehyde solution in PBS, and incubate for 10 min at RT on an orbital shaker with gentle shaking.
NOTE: Methanol can disrupt actin during the fixation process. Therefore, it is best to avoid any methanol-containing fixatives. The preferred fixative is methanol-free formaldehyde. The orbital shaker used in this step of the protocol was set at a power setting of 3 out of 10. - Wash twice with 200 µL/well of PBS, permeabilize the cells with 100 µL/well of 0.1% Triton X-100 solution diluted in PBS, and incubate for 10 min at RT on an orbital shaker with gentle shaking.
- Wash twice with 200 µL/well of PBS. To block non-specific binding and increase the signal, add 100 µL/well of blocking solution made with 2% bovine serum albumin (BSA)/PBS solution. Incubate for 20 min at RT on an orbital shaker with gentle shaking.
- Remove the blocking solution, and add 100 µL/well of fluorescently conjugated phalloidin solution diluted in 2% BSA/PBS solution. Incubate for 20 min at RT on an orbital shaker with gentle shaking and protected from light.
NOTE: Adjust the concentration of the phalloidin dye according to the manufacturer's recommendations. - Wash twice with 200 µL/well of PBS, stain the nuclei with 100 µL/well of a solution of PBS containing 300 nM DAPI, and incubate for 10-15 min at RT on an orbital shaker with gentle shaking and protected from light.
NOTE: DAPI is diluted in distilled water to make a 14.3 mM (5 mg/mL) DAPI stock solution. The stock solution is further diluted to the final concentration of 300 µM. Finally, the 300 µM DAPI solution is diluted one more time in PBS to a final concentration of 300 nM. - After 10-15 min, remove the DAPI solution, and replace it with 100 µL/well PBS.
NOTE: Depending on the chamber slides chosen, either store with an appropriate volume of PBS (100 µL/well for 18-well chamber slides), or mount the slide with coverslips and an appropriate mounting medium. The chamber slides can be stored for up to 1 week in the fridge. For 18-well chamber slides, use a 50-100 µL staining volume and 200-300 µL for washing. Scale up for the other chamber slide sizes accordingly. To avoid evaporation, keep the coverslips inside a covered container during the incubation times. The use of an orbital shaker is recommended but not essential. - Visualize the staining using appropriate immunofluorescence or confocal microscopes and magnifications between 4x to 40x.
8. Enrichment of mature OCs and OC precursors via flow cytometry sorting
- Resuspend the freshly enriched CD14+ monocytes at 1 x 106 cells/mL, and differentiate them into mature OCs in the presence of M-CSF and RANKL in the same way as described above (steps 4.1-4.8).
NOTE: When scaling up from a 96-well plate to a bigger plate size, follow Table 1; these volumes are calculated starting from a 1 x 106 cells/mL solution and provide an optimum density for cell-cell fusion. - On day 7, wash the wells once with warm PBS, and add 50 µL to 1 mL (volume determined by the plate size used) of accutase. Incubate the cells at 37 ˚C with 5% CO2 for 20 min.
- After incubation, check the plates under a light microscope to see if the cells have detached. Further, detach the cells by tapping the plates on all sides and pipetting them up and down.
- Collect the cell suspension in a 15 mL conical tube. Wash the wells with warm PBS (no Ca2+, no Mg2+), and combine them with the cell suspension. Repeat steps 8.2-8.3 once or twice until most of the cells have detached.
NOTE: Accutase, the recommended method prior to surface staining for flow cytometric analysis, does not detach very large OCs. The recovery rate is ~50%-70%. - Centrifuge the cells at 300 x g for 5 min, resuspend the cell pellet in 1 mL of PBS, and count the cells by trypan blue exclusion.
- Resuspend the cells at 1 x 106 cells/mL, remove 100 µL corresponding to 1 x 105 cells, and transfer those cells into a new polypropylene test tube. Add 200 µL of the cell sorting buffer, and set this aside on ice as the unstained control.
- Stain the remainder of the cells with live/dead dye diluted at 1:750 for 10 min at RT and protected from light.
NOTE: Live/dead staining needs to be performed in the absence of FBS to avoid high background staining. - Top up the 15 mL collection tube containing the live/dead stained cell suspension with warm cell sorting buffer (1x PBS, no Ca2+, no Mg2+, 1% FBS, and 5 mM EDTA), and centrifuge at 300 x g for 5 min to pellet the cells.
NOTE: A high concentration of EDTA and a low concentration of FBS are recommended in the sorting buffer to prevent cell clumps. - Remove a volume corresponding to 1 x 105 cells, and transfer them into a new polypropylene test tube for the OSCAR isotype control. Transfer all the remaining cells into another polypropylene test tube for staining and cell sorting.
NOTE: Polypropylene test tubes are used for sorting as the cells are less likely to adhere to these tubes than to polystyrene tubes. - Spin the tubes at 400 x g for 5 min to pellet the cells, and discard the excess supernatant by inversion.
- Resuspend the cell pellet in an antibody master mix solution prepared following Table 2. Stain the OSCAR isotype control tube with CD14 antibody and OSCAR isotype control in place of the OSCAR antibody.
- Incubate the cells at 4 ˚C protected from the light for 30 min.
- After 30 min, wash the cells by adding five volumes of the cell sorting buffer, and centrifuge at 400 x g for 5 min at 4 ˚C.
- Resuspend the cells in 300-1,000 µL of cold cell sorting buffer, and acquire the cells using a flow cytometry sorting machine fitted with a 100μM nozzle.
NOTE: OCs are very sticky cells, so it is important to filter them through a sterile 70 µm membrane prior to sorting. - Gate the OCs and pre-OCs as CD14−OSCAR+. Set the OSCAR+ gate based on the OSCAR isotype control tube.
- Collect the sorted cells in polypropylene test tubes containing complete α-MEM supplemented with 20% FBS at 8 ˚C.
- After sorting, pellet the cells by centrifugation at 300 x g for 5 min at RT, count the cells, and resuspend for downstream applications.
NOTE: Usually, to get ~1 x 105 sorted pre-OCs/OCs, start from ~10 x 106 cells plated at day 0. The low recovery rate is influenced by cell loss during detachment with accutase and by the processing for staining and sorting. It is recommended to carry out the whole procedure using sterile buffers and reagents, and work under sterile conditions.
9. ATP assay for mitochondrial activity
- Incubate the enriched CD14+ monocytes in the presence of M-CSF and RANKL in a 96-well plate in the same way as described before (steps 4.1-4.8). Plate four extra conditions in triplicates to use as controls.
- Conduct the ATP assay with the luminescence ATP detection assay kit according to the manufacturer's manual. Briefly, to prepare the ATP solution, add 10 mL of the substrate buffer solution to the lyophilized substrate, and leave it to incubate at RT for 30 min.
NOTE: Different methods can be used to measure the intracellular ATP production. Herein, we have used the detection of ATP production by luminescence. - During the incubation, prepare and add the controls directly to the control wells as follows: 2-Deoxy-D-glucose (2DG) at 10 mM and 100 mM, oligomycin at 1 µM, and 100 mM 2DG in combination with 1 µM oligomycin. Incubate for 30 min at 37 °C with 5% CO2.
NOTE: The 2DG blocks glycolysis, while oligomycin is an inhibitor of oxidative phosphorylation. Combining these two inhibitors results in the complete loss of ATP production via glycolysis and oxidative phosphorylation, thus meaning they serve as an internal control for the ATP assay. For 10 mM and 100 mM 2DG controls, add 0.5 µL and 5 µL of 2M 2DG stock solution per 100 µL culture well, respectively. For 1 µM oligomycin, dilute the 5 mM stock solution 1:100 in medium, and add 2 µL/well to the dedicated control wells. For the last control, add 5 µL of 2DG and 2 µL of diluted oligomycin solution per well. - Add 50 µL of the ATP solution to each well to stop the reaction, and incubate at RT on a shaker at 700 rpm for 5-10 min protected from light.
- Transfer 100 µL of the supernatant to a 96-well white-bottom plate specific for the ATP assay, and read the plate using a luminescence reader.
Results
OC generation from CD14+ monocytes
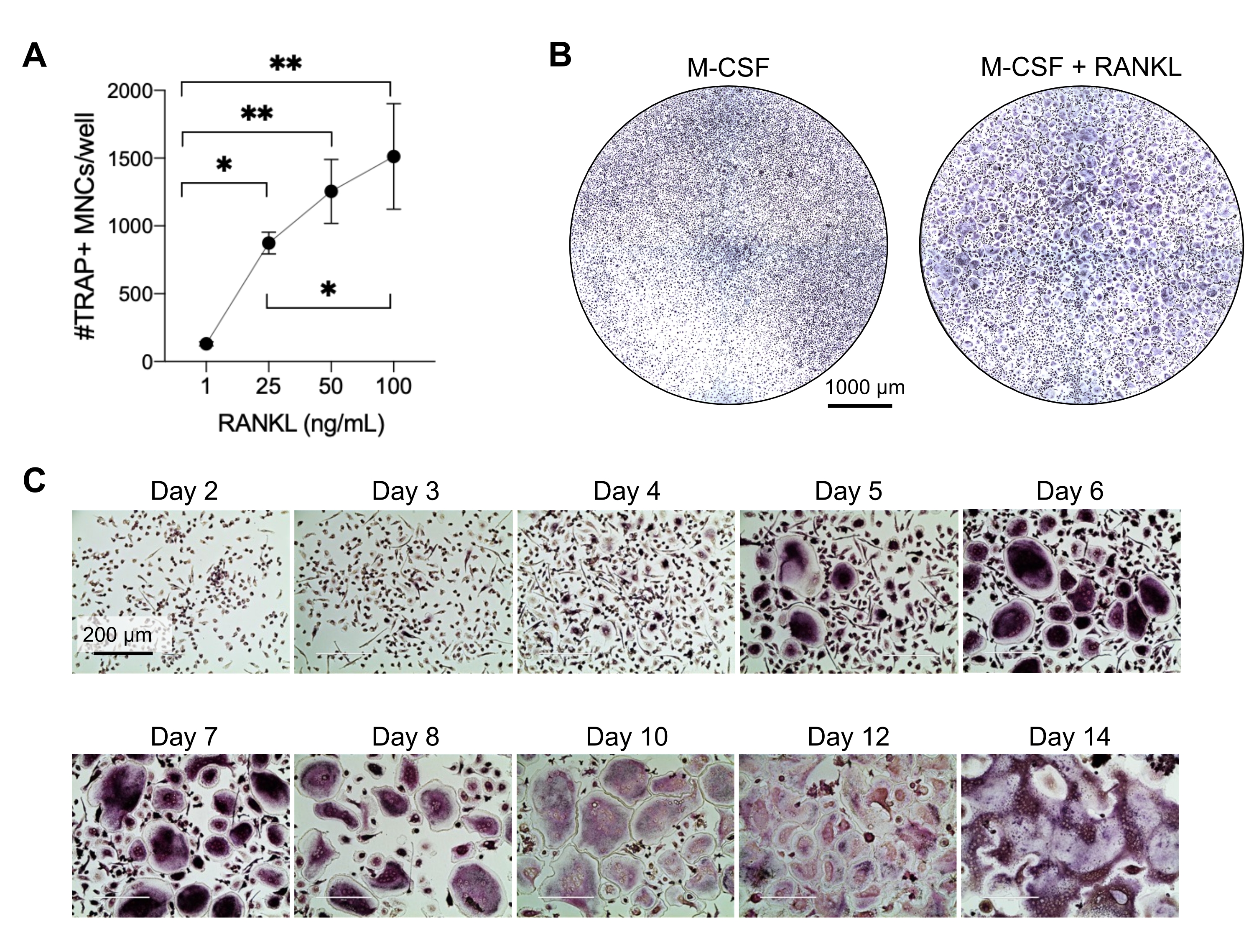
This method aimed to easily differentiate a large number of OCs from human peripheral blood CD14+ monocytes in vitro, typically in 1 week. Firstly, CD14+ monocytes were enriched from PBMCs and primed with M-CSF overnight to upregulate RANK, as previously reported15. Following monocyte priming, to determine the optimum concentration of RANKL for OC differentiation and maturation, RANKL concentrations of 1 ng/mL, 25 ng/mL, 50 ng/mL, and 100 ng/mL, along with 25ng/mL M-CSF, were used. The addition of RANKL produced increasing numbers of large TRAP-positive multinucleated OCs in a dose-dependent manner, and this was assessed using TRAP staining. Mature OCs are defined as TRAP-positive cells with multiple nuclei (typically more than three; Figure 2A,B and Supplementary Figure 1). Furthermore, the kinetics of OC differentiation from monocytes were investigated using TRAP staining and light microscopy over a 2-14 day culture period. In this instance, OC differentiation using an intermediate concentration of 50 ng/mL RANKL was chosen to assess how fast OCs differentiated in culture. In these culture conditions, multinucleated OCs were visible from day 5 onward, and optimal differentiation was reached on day 7 (Figure 2C). The prolonged incubation of cultures beyond 10 days on plastics resulted in abnormally giant fused cells. In this protocol, days 6-8 are usually used as the optimal endpoint of OC generation. The OCs can be quantified or used for downstream assays.
Functional assessment of differentiated OCs
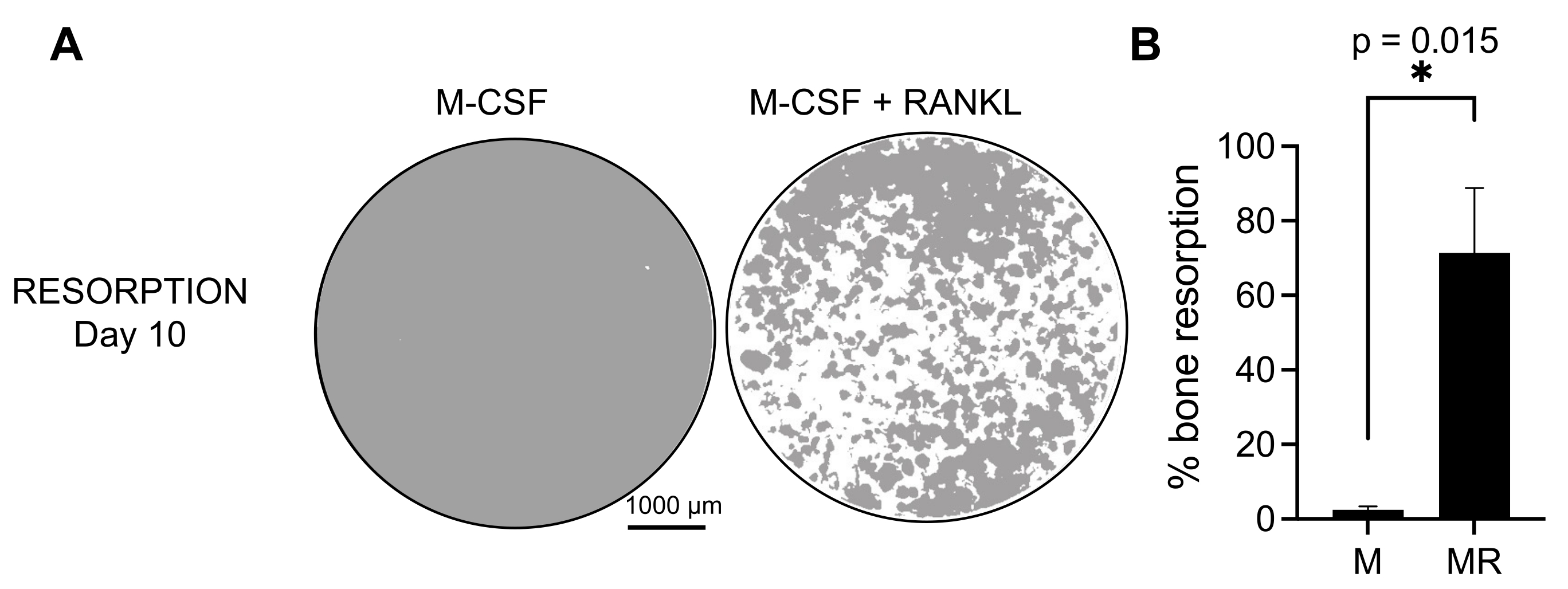
To determine the functional activity of the generated OCs, we examined their resorptive activity by differentiating the OCs on a mineralized surface. As large OCs are only generated after a 7 day culture period, and to allow sufficient time to resorb the mineral substrate, the cultures were maintained until day 10. The formation of round holes, or resorption pits, was observed only on the mineralized surfaces of wells containing cells that had been treated with both M-CSF and RANKL (Figure 3). Thus, the percentage of dissolved mineralized surface (resorption pits) allows for determining the OC resorptive capacity. Additionally, the OCs differentiated following this protocol up to day 7, both on plastic and glass chamber slides, displayed a well-organized actin ring structure that could be visualized by immunofluorescent staining (Supplementary Figure 2).
Effect of an inhibitor on mature OC function
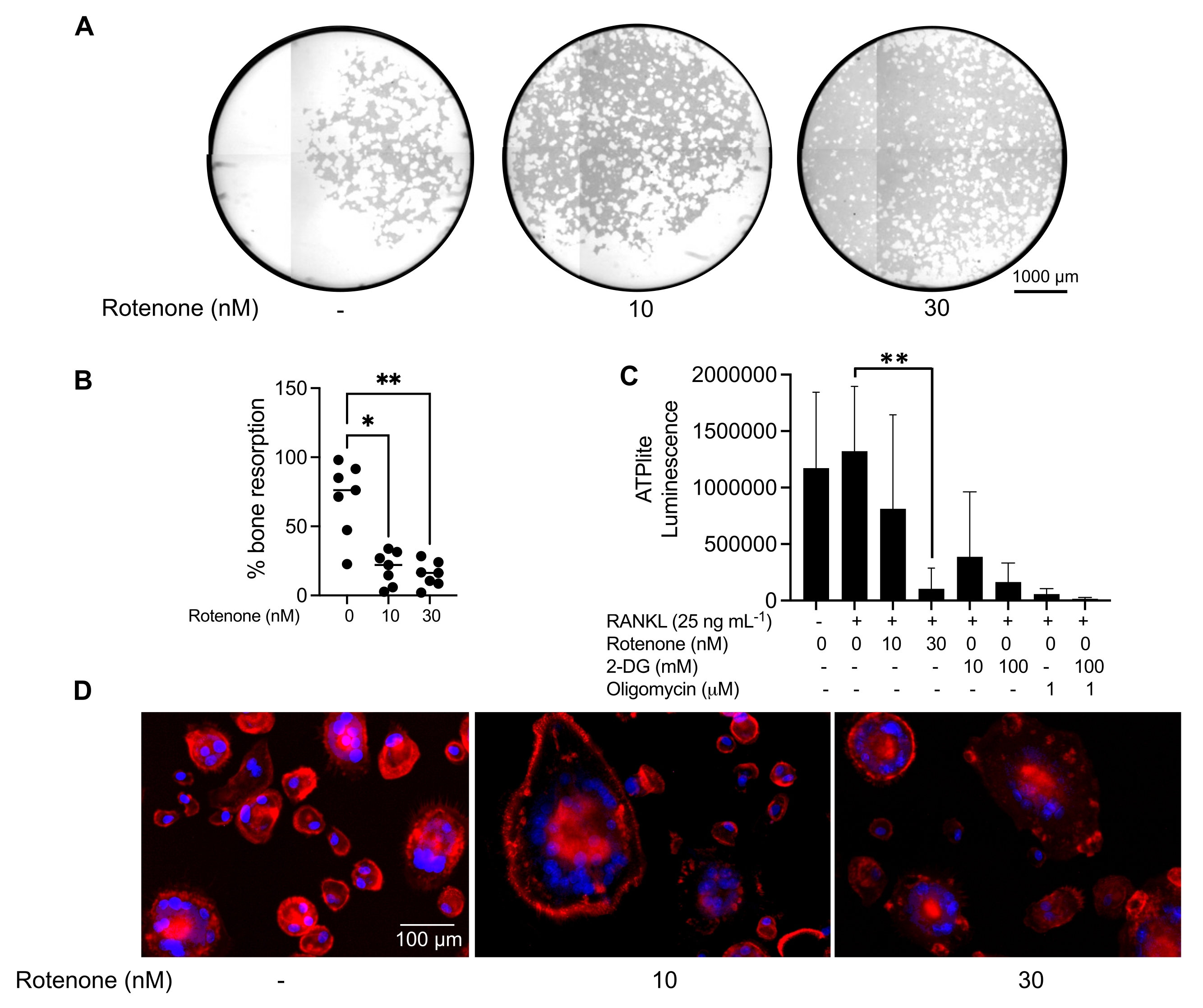
The above mentioned culturing conditions were utilized to determine the functional capability of the in vitro generated OCs in the presence of the known OC inhibitor, rotenone34. The OCs were differentiated for 6-8 days, and CD14−OSCAR+ OCs and OC precursors were enriched via flow cytometry (Figure 4). The enriched cells were then plated at 50,000 cells/per well onto a mineral-coated 96-well plate in pro-osteoclastogenic medium (25 ng/mL M-CSF and RANKL) for 3 days. Treatment with rotenone (Figure 5A,B) dose-dependently inhibited the resorption of the mineralized surface in comparison with the untreated control well, consistent with previous studies34. Additionally, OC functionality was assessed via ATP production and actin ring formation. The rotenone-dependent inhibition of OC resorption was associated with the inhibition of ATP production (Figure 5C). Resorbing OCs are highly polarized cells that regulate their resorptive capacity by promoting cytoskeletal organization. Alexa fluor 647 conjugated phalloidin was used to label the F-actin cytoskeleton of the mature OCs cultured in the presence or absence of rotenone. Rotenone caused the fragmentation of the RANKL-derived actin ring of the mature OCs (Figure 5D).

Figure 2: OCs efficiently differentiating from CD14+ monocyte precursors. CD14+ monocytes were magnetically enriched, plated at 1 x 105 cells/well in 96-well plates, and incubated overnight with 25 ng/mL M-CSF. (A) M-CSF-primed monocytes were stimulated with increasing concentrations of RANKL (1 ng/mL, 25 ng/mL, 50 ng/mL, and 100 ng/mL), fixed, and stained for TRAP on day 7. Images were acquired, and the TRAP+ multinucleated cells (MNCs) were counted. Representative images of TRAP staining are shown in Supplementary Figure 1. The error bars show mean ± SD (n = 3). The data were analyzed with a one-way ANOVA and Holm-Sidak's multiple comparisons test for paired data; * P ≤ 0.05 and ** P ≤ 0.005. (B) Representative image of a TRAP-stained well of a 96-well plate showing the typical amount of OCs/well expected and their morphology under 25 ng/mL RANK-L in comparison to M-CSF-derived macrophages at day 7. Scale bars: 1000 µm. (C) Representative images of OC formation under 50 ng/mL RANKL assessed via TRAP staining from day 2 to day 14. OCs are visible from day 5 onward. Giant abnormally fused OCs are present after 10 days. Scale bars: 200 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Resorptive OCs differentiated from CD14+ monocytes. CD14+ cells isolated from PBMCs were differentiated for 10 days into OCs in the presence of 25 ng/mL M-CSF (M) and RANKL (R) on mineral assay surface (osteo-assay) plates. (A) Images of representative reconstructed wells taken at 10x magnification to analyze the resorption on day 10 (mineral substrate in gray; resorption pits in white). Scale bars: 1000 µm. (B) Quantification of the percentage of resorbed area. The resorption data were analyzed with a Wilcoxon paired analysis. The error bars show mean ± SD (n = 7). Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Flow cytometry enrichment of CD14−OSCAR+ OCs. CD14+ monocytes were enriched from PBMCs, and the OCs were differentiated as previously described. Adherent OC cultures were detached with accutase and stained for flow cytometry. (A-C) OCs at day 8 were sorted based on CD14 and OSCAR expression. (A) Representative sorting gating strategy. The cells were gated as singlets, negative for dead staining, and the CD14+ OSCAR+ (red) and CD14− OSCAR+ (blue) subsets were sorted. (B) Representative plots showing the overlapping OSCAR staining of RANKL-derived OCs (cyan) and control M-CSF-derived macrophages (orange). In red is the OSCAR isotype-stained control of RANKL-derived OCs. (C) The sorted populations were plated on plastic and allowed to adhere for 2 h in pro-OC medium (25 ng/mL M-CSF and 50 ng/mL RANKL), followed by TRAP staining and visualization. The representative images show a lack of TRAP+ cells in the CD14+ subset (red) and mono- and multinucleated TRAP+ pre-OCs and OCs in the CD14− subset (blue). Scale bars: 200 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Assays to assess the function of mature OCs. To evaluate the function of mature OCs, CD14+ cells isolated from PBMCs were cultured either with M-CSF (M) alone or combined with RANKL (R) for 7 days, the OCs were enriched via flow cytometry, and the OCs were then treated with the inhibitor rotenone for 24 h. (A) Mature OCs were sorted via flow cytometry (CD14−OSCAR+) and were cultured on a mineral assay surface in the presence or absence of rotenone for 3 days, after which the cells were bleached and imaged at 10x to reveal the resorbed area (resorption pits in white). (A) Representative reconstructed images of wells. Scale bars: 1000 µm. (B) The quantification of the percentage of resorbed area. The data in (B) were analyzed with a one-way ANOVA with Dunn's multiple comparisons test (n = 7); * P ≤ 0.05 and** P ≤ 0.01. The error bars show the mean ± SD. (C) Total intracellular ATP content of undifferentiated and day 7 differentiated mature OCs differentiated with RANKL and treated with either vehicle or rotenone (10 nM and 30 nM). Here, 2DG and oligomycin were used as positive controls for the assay and were added 30 min prior to cell lysis and ATP quantification. The error bars show the mean ± SD (n = 4). The data were analyzed with a one-way ANOVA and Dunnett's multiple comparison test for paired data. ** P ≤ 0.01. (D) A representative 20x image of mature OCs stained for actin ring formation (red) and nuclei (blue), showing the loss of the actin ring with the inhibitor. Scale bars: 100 µm. Please click here to view a larger version of this figure.
{kind=link}
| Plate format | 96 well-plate | 48 well-plate | 24 well-plate | 12 well-plate | 6 well-plate |
| volume | 100 µL | 225–250 µL | 450–500 µL | 0.8–1 mL | 1.8–2 mL |
Table 1: Volume of cell suspension for different plate formats. The volumes are calculated starting from a 1 x 106 cells/mL solution and provide an optimum density for cell-cell fusion.
| Fluorophore, clone | Volume (μL) per 106 cells | |
| CD14 | PE/Cyanine7, HCD14 | 5 μL |
| OSCAR | FITC, REA494 | 10 μL |
| Cell sorting buffer | 80 μL |
Table 2 : Antibody master mix solution.
Supplementary Figure 1: TRAP staining of the RANKL dose response. CD14+ monocytes were magnetically enriched, plated at 1 x 105 cells/well in 96-well plates, and incubated overnight with 25 ng/mL M-CSF, as in Figure 2. Representative images of TRAP staining show MCSF-primed monocytes stimulated with increasing concentrations of RANKL (1 ng/mL, 25 ng/mL, 50 ng/mL, and 100 ng/mL), fixed, and stained for TRAP on day 7. Scale bars: 400 µm. Please click here to download this File.
Supplementary Figure 2: Acting ring staining in fully differentiated OCs. (A) A 10x magnification of OCs differentiated on TC plastic and stained with AF647 phalloidin (in red). Scale bar: 400 µm. (B) A 40x magnification of OCs differentiated on glass chamber slides and stained with AF488 phalloidin (in yellow). Scale bar: 100 µm.The nuclei are stained with DAPI, shown in blue in (A) and in cyan in (B). Please click here to download this File.
Supplementary Figure 3: Effect of different FBS batches on OC differentiation efficiency. OCs were differentiated from CD14+ monocytes in the presence of 25 ng/mL M-CSF and 50 ng/mL RANKL (MR) for 7 days. The control wells had M-CSF only (M). (A) Representative 10x magnifications (scale bars: 400 µm) and (B) quantification of TRAP-stained OCs differentiated from one donor in two different batches of FBS. The error bars show the mean ± SD of three technical replicates. Please click here to download this File.
Discussion
The easy culture and isolation of large numbers of functional OCs in vitro are important for advancing the understanding of bone biology and OC-mediated diseases. Classically, OCs were generated in co-cultures with osteoblasts or stromal cells and hematopoietic cells from the spleen or bone marrow38,39. A significant breakthrough in the understanding of osteoclastogenesis was the identification of RANKL as the major regulator of OC formation, differentiation, and survival40. Early protocols of RANKL-dependent culture systems utilized PBMCs for OC generation21,41,42. However, these mixed cultures are lengthy and present many confounding factors that limit the ability to test the direct effects on OC differentiation and function. This protocol describes an efficient and reliable in vitro model of osteoclastogenesis from human peripheral CD14+ monocytes in which optimal osteoclastogenesis can be obtained within 7 days (Figure 1 and Figure 2), which is considerably faster when compared to some other protocols43,44,45,46. The main distinguishing features of this protocol are (1) the use of purified CD14+ monocytes, (2) the priming of the monocytes with M-CSF prior to exposure to RANKL, (3) the length of the culture (<7 days), and (4) the reliable detection of the inhibition of OC formation (TRAP staining) and function (resorption, ATP production, actin ring reorganization) with inhibitors.
During the optimization of the methodology, several critical points were identified. It has been observed that the in vitro differentiation of OCs is largely dependent on the seeding density of the CD14+ monocytes. Thus, in this protocol, the cells are seeded at a high density (1 x 105 cells/well of a 96-well plate, in 100 µL of medium), as it is essential for the cells to be able to interact with each other and to be in proximity to fuse and become mature OCs. Similarly, seeding cells at a density that is too high limits their differentiation and growth due to medium limitations and a lack of the required space. Furthermore, to achieve maximum success with this protocol, it is important to perform the density gradient separation carefully and to ensure that the enriched population of CD14+ cells is as pure as possible. For example, inadequate washing steps result in a lack of removal of platelets, which consequently inhibits OC differentiation47,48. Similarly, the presence of minor T cell contamination in isolated CD14+ preparations stimulated with M-CSF alone can result in OC differentiation, potentially via RANKL secretion by T cells49. Therefore, it is important to include an M-CSF control for every experiment. A routine purity check, especially when using a new isolation kit, is also recommended to ensure the purity of the sample.
Optimum OC numbers (range: ~200-1,600 OCs/well) are achieved using α-MEM medium enriched with nucleosides and L-glutamine. Other conventional culture media, including Dulbecco's modified eagle medium (DMEM) and Roswell Park Memorial Institute (RPMI) 1640 medium, affect the OC yield. The source of FBS can also influence osteoclastogenesis. Different batches of FBS can lead to reduced RANK-L-derived osteoclastogenesis, as well as the appearance of low numbers of TRAP+ multinucleated cells in the M-CSF controls (Supplementary Figure 3). Therefore, to achieve consistent results, it is advised to test new FBS batches before use and to continue with the same batch throughout the experiments to minimize variations in the differentiation process. Additionally, donor-to-donor variability, in terms of total numbers of differentiated OCs obtained at the end time point, constitutes a limitation when using this protocol to compare, for instance, healthy donors to patients. In these cases, it is imperative to use exactly the same conditions and the same lot of medium, FBS, and other reagents.
Another necessary step for optimum OC differentiation and maturation is priming the monocytes with M-CSF before the RANKL addition. The exposure of the cells to M-CSF 18-24 h prior to RANKL primes the monocytes to upregulate RANK expression15,26. The addition of RANKL at this time point ensures optimal OC differentiation in a dose-dependent manner. The degree of OC differentiation varies from donor to donor; however, 25 ng/mL RANKL is usually sufficient to differentiate a high number of OCs in most donors. Additionally, 25 ng/mL RANKL can be used in assays for the initial screening of compounds, as it facilitates the evaluation of both the enhancing and inhibitory effects of the test compounds. Other culture systems have used longer M-CSF pre-incubation times prior to RANKL addition, but this results in a longer culturing time for osteoclastogenesis50. In addition, leaving the primed monocytes to incubate overnight allows them to attach to the plate, albeit not in a fully adherent state. Therefore, when RANKL is introduced for the first time, the medium must be half-changed very carefully rather than completely changed to prevent the detachment and loss of the primed monocytes. The medium also needs to be refreshed every 3-4 days to avoid medium depletion and prevent cell death. Moreover, due to the low volume used in this assay (100 µL/well in a 96-well plate), it is of the utmost importance to have a frame of empty wells that are filled with an aqueous solution (i.e., sterile distilled H2O or PBS) around the assay wells. This prevents medium evaporation and edge effects.
Finally, for metabolic assays (e.g., ATP assays), it is imperative that the cells are viable to avoid huge standard deviation between replicates (Figure 5). High viability of the cells is also important for sorting the cells and for the further culturing of the sorted OCs (Figure 4). This method, however, has several limitations. Fully mature OCs are very adherent and difficult to detach from the plates. The larger OCs are often impossible to detach, which can lead to a lower cell yield. Therefore, the cells need to be counted after sorting and prior to plating at the required concentration. Furthermore, in the present protocol, a non-enzymatic method (accutase) to detach the OCs is used to prevent membrane alterations in downstream surface staining for flow cytometry. The use of cell scrapers (both with soft or hard endings) was also tested and led to high cell death. Enzymatic detachment using 0.05% Trypsin/EDTA solutions can be used for a higher yield of detached OCs when membrane integrity is not required for downstream applications. Additionally, to prevent the OCs from clumping together, the use of a high concentration of EDTA in all the buffers following cell detachment, as well as appropriate filtering prior to flow cytometry acquisition, are highly recommended. It is important to note that OC cultures are a heterogeneous population of cells consisting of mature OCs, OC precursors, and macrophages. Macrophages can be easily distinguished from OCs, although both mononuclear pre-OCs and multinuclear OCs express OSCAR and cannot be distinguished with the present method (Figure 4). Indeed, this latter issue constitutes the main limitation of this method. In addition, a low expression of OSCAR is also present in M-CSF cultures (Figure 4B) and might indicate macrophages that are primed for OC lineage commitment. It is important to set the gate for OSCAR+ cells based on the FMO staining signal, as shown in Figure 4B.
In summary, this protocol describes an optimized and robust method for the efficient production of active and functionally mature OCs from circulating primary human monocytes. The strength of this protocol is its ability to generate OCs in a short time duration and yield high numbers of differentiated OCs. This method opens the way for investigating the basic mechanisms underlying OC differentiation and function.
Disclosures
The authors declare that they have no competing interests.
Acknowledgements
The authors gratefully acknowledge the Flow Core Facility and the Glasgow Imaging Facility (GIF) within the School of Infection and Immunity for their support and assistance in this work.
Materials
| Name | Company | Catalog Number | Comments |
| µ-Slide 18 well chamber slides | ibidi | 81816 | |
| 8-well glass chamber slides | Ibidi | 80807 | |
| 96-well TC plate | Corning | 3596 | |
| 96-well osteo assay stripwell plate | Corning | 3989 | |
| Acetate solution | Sigma Aldrich | 386-3 | from kit Cat No. 387A-1KT |
| Acetone | VWR | 20066.330 | |
| Acid phosphatase, Leukocyte (TRAP) kit | SIGMA-ALDRICH | 387A-1KT | |
| Alexa Fluor 488 Phalloidin | Theremo Fisher - Invitrogen | A12379 | AF488 |
| Alexa Fluor 647 Phalloidin | Thermo Fisher - Invitrogen | A22287 | AF647 |
| Alfa Aesar 2-Deoxy-D-glucose | Fisher Scientific | 11321867 | 2DG, 98% |
| Alpha minimum essential medium | gibco | 22571-020 | |
| ATPlite 1step | PerkinElmer | 6016731 | Luminiscence ATP detection assay system |
| BD FACSAria III cell sorter | BD Biosciences | ||
| Bovine serum albumin (BSA) | Sigma-Aldrich | A9418-100G | |
| Cell culture microplate, 96-well, PS, F-bottom | Greiner bio-one | 655083 | White-bottom plates |
| Citrate solution | Sigma Aldrich | 91-5 | from kit Cat No. 387A-1KT |
| Corning 6ml round-bottom polystyrene test tubes | Fisher Scientific | 352054 | |
| Corning osteo assay surface multiple well plate | Sigma-Aldrich | CLS3989 | |
| Corning osteo assay Surface multiple well plate 1 x 8 stripwell | Corning | CLS3989-2EA | |
| DAPI | Theremo Fisher | D3571 | |
| EasySep human CD14 positive selection kit | STEMCELL Technologies | 17858 | |
| EasySep red blood cell lysis buffer (10x) | StemCell Technologies | 20110 | |
| eBioscience fixable viability dye eFluor 780 | Theremo Fisher - Invitrogen | 65-0865-14 | |
| Ethylenediaminetetraacetic acid | Sigma-Aldrich | E7889-100ML | |
| EVOS FL auto imaging system | Thermo Fisher | A32678 | |
| Falcon round-bottom polypropylene test tubes with cap | Fisher Scientific | 10314791 | |
| Falcon tubes 15 mL | Corning | 430790 | |
| Falcon tubes 50 mL | Corning | 430828 | |
| Fast Garnet GBC base solution | Sigma Aldrich | 387-2 | from kit Cat No. 387A-1KT |
| Fetal bovine serum | gibco | 10500-064 | FBS |
| Ficoll-Paque Plus | cytiva | 17144003 | |
| Formaldehyde | Sigma-Aldrich | F-8775 | |
| Human sRANK ligand | PEPROTECH | 310-01-100UG | Receptor activator of nuclear factor kappa-B ligand (RANKL) |
| ImageJ Image analysis software | Image J | version 2.9.0 | |
| L-glutamine | gibco | 25030-024 | |
| Lithium heparin tubes (9 mL) | VACUETTE | 455084 | |
| Macrophage colony-stimulating factor | PEPROTECH | 300-25-100UG | M-CSF |
| Napthol AS-BI phosphoric acid solution | Sigma Aldrich | 387-1 | from kit Cat No. 387A-1KT |
| Neubauer hemacytometer counting chamber | Camlab | SKU 1127885 | |
| Oligomycin from Streptomyces Diastatochromogenes | Sigma-Aldrich | Q4876-5MG | |
| OSCAR Antibody, anti-human, Vio Bright FITC, REAfinit | Miltenyi Biotec | 130-107-661 and 130-107-617 | Clone REA494 |
| PE/Cyanine7 anti-human CD14 antibody | Biolegend | 325618 | Clone HCD14 |
| Penicilin/streptomycin | SIGMA | P0781 | |
| PHERAstar machine and software | BMG LABTECH | ||
| Phosphate-buffered saline (DPBS, 1x) | gibco | 14190-094 | |
| REA control antibody (S), human IgG1, Vio Bright FITC, REAfinity | Miltenyi Biotec | 130-113-443 | |
| Sodium hypochlorite solution | Sigma-Aldrich | 425044-1L | |
| Sodium nitrite solution | Sigma Aldrich | 91-4 | from kit Cat No. 387A-1KT |
| Tartrate solution | Sigma Aldrich | 387-3 | from kit Cat No. 387A-1KT |
| Triton X-100 | Sigma-Aldrich | 9002-93-1 | |
| Trypan blue | Sigma-Aldrich | T8154-100ML |
References
- Boyle, W. J., Simonet, W. S., Lacey, D. L. Osteoclast differentiation and activation. Nature. 423 (6937), 337-342 (2003).
- Boyce, B. F., Xing, L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Archives Of Biochemistry and Biophysics. 473 (2), 139-146 (2008).
- Jacome-Galarza, C. E., et al. Developmental origin, functional maintenance and genetic rescue of osteoclasts. Nature. 568 (7753), 541-545 (2019).
- Agemura, T., Hasegawa, T., Yari, S., Kikuta, J., Ishii, M. Arthritis-associated osteoclastogenic macrophages (AtoMs) participate in pathological bone erosion in rheumatoid arthritis. Immunological Medicine. 45 (1), 22-26 (2022).
- Hasegawa, T., et al. Identification of a novel arthritis-associated osteoclast precursor macrophage regulated by FoxM1. Nature Immunology. 20 (12), 1631-1643 (2019).
- Walsh, N. C., Crotti, T. N., Goldring, S. R., Gravallese, E. M. Rheumatic diseases: The effects of inflammation on bone. Immunological Reviews. 208 (1), 228-251 (2005).
- Gravallese, E. M., et al. Identification of cell types responsible for bone resorption in rheumatoid arthritis and juvenile rheumatoid arthritis. The American Journal of Pathology. 152 (4), 943-951 (1998).
- Bromley, M., Woolley, D. E. Chondroclasts and osteoclasts at subchondral sites of erosion in the rheumatoid joint. Arthritis & Rheumatism. 27 (9), 968-975 (1984).
- Kleyer, A., Schett, G. Arthritis and bone loss: A hen and egg story. Current Opinion in Rheumatology. 26 (1), 80-84 (2014).
- Kawai, V. K., Stein, C. M., Perrien, D. S., Griffin, M. R. Effects of anti-tumor necrosis factor α (anti-TNF) agents on bone. Current Opinion in Rheumatology. 24 (5), 576-585 (2012).
- Siebert, S., Tsoukas, A., Robertson, J., McInnes, I. Cytokines as therapeutic targets in rheumatoid arthritis and other inflammatory diseases. Pharmacological Reviews. 67 (2), 280-309 (2015).
- Smiljanovic, B., et al. Monocyte alterations in rheumatoid arthritis are dominated by preterm release from bone marrow and prominent triggering in the joint. Annals of the Rheumatic Diseases. 77 (2), 300-308 (2018).
- Anderson, J. R., et al. 1H NMR metabolomics identifies underlying inflammatory pathology in osteoarthritis and rheumatoid arthritis synovial joints. Journal of Proteome Research. 17 (11), 3780-3790 (2018).
- McGarry, T., et al. Rheumatoid arthritis CD14+ monocytes display metabolic and inflammatory dysfunction, a phenotype that precedes clinical manifestation of disease. Clinical & Translational Immunology. 10 (1), 1237(2021).
- Ansalone, C., et al. TNF is a homoeostatic regulator of distinct epigenetically primed human osteoclast precursors. Annals of the Rheumatic Diseases. 80 (6), 748-757 (2021).
- Yokota, K., et al. Characterization and function of tumor necrosis factor and interleukin-6-induced osteoclasts in rheumatoid arthritis. Arthritis and Rheumatology. 73 (7), 1145-1154 (2021).
- Allard-Chamard, H., et al. Osteoclasts and their circulating precursors in rheumatoid arthritis: Relationships with disease activity and bone erosions. Bone Reports. 12, 100282(2020).
- Takegahara, N., et al. Involvement of receptor activator of nuclear factor-κB ligand (RANKL)-induced incomplete cytokinesis in the polyploidization of osteoclasts. The Journal of Biological Chemistry. 291 (7), 3439-3454 (2016).
- Jansen, I. D. C., Vermeer, J. A. F., Bloemen, V., Stap, J., Everts, V. Osteoclast fusion and fission. Calcified Tissue International. 90 (6), 515-522 (2012).
- McDonald, M. M., et al. Osteoclasts recycle via osteomorphs during RANKL-stimulated bone resorption. Cell. 184 (5), 1330-1347 (2021).
- Lacey, D. L., et al. Bench to bedside: Elucidation of the OPG-RANK-RANKL pathway and the development of denosumab. Nature Reviews Drug Discovery. 11 (5), 401-419 (2012).
- Zhao, B., Grimes, S. N., Li, S., Hu, X., Ivashkiv, L. B. TNF-induced osteoclastogenesis and inflammatory bone resorption are inhibited by transcription factor RBP-J. The Journal of Experimental Medicine. 209 (2), 319-334 (2012).
- Zhao, B. Does TNF promote or restrain osteoclastogenesis and inflammatory bone resorption. Critical Reviews in Immunology. 38 (4), 253-261 (2018).
- Crotti, T. N., et al. Receptor activator NF-κB ligand (RANKL) expression in synovial tissue from patients with rheumatoid arthritis, spondyloarthropathy, osteoarthritis, and from normal patients: semiquantitative and quantitative analysis. Annals of the Rheumatic Diseases. 61 (12), 1047-1054 (2002).
- Kim, H. R., et al. Reciprocal activation of CD4+ T cells and synovial fibroblasts by stromal cell-derived factor 1 promotes RANKL expression and osteoclastogenesis in rheumatoid arthritis. Arthritis and Rheumatology. 66 (3), 538-548 (2014).
- Arai, F., et al. Commitment and differentiation of osteoclast precursor cells by the sequential expression of c-Fms and receptor activator of nuclear factor κB (RANK) receptors. Journal of Experimental Medicine. 190 (12), 1741-1754 (1999).
- Hayman, A. Tartrate-resistant acid phosphatase (TRAP) and the osteoclast/immune cell dichotomy. Autoimmunity. 41 (3), 218-223 (2008).
- Nedeva, I. R., Vitale, M., Elson, A., Hoyland, J. A., Bella, J. Role of OSCAR signaling in osteoclastogenesis and bone disease. Frontiers in Cell and Developmental Biology. 9, 780(2021).
- Boyce, B. F., Yoneda, T., Lowe, C., Soriano, P., Mundy, G. R. Requirement of pp60c-src expression for osteoclasts to form ruffled borders and resorb bone in mice. The Journal of Clinical Investigation. 90 (4), 1622-1627 (1992).
- Matsubara, T., et al. Regulation of osteoclast differentiation and actin ring formation by the cytolinker protein plectin. Biochemical and Biophysical Research Communications. 489 (4), 472-476 (2017).
- Roscher, A., et al. The F-actin modulator SWAP-70 controls podosome patterning in osteoclasts. Bone Reports. 5, 214-221 (2016).
- Jurdic, P., Saltel, F., Chabadel, A., Destaing, O. Podosome and sealing zone: Specificity of the osteoclast model. European Journal of Cell Biology. 85 (3-4), 195-202 (2006).
- Francis, M. J. O., et al. ATPase pumps in osteoclasts and osteoblasts. The International Journal of Biochemistry & Cell Biology. 34 (5), 459-476 (2002).
- Kwak, H. B., et al. Inhibition of osteoclast differentiation and bone resorption by rotenone, through down-regulation of RANKL-induced c-Fos and NFATc1 expression. Bone. 46 (3), 724-731 (2010).
- Massey, H. M., Flanagan, A. M. Human osteoclasts derive from CD14-positive monocytes. British Journal of Haematology. 106 (1), 167-170 (1999).
- Xue, J., et al. CD14+CD16-monocytes are the main precursors of osteoclasts in rheumatoid arthritis via expressing Tyro3TK. Arthritis Research and Therapy. 22 (1), 221(2020).
- Marco-Casanova, P., et al. Preparation of peripheral blood mononuclear cell pellets and plasma from a single blood draw at clinical trial sites for biomarker analysis. Journal of Visualized Experiments. (169), e60776(2021).
- Takahashi, N., et al. Osteoblastic cells are involved in osteoclast formation. Endocrinology. 123 (5), 2600-2602 (1988).
- Yasuda, H., et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proceedings of the National Academy of Sciences of the United States of America. 95 (7), 3597-3602 (1998).
- Lacey, D. L., et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 93 (2), 165-176 (1998).
- Matsuzaki, K., et al. Osteoclast differentiation factor (ODF) induces osteoclast-like cell formation in human peripheral blood mononuclear cell cultures. Biochemical and Biophysical Research Communications. 246 (1), 199-204 (1998).
- Shalhoub, V., et al. Osteoprotegerin and osteoprotegerin ligand effects on osteoclast formation from human peripheral blood mononuclear cell precursors. Journal of Cellular Biochemistry. 72 (2), 251-261 (1999).
- Neale, S. D., Smith, R., Wass, J. A. H., Athanasou, N. A. Osteoclast differentiation from circulating mononuclear precursors in Paget's disease is hypersensitive to 1,25-dihydroxyvitamin D3 and RANKL. Bone. 27 (3), 409-416 (2000).
- Abdallah, D., et al. An optimized method to generate human active osteoclasts from peripheral blood monocytes. Frontiers in Immunology. 9, 632(2018).
- Komano, Y., Nanki, T., Hayashida, K., Taniguchi, K., Nobuyuki, M. Identification of a human peripheral blood monocyte subset that differentiates into osteoclasts. Arthritis Research and Therapy. 8 (5), 152(2006).
- Kylmäoja, E., et al. Peripheral blood monocytes show increased osteoclast differentiation potential compared to bone marrow monocytes. Heliyon. 4 (9), 00780(2018).
- Wang, D., et al. Platelet-rich plasma inhibits RANKL-induced osteoclast differentiation through activation of Wnt pathway during bone remodeling. International Journal of Molecular Medicine. 41 (2), 729-738 (2018).
- Cenni, E., Avnet, S., Fotia, C., Salerno, M., Baldini, N. Platelet-rich plasma impairs osteoclast generation from human precursors of peripheral blood. Journal of Orthopaedic Research. 28 (6), 792-797 (2010).
- D'Amico, L., Roato, I. Cross-talk between T cells and osteoclasts in bone resorption. BoneKEy Reports. 1 (6), 82(2012).
- Quinn, J. M. W., Elliott, J., Gillespie, M. T., Martin, T. J. A combination of osteoclast differentiation factor and macrophage-colony stimulating factor is sufficient for both human and mouse osteoclast formation in vitro. Endocrinology. 139 (10), 4424-4427 (1998).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved