
Method Article
单细胞分辨率下小胶质细胞 Ca2+ 活性的时空分析
摘要
在本文中,我们描述了一种用于小胶质细胞 Ca2+ 活性的体内成像方案,并随后分析其时空动力学。这种方法能够彻底表征小胶质细胞如何响应大脑环境的变化,适当地捕捉此类事件发生的精细时空尺度。
摘要
小胶质细胞是中枢神经系统中唯一的常驻免疫细胞。它们的形态具有高度可塑性,根据其活动而变化。在稳态条件下,小胶质细胞具有高度分叉的形态。这有助于他们通过不断扩展和缩回其流程来监控周围环境。然而,在脑损伤和炎症期间,小胶质细胞被激活并发生剧烈的形态变化,收缩其分叉过程并使其细胞体肿胀。这促进了迁移和吞噬作用等活动,小胶质细胞承担这些活动是为了将大脑环境导航至不那么病理的状态。
小胶质细胞形态与其活性变化之间的这种密切关系使人们对各种小胶质细胞功能有了相当大的了解。然而,这种形态和活性变化本身就是可以由任意数量的细胞内信号通路引起的现象。此外,刺激和反应之间的时间滞后,以及小胶质细胞高度区室化的形态,使得很难分离支撑功能的致病机制。为了解决这个问题,我们开发了一种转基因小鼠系,其中高度敏感的荧光 Ca2+ 指示蛋白在小胶质细胞中特异性表达。
在描述了 体内 小胶质细胞 Ca2+ 成像的方法之后,本文提出了一种结构化分析方法,该方法将这种 Ca2+ 活性分类为合理定义的亚细胞区域,从而确保有意义地提取编码信息的空间和时间维度。我们相信这种方法将提供对细胞内信号传导规则的详细理解,这些规则控制着与高级大脑功能和病理状况相关的各种小胶质细胞活动。
引言
小胶质细胞是中枢神经系统 (CNS) 中的常驻免疫细胞,在维持稳态脑环境和在大脑发育过程中调节神经回路形成方面发挥着重要作用 1,2。CNS 中小胶质细胞的一个独特特征是它们的形态具有高度可塑性;然而,不同的形态表型可能与特定功能相关。此外,形态表型之间的转化是高度动态的,响应周围环境的变化在快速的时间尺度上发生 3,4。
在稳态生理条件下,小胶质细胞呈现高度分叉的形态,具有多个过程向各个方向向外辐射。这些分叉过程本身表现出高运动性,不断伸展和收缩 3,4。这种活动主要针对与神经元突触、轴突和胞体的周期性接触,以监测神经元活动 5,6,7,8,9。然而,当大脑受伤时,小胶质细胞会迅速检测到这种异常,并作为它们适应性反应的第一步,将其过程向相应的区域延伸 3,4。当小胶质细胞需要对死细胞和代谢物进行吞噬作用时,它们呈现出类似变形虫的形态,缩短了它们的过程并扩大了它们的细胞体,作为它们向免疫激活表型过渡的一部分10,11。
然而,虽然小胶质细胞过程的剧烈形态变化很容易检测到,但细胞胞体的更精细尺度的变化明显更难捕获,尤其是在生理相关的时间分辨率下。此外,形态变化本身仅代表任意数量的细胞内信号通路的综合结果。这对于跟踪功能活动并机械地将刺激与其引发的最终反应联系起来的目标来说是有问题的。
鉴于其作为第二信使的广泛作用,在研究动态细胞过程时,检查细胞内 Ca2+ 动力学可以更好地捕获相关的时空信息。这种方法适用于小胶质细胞,因为它们表达与下游细胞内 Ca2+ 升高相关的各种离子型和代谢型受体。事实上, 体内 Ca2+ 成像已被用于实时表征小胶质细胞活动的时空方面,成功地将小胶质细胞 Ca2+ 活性的变化与脑损伤、炎症以及神经元的过度活跃和低活跃相关联12、13、14、15、16。例如,响应于高/低活性神经元活动而与小胶质细胞过程延伸相关的 Ca2+ 升高可能反映了潜在的 Ca2+ 依赖性肌动蛋白聚合过程16。此外, 体内 Ca2+ 成像也可以很容易地与药理学方法相结合。例如,虽然小胶质细胞同时表达 P2X(离子型)和 P2Y(代谢型)受体,但 P2Y 激动剂的局部应用模拟并随后使小胶质细胞 Ca2+ 对受损邻近神经元的反应脱敏13,从而意味着 P2Y 信号转导与神经元损伤检测的相关性更大。
迄今为止,以前研究小胶质细胞 Ca2+ 活性的报告采用了基于感兴趣区域 (ROI) 的分析方法。这些方法的一个缺点是它们仍然太粗糙,无法在单个小胶质细胞过程的水平上解析 Ca2+ 活性的时空动力学。因此,该协议描述了用于分析小胶质细胞 Ca2 + 活性的基于 ROI 的常规方法和更新的基于事件的方法,这些方法可以提取小胶质细胞过程中的单个 Ca2 + 事件。在此之前,我们提供了 体内 双光子成像的一般指南,以适当捕获小胶质细胞 Ca2+ 活性以进行详细分析。
研究方案
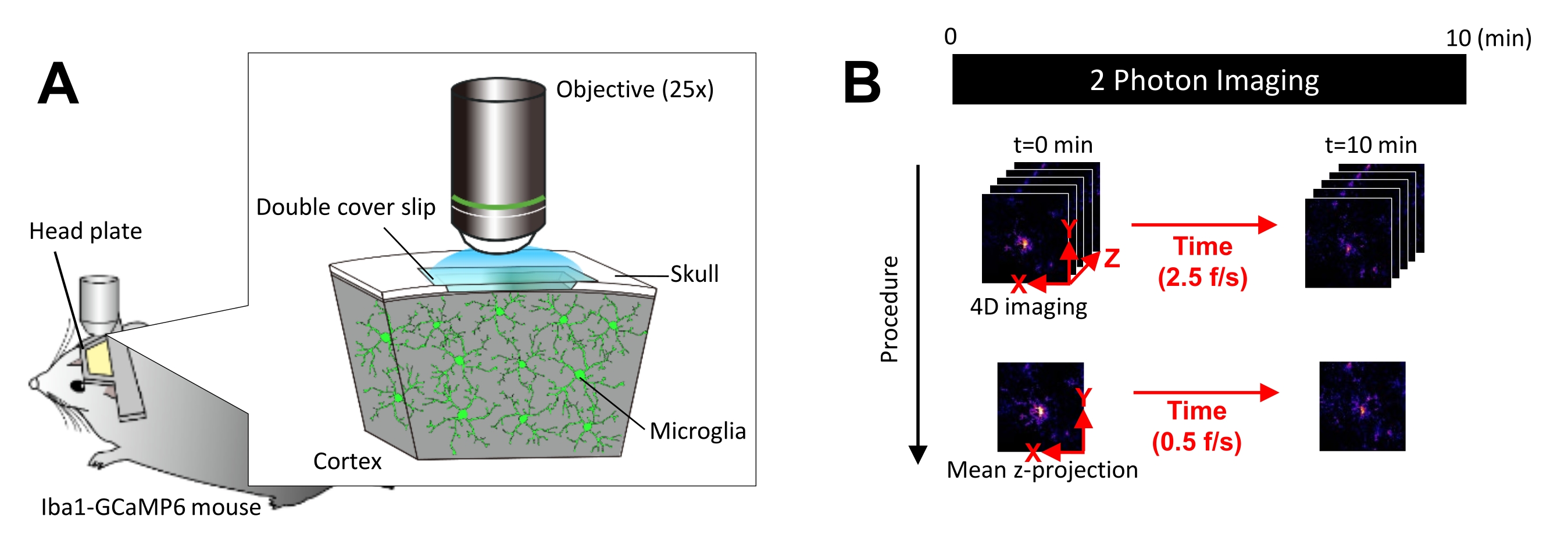
所有动物实验均已获得美国国家生理科学研究所动物研究委员会的批准,并符合美国国立卫生研究院的指南。对于所有实验,在 12/12 小时的光照/黑暗周期下饲养 8-10 周龄雄性小鼠,随意获取食物和水。为了观察小胶质细胞中的 Ca2+ 活性,将电离的 Ca2+ 结合接头分子 1 (Iba1)-四环素反式激活因子 (Iba1-tTA) 小鼠与四环素纵子-GCaMP6 (tetO-GCaMP6) 小鼠杂交17,18。因此,在没有四环素类似物补充剂的情况下,Iba1 启动子仅在小胶质细胞中驱动 GCaMP6 的表达。对于所有实验,多西环素膳食补充剂在出生后 6 周停止。在所有实验结束时,通过异氟醚过量对小鼠实施安乐死,然后进行颈椎脱位。有关本协议中使用的所有材料、动物和试剂的详细信息,请参阅材料表。
1. 小鼠 体内双光 子成像的手术准备;第 1 天
- 在层流式空气流通柜内执行所有外科手术,以保持无菌的工作条件。开始手术前,用紫外线对橱柜内部消毒 5 分钟。
- 用 70% 乙醇擦拭所有工作台面、手术架和立体定位器械,对它们进行消毒。
- 通过将所有手术器械(剪刀、镊子、剃须刀片、镊子)和定制的头板浸入 1% 葡萄糖酸氯己定溶液中,对它们进行消毒。
- 用氯胺酮(7.4 mg kg -1,腹膜内 [ip])和甲苯噻嗪(10 mg kg -1,ip)麻醉小鼠。将其放回笼子中,直到麻醉生效。通过脚趾捏反射的丧失确认麻醉的完全诱导。
- 用 1% 葡萄糖酸氯己定对头皮进行消毒。用剃须刀片剃掉皮毛。
- 通过立体定位器械将鼠标固定在手术框架内。
- 在麻醉下将兽医软膏涂抹在眼睛上,以防止干燥。
- 将 2% 赛洛卡因果冻涂抹在头皮上以控制疼痛。等待 5 分钟。
- 用剪刀去除头皮,露出头骨。用棉签摩擦清洁骨膜并擦干裸露的颅骨表面。
注意:裸露的颅骨区域必须完全干燥,以确保与定制的头板牢固粘合。 - 用牙科水泥将定制的头板固定在颅骨上。
- 一旦水泥凝固,用额外的牙科水泥填充颅骨表面和定制头板边缘之间的任何间隙。
- 通过涂覆丙烯酸基牙科胶粘剂树脂胶粘剂对水泥和颅骨表面进行防水处理。
- 将鼠标放回其固定架上,将其放在加热垫上。监测小鼠,直到它恢复足够的意识以维持胸骨卧位(2 小时内)。
注意:小鼠应在第二天完全康复,然后可以与其他动物一起饲养。
2. 小鼠 体内双光 子成像的手术准备;第 2 天
- 在层流式空气流通柜内执行所有外科手术,以保持无菌的工作条件。开始手术前,用紫外线对橱柜内部消毒 5 分钟。
- 用 UV 固化光学级树脂将两个不同尺寸的玻璃盖玻片(顶部玻璃:3 mm × 3 mm;底部玻璃:2 mm × 2 mm)层压在一起。
注意:双盖玻片为颅窗暴露的大脑区域提供长期保护,同时实现长期光学访问。因此,可以改变其尺寸以适应要成像的大脑区域。 - 用 70% 乙醇擦拭所有工作台面和手术架,对它们进行消毒。
- 将所有手术器械(钢钻、镊子、手术针钩)浸入 1% 葡萄糖酸氯己定溶液中,对它们进行消毒。
- 用异氟醚 (4% 诱导,1.2%-1.5% 维持) 麻醉小鼠。 通过 鼠标的头板将鼠标固定在手术框架内。
- 要在初级运动皮层上制作颅窗,请在前囟头骨标志前 0.2 毫米和外侧 0.2 毫米处标记一个尺寸为 2 毫米× 2 毫米的正方形。
- 使用钢钻沿标记方块的边界将头骨变薄。
注意:当接近所需的厚度时,用盐水润湿时,变薄的头骨区域会显得透明,并且会开始出现发际线裂纹。 - 在确认标记正方形的整个边界已适当变薄后,小心地将手术针钩插入颅骨表面正下方,使其尖端朝向正方形的中心。用钩子轻轻提起方形头骨片,然后用镊子将其从头骨的其余部分剥落。如果发生出血,请不断用生理盐水清洗裸露的大脑表面,直到它完全消退。
- 将双盖玻片放在暴露的大脑表面上,将带有较小盖玻片的一侧朝向大脑。确保较大的盖玻片的边缘与颅窗的边缘接触。
- 使用安装在机械手中的硅尖玻璃棒,轻轻按下双盖玻片,确保它与大脑表面接触良好。
- 用 UV 固化树脂填充双盖玻片、颅骨和脑表面之间的间隙,并用 UV 光照射直至硬化 (~20 s)。慢慢地将硅尖玻璃棒从双盖玻片上抬起。
- 让鼠标恢复,将其放在加热板上。监测小鼠,直到它恢复足够的意识以维持胸骨卧位(30 分钟)。
- 继续执行步骤 3 或将鼠标放回其原位固定架。
注意:熟练手术后,硬脑膜和所有底层血管将完全完好无损,无出血。在这种情况下,炎症很小,可以立即进行第 3 步。如果不确定,请等待 1-3 周以确保任何炎症已完全消退。
3. 使用 体内双 光子成像进行数据收集
- 提前打开成像设置,以确保激光器有足够的时间预热和稳定。调整激光器以 920 nm 的波长发射,这是最佳激发 GCaMP6 荧光团的双光子光谱。
- 将鼠标置于 25 倍显微镜物镜下并使其习惯 30 分钟。如有必要,在成像过程中用异氟醚麻醉小鼠(4% 诱导,1.2%-1.5% 维持)。
注意:异氟醚可能不适用于某些研究应用,因为它会影响小胶质细胞过程的运动性和 Ca2+ 活性 19,20,21。 - 将缩放设置为 1 倍,然后在颅窗区域搜索表达 GCaMP6 的小胶质细胞。在大脑表面以下 100 μm 至 300 μm 的深度进行搜索。
- 找到合适的 GCaMP6 表达小胶质细胞后,最大化缩放,以便以单细胞分辨率捕获 Ca2+ 活性。
- 目视确认 Ca2+ 活性被清晰捕获,并在必要时调整激光功率和成像增益。
注意:应尽可能减少激光功率,以避免光漂白和对小胶质细胞的伤害。 - 按如下方式执行四维 (4D) 图像采集(步骤 3.6.1-3.6.3; 图 1B):
- 将图像尺寸设置为 XY 区域 = 512 × 512 像素,0.25 μm/像素;Z 面积 = 五个 z 平面,3 μm z 步长(图 1B)。
- 将采集速率设置为 2.5 帧/秒。
注意:这种速度的 Z 扫描是由压电纳米定位系统实现的。 - 采集数据,直到观察到至少 10 个单独的 Ca2 + 事件(通常为 10 分钟)。
4. 分析准备(运动校正、平均值/最大 z 投影)
- 在继续分析小胶质细胞 Ca2+ 活性之前,在 MATLAB 编程环境 (R2020a) 中使用 ECC 图像对齐算法22 校正 4D 图像中的运动相关伪影(图像配准)。从以下位置下载代码:https://www.mathworks.com/matlabcentral/fileexchange/27253-ecc-image-alignment-algorithm-image-registration。
- 通过使用 imregister MATLAB作(标准图像处理工具箱)在第一个时间范围内注册所有图像来创建参考 3D 图像。
- 注册所有后续图像,使用 ecc 函数(ECC 图像对齐算法库)将它们与参考 3D 图像的相应 z 平面匹配。
- 使用 平均 MATLAB 运算从注册图像生成平均强度 z 投影。如果成像信号太弱,则使用 最大 MATLAB 运算生成最大强度的 z 投影。
注意:使用最大强度 z 投影时,信噪比会更差。 - 继续执行步骤 5 或 6。使用步骤 4 中生成的 z 投影作为分析目标,在步骤 5 和 6 中绘制 Ca2+ 活动的时空动态。
5. 基于 ROI 的分析
- 使用步骤 4 中生成的 z 投影,确定在整个成像期间保持稳定区域剖面(稳定区域)的小胶质细胞突起,以便随后分析 Ca2+ 活性,如下所示(步骤 5.3-5.6):
- 使用 最大 MATLAB作,从成像周期开始和结束时采集的 2 分钟样本中生成注册的 4D 图像的单独最大强度 t 投影。
- 使用 imbinarize MATLAB作将 t 投影二值化,以生成对应于成像周期开始和结束的小胶质细胞形态的多边形。使用自动设置的默认阈值。
注意:如果 Ca2+ 信号在成像周期的开始和结束时较弱,则二值化可能会生成缺少边界的多边形。在这种情况下,请使用 ImageJ 中的 铅笔工具 手动绘制缺失的边框。 - 使用 imadd MATLAB作叠加 t 投影多边形。重叠区域代表稳定区域(图 2B)。
- 在确定的稳定区域内,使用 drawpolygon MATLAB作手动定义和跟踪每个主要小胶质细胞过程和任何明显的二阶子分支的 ROI。
- 跟踪所有时间范围内单个 ROI 的平均绝对荧光强度(图 2C、D)。
- 根据绝对荧光强度的时间序列,根据方程 (1) 计算荧光强度 (ΔF/F) 时间序列的相对变化。该时间序列表示单个 ROI 水平的归一化小胶质细胞 Ca2+ 动力学。
ΔF/F = (F(t) - F0) / F0 (1)
其中 F(t) 是在给定时间记录的荧光强度,F0 是所有时间范围内荧光强度的第 10 个百分位数(图 2E)。 - 按如下方式确定给定单个 ROI 中发生的候选 Ca2+ 触发事件(步骤 5.5.1-5.5.3):
- 使用 fir1 MATLAB 运算对 ΔF/F 时间序列应用 I 型有限脉冲响应 (FIR) 低通滤波。将截止频率设置为 Nyquist 值(采样率的一半)。
- 目视检查过滤后的迹线,以确认单个迹线峰对应于注册的 4D 图像中 Ca2+ 活度的突发(图 2F)。
- 将候选 Ca2+ 触发事件识别为ΔF/F 时间序列轨迹中的凹下拐点。将基线阈值定义为上限和下限内的中值 ΔF/F 值,不包括所有时间范围内的最大和最小 10% 振幅。定义高于基线阈值的三个 SD 的检测阈值(图 2F,H)。
- 按如下方式从候选对象中识别真正的 Ca2+ 触发事件(步骤 5.5.1-5.5.2):
- 通过使用数值梯度 MATLAB 运算对相应的过滤 ΔF/F 时间序列帧进行微分,计算每个候选事件的斜率。
- 随后,根据配置文件的上升时间识别真正的 Ca2+ 事件。将基线阈值定义为所有候选事件的坡度值的平均值。定义高于基线阈值的三个 SD 的检测阈值(图 2G,H)。
- 按如下方式描述真实的 Ca2+ 事件(步骤 5.6.1-5.6.3):
注:以下不是可用于表征小胶质细胞 Ca2+ 事件的参数的详尽列表。感兴趣的参数取决于研究的目的。- 将真实 Ca2+ 触发事件的最大振幅推导出为相应 ΔF/F 时间序列帧的 ΔF/F 值。
- 将真实 Ca2+ 触发事件的平均振幅推导出为整个相应 ΔF/F 时间序列帧子集的平均 ΔF/F 值。
- 将真实 Ca2+ 事件的频率推导出为事件数除以成像时间。
6. 基于事件的分析
- 在 MATLAB 编程环境中使用 AQuA 库对步骤 4 中的 z 投影执行基于事件的分析。从以下位置下载代码:https://github.com/yu-lab-vt/AQuA。
注意:AQuA 库的常规使用演练可从以下网址获得:https://drive.google.com/file/d/1a3lhe0dUth-5J1-S2fZlPOCZlPbeuvUr/view。AQuA 库的详细文档可从以下网址获得:https://drive.google.com/file/d/1CckDLbrkw16b7MPlOQdYpZciIz80Snm_/view。 - 启动 MATLAB 后,使用 cd 作从默认工作目录文件夹切换到 AQuA 的指定工作目录文件夹。
- 按如下方式将注册的 4D 图像加载到 AQuA 分析管道中(步骤 6.3.1-6.3.2):
- 启动 AQuA GUI。在 AQuA 的指定工作目录文件夹中的 MATLAB 命令窗口中键入 aqua_gui 。
- 在 AQuA GUI 中,单击 New project 并选择要分析的 已注册图像 。指定 数据类型 (GCaMPInVivo_cyto_Lck_) 和 成像参数 (每帧时间分辨率秒数 = 1.993;空间分辨率每像素 μm = 0.25;不包括短于此边界距离的像素 = 5)。单击 open 以加载数据。
- 通过追踪感兴趣区域来定义后续分析管道的地标。通常,特征点基于单元边界和单元体的面积。
- 随后,通过运行以下参数的自动分析管道来检测候选 Ca2+ 事件:活动信号、超级体素、事件检测、清理事件和合并事件。
注意:AQuA 演练中提供了有关每个参数及其候选分数输出的详细说明。简而言之,这些参数调整 Ca2+ 事件检测阈值如下:活动信号 = 荧光振幅,超级体素 = 3D 空间中荧光的聚集,事件检测 = 上升/衰减动力学,干净事件 = 信噪比,合并事件 = 事件的时间分离。 - 目视检查检测到的 Ca2+ 事件(自动叠加到原始注册图像上)。如有必要,根据上述参数考虑图像质量来调整分析管道的参数设置;如果给定参数的图像质量良好,则可以设置更高的阈值,反之亦然。
- 正确检测到所有 Ca2+ 事件后,在主 MATLAB 环境中按如下方式描述这些事件(步骤 6.8-6.14):
- 导出 AQuA 分析输出文件。
注意:AQuA 文档中提供了有关输出文件、用于定义 Ca2+ 事件的提取特征以及这些提取特征的基础参数的详细说明。 - (可选)将所有 Ca2+ 事件分为两组:1) 从 soma 开始的事件,以及 2) 从进程开始的事件。
- 在 AQuA 分析输出文件(.mat 文件)的 res.dffMat MATLAB 结构中访问单个 Ca2+ 事件的振幅。
- 使用 res.fts.loc.x3D MATLAB 运算(AQuA 库)推导单个 Ca2+ 事件的频率。
- 使用 res.fts.curve.width11 MATLAB 运算(AQuA 库)推导单个 Ca2+ 事件的持续时间。
- 使用 res.fts.basic.area MATLAB 运算(AQuA 库)推导单个 Ca2+ 事件的面积。
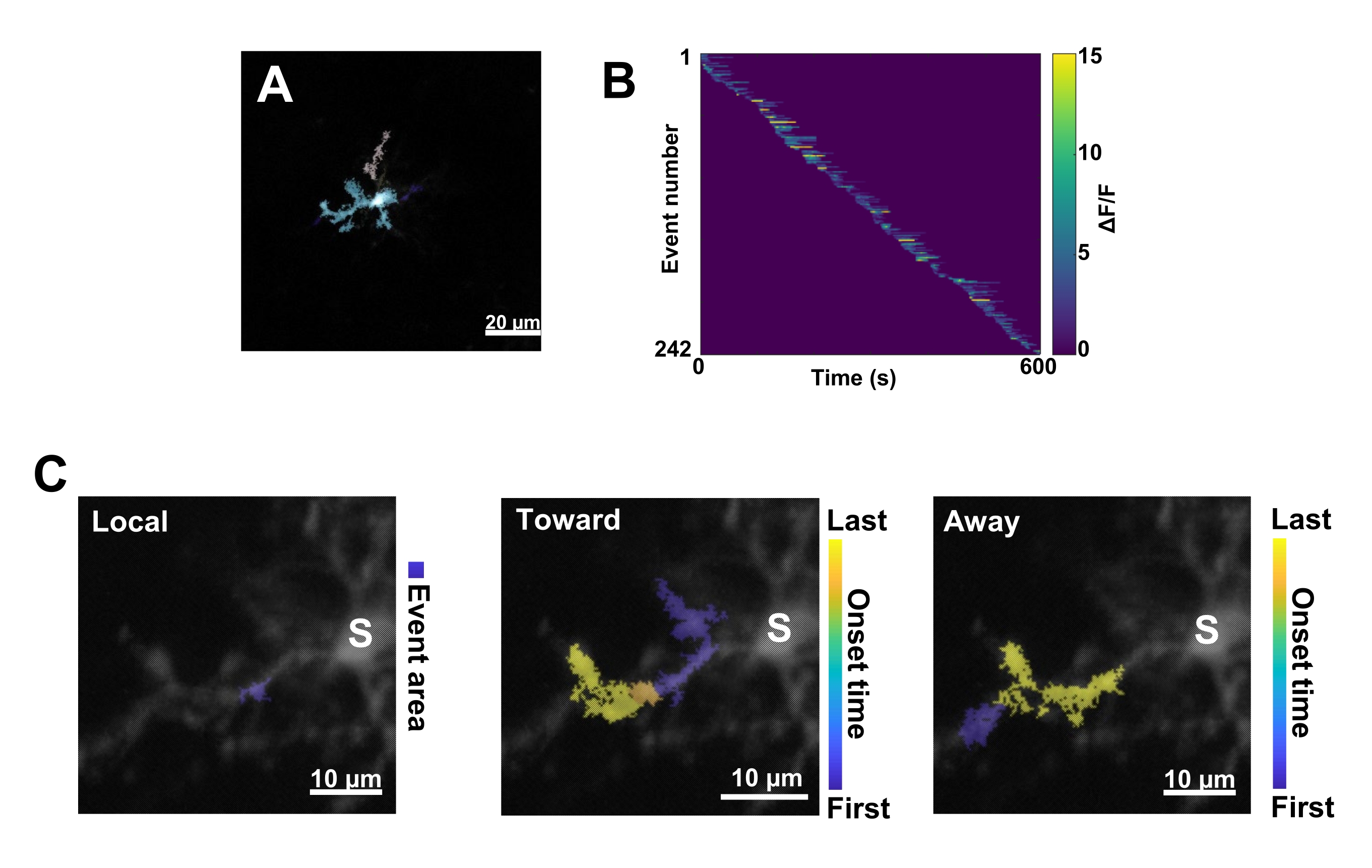
- 将所有 Ca2+ 事件分类为:1) 局部事件,2) 向 soma 传播的传播事件,以及 3) 远离 soma 传播的传播事件。为此,请使用 res.fts.region.landmarkDir.chgTowards 和 res.fts.region.landmarkDir.chgAway MATLAB作(AQuA 库)(图 3C)。
结果
在小胶质细胞中专门表达 GCaMP6 (Ca2+ 敏感荧光蛋白) 的转基因小鼠中,我们通常会观察到小胶质细胞 Ca2+ 活性的不同模式(图 2A)。重要的是,即使在单个小胶质细胞中,Ca2+ 活性的模式也会因过程而异。
为了量化小胶质细胞 Ca2 + 活性时空动力学中的这种过程间差异,必须首先确定稳定区域,然后划分为精细分割的 ROI(图 2B,C)。对于每个 ROI,必须通过从荧光强度时间序列中提取局部振幅和迹线斜率等特征来推导出和量化 Ca2+ 活性的参数,例如振幅和频率(图 2D-G)。
接下来,必须通过应用准确的定量算法 AQuA 来检查单个 Ca2 + 事件(图 3A)。从这种基于事件的分析中,通常会观察到单个 Ca2+ 事件的来源、振幅、持续时间、位置和流向特征的巨大差异(图 3B)。如果专注于分析小胶质细胞过程中的 Ca2 + 活动动力学,则局部事件、流向胞体的事件和远离胞体的事件的分类方案是有用的(图 3C)。

图 1:体内小胶质细胞 Ca2+ 成像的实验装置。 (A) 实验装置。具有小胶质细胞特异性 GCaMP6 表达的 Iba1-tTA × tetO-GCaMP6 小鼠。通过在小鼠的颅骨中插入颅窗,可以使用双光子显微镜在体内观察到小胶质细胞 Ca2+ 活性。(B) 实验时间表和分析程序。4D 图像以五帧 z 堆栈的形式采集至少 10 分钟。帧采集速率为 2.5 帧/秒。在分析小胶质细胞 Ca2 + 活性之前,通过采用平均(或最大)强度将五帧 z 堆栈转换为 2D z 投影。z 投影播放速率为 0.5 帧/秒。请单击此处查看此图的较大版本。
{kind=link}

图 2:基于 ROI 的小胶质细胞 Ca2+ 活性分析。 (A) 单个小胶质细胞在 10 分钟内的平均 GCaMP6 强度投影。(B) 稳定区域(白色)由二值化的最大 GCaMP6 强度 t 投影叠加定义,这些投影来自在成像周期开始(洋红色)和结束(绿色)采集的 2 分钟样品。(C) 稳定区域进一步细分为区域 ROI。单个颜色表示单个 ROI。(D) C 中所有单个 ROI 的 ΔF/F 迹线。请注意 ROI 之间活动模式的变化。(E) 从单个 ROI 的绝对强度值得出的 ΔF/F 时间序列的原始迹线。(F) 低通滤波后相同的 ΔF/F 时间序列。候选 Ca2+ 事件通过定义为基线 + 三个 SD 的振幅截止阈值(红线)检测。基线(绿线)定义为上限和下限内整个 ΔF/F 时间序列的中位数,不包括 ΔF/F 值的最大值和最小值 10%。(G) 从过滤后的 ΔF/F 时间序列得出的斜率轨迹(以 F 为单位)。真正的 Ca2+ 事件根据定义为基线 + 三个 SD 的斜率截止阈值(红线)从候选 Ca2+ 事件中排序。基线(绿线)定义为上限和上限下限内整个坡度时间序列的平均值,其中不包括坡度值的最大值和最小值 10%。(H) 由 F 中的振幅标准鉴定的候选 Ca2+ 事件以橙色表示。True Ca2+ 事件按红色 G 中的斜率标准从候选 Ca2+ 事件中排序。请注意,一些候选 Ca2+ 事件已根据坡度标准进行合并。相应的筛选 ΔF/F 时间序列叠加在下面以供参考。黑线表示零振幅 (ΔF/F)。真实 Ca2+ 事件的均值和最大振幅是它们在过滤后的 ΔF/F 时间序列中相应峰的均值和最大值。频率(事件/分钟)的计算方法是真正的 Ca2+ 触发事件的数量除以成像周期(10 分钟)。比例尺 = 20 μm (A,B),10 μm (C)。缩写:ROI = 感兴趣区域。请单击此处查看此图的较大版本。
{kind=link}

图 3:基于事件的小胶质细胞 Ca2+ 活性分析。 (A) 使用 AQuA 算法检测到的事件的代表性图像。这些颜色表示在某个特定时间点检测到的单个事件区域。(B) 单个事件中按发病顺序排序的代表性归一化 Ca2+ 活性 (ΔF/F)。右侧条形表示颜色指示的 ΔF/F。(C) 向胞体传播和远离胞体或局部事件的事件的代表性活动足迹。对于本地事件,事件形状以蓝色显示。对于传播事件,事件开始时间由蓝黄色刻度表示。由于 AQuA 最初单独检测 Ca2+ 事件,因此随后根据多个单个 Ca2+ 事件的重叠空间位置和时间序列来识别传播事件。请注意,这与图 2E-H 中用于 ROI 分析的树突分支相同。比例尺 = 20 μm (A),10 μm (C)。缩写:ROI = 感兴趣区域;S = 体细胞。请单击此处查看此图的较大版本。
{kind=link}
讨论
本文介绍了一种改进的高时空分辨率对小胶质细胞 Ca2+ 活性进行成像的方法。该方法足够灵敏,可以在单个分支过程的水平上检测不同类型的小胶质细胞 Ca2+ 活性,很容易区分局部和繁殖事件。
在小胶质细胞 Ca2+ 活性的体内双光子成像的一般方法中,必须特别注意以下几点,以最大限度地提高成像质量。首先,由于小胶质细胞对损伤极为敏感,因此在手术过程中尽量减少用手术工具直接接触大脑表面非常重要。手术已熟练进行的关键迹象是血管和硬脑膜完整以及手术过程中非常轻微的出血。其次,头板牢固地附着在小鼠的头骨上,并且双盖玻片和大脑表面之间的良好接触大大减少了成像时与运动相关的伪影。当以高时空分辨率成像和完全清醒的小鼠时,这一点尤其重要。虽然分析管道可靠地补偿了由心跳、呼吸和一般漂移引起的与运动相关的伪影,但在处理由突然的大运动引起的重大几何变形时,它的稳健性较差。
这里描述的两种分析方法具有不同的优势,适用于不同的研究问题。在基于 ROI 的分析中,用户预定义 ROI(例如单个流程),从而可以提取此 ROI 的 Ca2+ 活动的聚合动态。因此,它最适合于预期现象局限于具有明确形态边界和相对较大区域(即过程分支)的亚细胞区域的情况。在基于事件的分析中,单个事件是根据小胶质细胞 Ca2+ 活动本身的时空动力学定义的,然后必须将其放置在小胶质细胞内用户定义的标志的上下文中,以便解释其功能。因此,它最适合于无法对现象定位做出假设或感兴趣区域相对较小(即工艺尖端)的情况。因此,与以前的方法相比,基于事件的分析在表征小胶质细胞 Ca2 + 活性时提供了改进的时空分辨率。
在这些小鼠中,小胶质细胞表达的唯一荧光标志物是 Ca2+ 指示剂 GCaMP6。因此,在 Ca2+ 活性较低的区域,必须通过组合多个时间框架来提取小胶质细胞形态,这会降低时间分辨率。然而,可以通过在小胶质细胞中表达单独的红色稳定荧光蛋白来克服这一限制。值得注意的是,最近已经描述了能够转染小胶质细胞的新型腺相关病毒 23,24,25。
小胶质细胞 Ca2+ 活性如何被周围环境改变是一个新兴的有趣话题。特别是,小胶质细胞 Ca2+ 活性似乎与神经元活动显示出显着的相关性,尽管其功能意义尚未完全表征。因此,将神经元活动作与此处介绍的小胶质细胞 Ca2+ 活性的成像和分析方法相结合,应该会对小胶质细胞生理学产生新的见解,并进一步促进我们对小胶质细胞在生理和病理状态中的作用的理解。
披露声明
作者声明与本手稿不存在利益冲突。
致谢
我们感谢 Kenji Tanaka 教授(日本东京庆应义塾大学)提供 Iba1-tTA 小鼠和 tetO-GCaMP6 小鼠。这项工作得到了青年科学家助学金 (B) [16K19001 (to H.H.)]、早期职业科学家助学金 [18K14825 (to H.H.)]、科学研究助学金 (B) [21H03027 (to H.H.)]、变革性研究领域助学金 (A) [21H05639 (to H.H.)]、科学研究助学金 (A) [17H01530, 20H00500 (to J.N.)] 和 JST CREST 助学金 [JPMJCR1755 (to J.N.)], 日本。
材料
| Name | Company | Catalog Number | Comments |
| 2% xylocaine jelly | AstraZeneca, UK | ||
| B6(129S6)-Tg(Aif1-tTA)54Kftnk | RIKEN RBC, Japan | RBRC05769 | Iba1-tTA mice |
| B6;129-Actb(tm3.1(tetO-GCaMP6)Kftnk) | RIKEN RBC, Japan | RBRC09552 | tetO-GCaMP6 mice |
| Forceps | Fine science tools, US | 13008-12 | |
| G-CEM ONE | GC corporation, Japan | ||
| Glass capillary | Narishige, Japan | GDC-1 | |
| ImageJ | NIH, US | ||
| Isofulrane | Pfizer, US | ||
| Ketamin | Daiichi-Sankyo, Japan | ||
| Kwik-sil | World Precision Instruments, US | KWIK-SIL | |
| MATLAB, 2020a | MathWorks, US | ||
| Micro cover glass (2 x 2 mm, No.3) | Matsunami, Japan | custum-made | Bottom glass for cranial window |
| Micro cover glass (3 x 3 mm, No.0) | Matsunami, Japan | custum-made | Upper glass for cranial window |
| N25X-APO-MP | Nikon, Japan | N25X-APO-MP | Objective lens (25x) |
| Norland optical adhesive | Edmund optics, US | 6101 | |
| Piezo nano-positioning system, Nano-Drive | Mao City Labs, US | ||
| Razor blade | Feather, Japan | FA-10 | |
| Scissors | Fine science tools, US | 14060-11 | |
| Steel drill | Minitor, Japan | BS1201 | |
| Stereotaxic instruments | Narishige, Japan | SR-5M-HT | |
| Super-bond (C&B kit) | Sun Medical, Japan | 4560227797382 | |
| Surgical needle hook | Fine science tools, US | 10065-15 | |
| Ti:Sappire laser, MaiTai DeepSee | Spectra Physics, US | Mai Tai eHP DS | |
| Tweezers | Fine science tools, US | 11051-10 | |
| Tweezers | Fine science tools, US | 11255-20 | |
| Two-photon microscope | Nikon, Japan | A1R-MP | |
| UV craft resin | Kiyohara, Japan | UVR | |
| Xylazine | Bayer, Germany |
参考文献
- Miyamoto, A., et al. Microglia contact induces synapse formation in developing somatosensory cortex. Nature Communications. 7, 12540(2016).
- Schafer, D. P., et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent. Neuron. 74 (4), 691-705 (2012).
- Davalos, D., et al. ATP mediates rapid microglial response to local brain injury in vivo. Nature Neuroscience. 8 (6), 752-758 (2005).
- Nimmerjahn, A., Kirchhoff, F., Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 308 (5726), 1314-1318 (2005).
- Cserep, C., et al. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science. 367 (6477), 528-537 (2020).
- Kato, G., et al. Microglial contact prevents excess depolarization and rescues neurons from excitotoxicity. eNeuro. 3 (3), (2016).
- Li, Y., Du, X. -F., Liu, C. -S., Wen, Z. -L., Du, J. -L. Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Developmental Cell. 23 (6), 1189-1202 (2012).
- Tremblay, M. -E., Lowery, R. L., Majewska, A. K. Microglial interactions with synapses are modulated by visual experience. Plos Biology. 8 (11), 1000527(2010).
- Wake, H., Moorhouse, A. J., Jinno, S., Kohsaka, S., Nabekura, J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. The Journal of Neuroscience. 29 (13), 3974-3980 (2009).
- Oppenheim, R. W. Cell death during development of the nervous system. Annual Review of Neuroscience. 14 (1), 453-501 (1991).
- Sierra, A., Abiega, O., Shahraz, A., Neumann, H. Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Frontiers in Cellular Neuroscience. 7, 6(2013).
- Brawek, B., et al. A new approach for ratiometric in vivo calcium imaging of microglia. Scientific Reports. 7 (1), 6030(2017).
- Eichhoff, G., Brawek, B., Garaschuk, O. Microglial calcium signal acts as a rapid sensor of single neuron damage in vivo. Biochimica et Biophysica Acta. 1813 (5), 1014-1024 (2011).
- Liu, L., et al. Microglial calcium waves during the hyperacute phase of ischemic stroke. Stroke. 52 (1), 274-283 (2021).
- Pozner, A., et al. Intracellular calcium dynamics in cortical microglia responding to focal laser injury in the PC::G5-tdT reporter mouse. Frontiers in Molecular Neuroscience. 8, 12(2015).
- Umpierre, A. D., et al. Microglial calcium signaling is attuned to neuronal activity in awake mice. Elife. 9, e56502(2020).
- Ohkura, M., et al. Genetically encoded green fluorescent Ca2+ indicators with improved detectability for neuronal Ca2+ signals. Plos One. 7 (12), e51286(2012).
- Tanaka, K. F., et al. Expanding the repertoire of optogenetically targeted cells with an enhanced gene expression system. Cell Reports. 2 (2), 397-406 (2012).
- Liu, Y. U., et al. Neuronal network activity controls microglial process surveillance in awake mice via norepinephrine signaling. Nature Neuroscience. 22 (11), 1771-1781 (2019).
- Stowell, R. D., et al. Noradrenergic signaling in the wakeful state inhibits microglial surveillance and synaptic plasticity in the mouse visual cortex. Nature Neuroscience. 22 (11), 1782-1792 (2019).
- Sun, W. L., et al. In vivo two-photon imaging of anesthesia-specific alterations in microglial surveillance and photodamage-directed motility in mouse cortex. Frontiers in Neuroscience. 13, 421(2019).
- Evangelidis, G. D., Psarakis, E. Z. Parametric image alignment using enhanced correlation coefficient maximization. IEEE Transactions on Pattern Analysis and Machine Intelligence. 30 (10), 1858-1865 (2008).
- Rosario, A. M., et al. Microglia-specific targeting by novel capsid-modified AAV6 vectors. Molecular Therapy. Methods & Clinical Development. 3, 16026(2016).
- Maes, M. E., Wogenstein, G. M., Colombo, G., Casado-Polanco, R., Siegert, S. Optimizing AAV2/6 microglial targeting identified enhanced efficiency in the photoreceptor degenerative environment. Molecular Therapy. Methods & Clinical Development. 23, 210-224 (2021).
- Lin, R., et al. Directed evolution of adeno-associated virus for efficient gene delivery to microglia. Nature Methods. 19 (8), 976-985 (2022).
转载和许可
请求许可使用此 JoVE 文章的文本或图形
请求许可探索更多文章
This article has been published
Video Coming Soon
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。