
Method Article
Raum-zeitliche Analyse der Ca2+ -Aktivität von Mikroglia mit Einzelzellauflösung
In diesem Artikel
Zusammenfassung
In dieser Arbeit beschreiben wir ein Protokoll für die in vivo Bildgebung der mikroglialenCa2+ -Aktivität und die anschließende Analyse ihrer raumzeitlichen Dynamik. Diese Methode ermöglicht eine gründliche Charakterisierung, wie Mikroglia auf Veränderungen in der Gehirnumgebung reagieren, und erfasst die feinen raumzeitlichen Skalen, auf denen solche Ereignisse auftreten, angemessen.
Zusammenfassung
Mikroglia sind die einzigen ansässigen Immunzellen im zentralen Nervensystem. Ihre Morphologie ist sehr plastisch und ändert sich je nach ihrer Aktivität. Unter homöostatischen Bedingungen besitzen Mikroglia eine stark verzweigte Morphologie. Dies erleichtert ihnen die Überwachung der Umgebung durch das kontinuierliche Aus- und Einfahren ihrer Prozesse. Bei Hirnverletzungen und Entzündungen werden Mikroglia jedoch aktiviert und durchlaufen dramatische morphologische Veränderungen, wodurch sich ihre verzweigten Prozesse zurückziehen und ihr Zellkörper anschwillt. Dies erleichtert Aktivitäten wie Migration und Phagozytose, die Mikroglia unternehmen, um die Gehirnumgebung in einen weniger pathologischen Zustand zu versetzen.
Dieser enge Zusammenhang zwischen der Morphologie der Mikroglia und Veränderungen in ihrer Aktivität hat beträchtliche Einblicke in verschiedene Mikrogliafunktionen ermöglicht. Solche morphologischen und Aktivitätsänderungen sind jedoch selbst Phänomene, die aus einer Vielzahl von intrazellulären Signalwegen resultieren können. Darüber hinaus machen es die Zeitverzögerung zwischen Reiz und Reaktion sowie die stark kompartimentierte Morphologie der Mikroglia schwierig, die ursächlichen Mechanismen zu isolieren, die der Funktion zugrunde liegen. Um dieses Problem zu lösen, haben wir eine genetisch veränderte Mauslinie entwickelt, in der ein hochempfindliches fluoreszierendes Ca2+-Indikatorprotein spezifisch in Mikroglia exprimiert wird.
Nach der Beschreibung von Methoden für die in vivo mikroglialeCa2+ -Bildgebung wird in diesem Artikel ein strukturierter Analyseansatz vorgestellt, der diese Ca2+ -Aktivität in rational definierte subzelluläre Regionen einordnet und so sicherstellt, dass die räumlichen und zeitlichen Dimensionen der kodierten Informationen sinnvoll extrahiert werden. Wir glauben, dass dieser Ansatz ein detailliertes Verständnis der intrazellulären Signalregeln ermöglichen wird, die die vielfältigen Mikrogliaaktivitäten steuern, die sowohl mit höheren Gehirnfunktionen als auch mit pathologischen Zuständen verbunden sind.
Einleitung
Mikroglia sind die residenten Immunzellen im Zentralnervensystem (ZNS) und spielen eine wichtige Rolle bei der Aufrechterhaltung einer homöostatischen Gehirnumgebung und bei der Regulierung der Bildung neuronaler Schaltkreise während der Gehirnentwicklung 1,2. Ein einzigartiges Merkmal der Mikroglia im ZNS ist, dass ihre Morphologie sehr plastisch ist; Unterschiedliche morphologische Phänotypen können jedoch mit bestimmten Funktionen in Verbindung gebracht werden. Darüber hinaus ist die Transformation zwischen den morphologischen Phänotypen hochdynamisch und erfolgt auf schnellen Zeitskalen als Reaktion auf Veränderungen in der Umgebung 3,4.
Unter homöostatischen physiologischen Bedingungen nehmen Mikroglia eine stark verzweigte Morphologie an, wobei mehrere Fortsätze in alle Richtungen nach außen strahlen. Diese verzweigten Prozesse weisen selbst eine hohe Motilität auf, indem sie sich kontinuierlich aus- und zurückziehen 3,4. Eine solche Aktivität ist in erster Linie auf den periodischen Kontakt mit neuronalen Synapsen, Axonen und Somas gerichtet, um die neuronale Aktivität zu überwachen 5,6,7,8,9. Wenn jedoch das Gehirn verletzt wird, erkennen Mikroglia diese Anomalie schnell und lenken als ersten Schritt ihrer adaptiven Reaktion die Ausdehnung ihrer Prozesse auf die entsprechende Lokalisation 3,4. Dort, wo Mikroglia für die Phagozytose von toten Zellen und Metaboliten benötigt werden, nehmen sie eine amöbenähnliche Morphologie an, die ihre Fortsätze verkürzt und ihre Zellkörper vergrößert, als Teil ihres Übergangs in den immunologisch aktivierten Phänotyp 10,11.
Während die dramatischen morphologischen Veränderungen der Mikrogliaprozesse leicht zu erkennen sind, sind feinskaligere Veränderungen des Zellsomas deutlich schwieriger zu erfassen, insbesondere bei einer zeitlichen Auflösung, die physiologisch relevant ist. Darüber hinaus stellen morphologische Veränderungen selbst nur das integrierte Ergebnis einer beliebigen Anzahl intrazellulärer Signalwege dar. Dies ist problematisch für das Ziel, die funktionelle Aktivität zu verfolgen und einen Reiz mechanistisch mit der Endreaktion zu verknüpfen, die er hervorruft.
Aufgrund seiner weit verbreiteten Rolle als zweiter Botenstoff erfasst die Untersuchung der intrazellulären Ca2+-Dynamik die damit verbundenen raumzeitlichen Informationen bei der Untersuchung dynamischer Zellprozesse besser. Ein solcher Ansatz ist auf Mikroglia anwendbar, da sie eine Vielzahl von ionotropen und metabotropen Rezeptoren exprimieren, die mit der nachgeschalteten intrazellulären Ca2+-Erhöhung verbunden sind. In der Tat wurde die In-vivo-Ca2+-Bildgebung verwendet, um raumzeitliche Aspekte der Mikrogliaaktivitäten in Echtzeit zu charakterisieren und Veränderungen der Mikroglia-Ca2+-Aktivität erfolgreich mit Hirnverletzungen, Entzündungen und sowohl Hyper- als auch Hypoaktivität in Neuronen zu korrelieren 12,13,14,15,16. Zum Beispiel spiegeln Ca2+-Erhöhungen, die mit der Erweiterung des mikroglialen Prozesses als Reaktion auf hyper/hypoaktive neuronale Aktivität assoziiert sind, wahrscheinlich den zugrunde liegenden Ca2+-abhängigen Aktinpolymerisationsprozesswider 16. Darüber hinaus kann die in vivo Ca2+-Bildgebung auch gut mit pharmakologischen Ansätzen kombiniert werden. Während beispielsweise Mikroglia sowohl P2X- (ionotrope) als auch P2Y-Rezeptoren (metabotrope) exprimieren, ahmt die lokale Anwendung von P2Y-Agonisten die mikroglialeCa2+-Reaktion auf geschädigte benachbarte Neuronen nach und desensibilisiert sieanschließend 13, was die größere Relevanz der P2Y-Signalübertragung für die Erkennung neuronaler Schäden impliziert.
Bisher wurden in früheren Berichten, die die Ca2+ -Aktivität von Mikroglia untersuchten, auf Region of Interest (ROI) basierende Analysemethoden verwendet. Ein Nachteil dieser Ansätze ist, dass sie noch zu grob sind, um die raumzeitliche Dynamik der Ca2+ -Aktivität auf der Ebene einzelner Mikrogliaprozesse auflösen zu können. Daher beschreibt dieses Protokoll sowohl konventionelle ROI-basierte Methoden zur Analyse der mikroglialenCa2+ -Aktivität als auch neuere ereignisbasierte Ansätze, die einzelne Ca2+ -Ereignisse in Mikrogliaprozessen extrahieren können. Zuvor stellen wir einen allgemeinen Leitfaden für die in vivo Zwei-Photonen-Bildgebung zur Verfügung, um dieCa2+ -Aktivität der Mikroglia für eine detaillierte Analyse angemessen zu erfassen.
Protokoll
Alle Tierversuche wurden von den Tierversuchsausschüssen des National Institute for Physiological Sciences genehmigt und entsprachen den Richtlinien der National Institutes of Health. Für alle Experimente wurden 8-10 Wochen alte männliche Mäuse unter einem 12/12-stündigen Hell-Dunkel-Zyklus mit ad libitum Zugang zu Nahrung und Wasser aufgezogen. Um die Ca2+-Aktivität in Mikroglia sichtbar zu machen, wurden ionisierte Mäuse des Ca2+-Bindungsadaptermoleküls 1 (Iba1)-Tetracyclin-Transaktivator (Iba1-tTA) mit Mäusen des Tetracyclin-Operator-GCaMP6 (tetO-GCaMP6) gekreuzt17,18. In Ermangelung einer Tetracyclin-Analog-Supplementierung steuert der Iba1-Promotor die Expression von GCaMP6 ausschließlich in Mikroglia. Bei allen Experimenten wurde die Nahrungsergänzung mit Doxycyclin 6 Wochen nach der Geburt gestoppt. Am Ende aller Experimente wurden die Mäuse durch eine Überdosierung von Isofluran mit anschließender Zervixluxation euthanasiert. In der Materialtabelle finden Sie Einzelheiten zu allen Materialien, Tieren und Reagenzien, die in diesem Protokoll verwendet werden.
1. Chirurgische Vorbereitung von Mäusen für die in vivo Zwei-Photonen-Bildgebung; Tag 1
- Führen Sie alle chirurgischen Eingriffe in einem Laminarluftschrank durch, um sterile Arbeitsbedingungen zu gewährleisten. Sterilisieren Sie vor Beginn der Operation das Innere des Schranks 5 Minuten lang mit UV-Licht.
- Sterilisieren Sie alle Arbeitsflächen, den OP-Rahmen und die stereotaktischen Instrumente, indem Sie sie mit 70 % Ethanol abwischen.
- Sterilisieren Sie alle chirurgischen Instrumente (Schere, Pinzette, Rasierklinge, Pinzette) und die speziell angefertigte Kopfplatte, die am Schädel der Maus befestigt werden soll, indem Sie sie in 1%ige Chlorhexidingluconatlösung tauchen.
- Betäuben Sie die Maus mit Ketamin (7,4 mg kg-1, intraperitoneal [i.p.]) und Xylazin (10 mg kg-1, i.p.). Bringen Sie es in seinen Heimatkäfig zurück, bis die Narkose greift. Bestätigen Sie die vollständige Einleitung der Anästhesie durch Verlust des Zehenkneifreflexes.
- Sterilisieren Sie die Kopfhaut mit 1% Chlorhexidingluconat. Rasieren Sie das Fell mit einer Rasierklinge ab.
- Sichern Sie die Maus mit stereotaktischen Instrumenten innerhalb des Operationsrahmens.
- Tragen Sie Tierarztsalbe auf die Augen auf, um Trockenheit während der Narkose zu verhindern.
- Tragen Sie 2% Xylocain-Gelee zur Schmerzbehandlung auf die Kopfhaut auf. Warten Sie 5 Minuten.
- Entfernen Sie die Kopfhaut mit einer Schere und legen Sie den Schädel frei. Reinigen Sie das Periost und trocknen Sie die freiliegende Schädeloberfläche durch Reiben mit Wattestäbchen.
HINWEIS: Die freiliegenden Schädelbereiche müssen vollständig trocken sein, um eine starke Verbindung mit der maßgefertigten Kopfplatte zu gewährleisten. - Befestigen Sie die maßgefertigte Kopfplatte mit Zahnzement am Schädel.
- Sobald der Zement ausgehärtet ist, füllen Sie die Lücken zwischen der Schädeloberfläche und den Rändern der maßgefertigten Kopfplatte mit zusätzlichem Zahnzement.
- Imprägnieren Sie die Zement- und Schädeloberflächen durch Auftragen von Zahnhaftharzzement auf Acrylbasis.
- Bringen Sie die Maus wieder in ihren Heimatkäfig zurück und legen Sie sie auf ein Wärmekissen. Überwachen Sie die Maus, bis sie wieder genügend Bewusstsein erlangt, um die sternale Liege aufrechtzuerhalten (innerhalb von 2 Stunden).
HINWEIS: Die Mäuse sollten bis zum nächsten Tag vollständig genesen sein und können dann mit anderen Tieren untergebracht werden.
2. Chirurgische Vorbereitung von Mäusen für die in vivo Zwei-Photonen-Bildgebung; Tag 2
- Führen Sie alle chirurgischen Eingriffe in einem Laminarluftschrank durch, um sterile Arbeitsbedingungen zu gewährleisten. Sterilisieren Sie vor Beginn der Operation das Innere des Schranks 5 Minuten lang mit UV-Licht.
- Laminieren Sie zwei Glasdeckgläser unterschiedlicher Abmessungen (Oberglas: 3 mm × 3 mm; Unterglas: 2 mm × 2 mm) mit UV-härtendem optischem Harz.
HINWEIS: Das doppelte Deckglas bietet langfristigen Schutz für die Gehirnregion, die durch das Schädelfenster freigelegt wird, und ermöglicht gleichzeitig einen chronischen optischen Zugang. So können seine Abmessungen verändert werden, um der abzubildenden Gehirnregion gerecht zu werden. - Sterilisieren Sie alle Arbeitsflächen und den OP-Rahmen, indem Sie sie mit 70% Ethanol abwischen.
- Sterilisieren Sie alle chirurgischen Instrumente (Stahlbohrer, Pinzette, chirurgischer Nadelhaken), indem Sie sie in 1%ige Chlorhexidingluconatlösung tauchen.
- Betäuben Sie die Maus mit Isofluran (4 % Induktion, 1,2 %-1,5 % Erhaltung). Befestigen Sie die Maus über die Kopfplatte im Operationsrahmen.
- Um ein Schädelfenster über dem primären motorischen Kortex zu erstellen, markieren Sie ein Quadrat mit den Abmessungen 2 mm × 2 mm, das 0,2 mm vor und 1 mm lateral von der Landmarke des Bregma-Schädels zentriert ist.
- Dünne den Schädel entlang des Randes des markierten Quadrats mit dem Stahlbohrer aus.
HINWEIS: Wenn sie sich der gewünschten Dicke nähern, erscheinen die verdünnten schädelförmigen Bereiche transparent, wenn sie mit Kochsalzlösung benetzt werden, und es treten Haarrisse auf. - Nachdem Sie sich vergewissert haben, dass der gesamte Rand des markierten Quadrats ausreichend ausgedünnt wurde, führen Sie den chirurgischen Nadelhaken vorsichtig direkt unter der Schädeloberfläche ein und richten Sie seine Spitze in Richtung der Mitte des Quadrats aus. Hebe das quadratische Schädelstück vorsichtig mit dem Haken an und ziehe es mit einer Pinzette vom Rest des Schädels ab. Wenn Blutungen auftreten, waschen Sie die freiliegende Gehirnoberfläche kontinuierlich mit Kochsalzlösung, bis sie vollständig abklingt.
- Legen Sie das doppelte Deckglas auf die freiliegende Gehirnoberfläche und richten Sie die Seite mit dem kleineren Deckglas zum Gehirn hin aus. Stellen Sie sicher, dass die Kanten des größeren Deckglases mit den Rändern des Schädelfensters in Kontakt kommen.
- Drücken Sie mit einem silikonbestückten Glasstab, der in einem Manipulator montiert ist, vorsichtig auf das doppelte Deckglas, um sicherzustellen, dass es einen guten Kontakt mit der Gehirnoberfläche hat.
- Füllen Sie den Spalt zwischen dem doppelten Deckglas, dem Schädel und der Gehirnoberfläche mit UV-härtendem Harz und bestrahlen Sie es mit UV-Licht, bis es ausgehärtet ist (~20 s). Heben Sie den silikonbestückten Glasstab langsam vom doppelten Deckglas weg.
- Lassen Sie die Maus sich erholen und legen Sie sie auf ein Wärmekissen. Überwachen Sie die Maus, bis sie wieder genügend Bewusstsein erlangt, um die Brustbeinlage aufrecht zu erhalten (30 Minuten).
- Fahren Sie mit Schritt 3 fort oder setzen Sie die Maus in ihren Heimatkäfig zurück.
HINWEIS: Nach einer fachmännischen Operation sind die Dura und alle darunter liegenden Blutgefäße vollständig intakt und es kommt zu Blutungen. In diesem Fall ist die Entzündung minimal und es ist möglich, sofort mit Schritt 3 fortzufahren. Wenn Sie sich nicht sicher sind, warten Sie 1-3 Wochen, um sicherzustellen, dass die Entzündung vollständig abgeklungen ist.
3. Datenerhebung mittels in vivo Zwei-Photonen-Bildgebung
- Schalten Sie die Bildgebung im Voraus ein, um sicherzustellen, dass der Laser genügend Zeit zum Aufwärmen und Stabilisieren hat. Stimmen Sie den Laser so ab, dass er bei einer Wellenlänge von 920 nm emittiert, dem Zwei-Photonen-Spektrum, das das GCaMP6-Fluorophor optimal anregt.
- Positionieren Sie die Maus unter einer 25-fachen Mikroskopobjektive und gewöhnen Sie sie 30 Minuten lang. Falls erforderlich, wird die Maus während der Bildgebung mit Isofluran anästhesiert (4 % Induktion, 1,2 %-1,5 % Erhaltung).
HINWEIS: Isofluran ist für einige Forschungsanwendungen möglicherweise nicht geeignet, da es die Beweglichkeit des Mikrogliaprozesses und die Ca2+-Aktivität beeinflusst 19,20,21. - Stellen Sie den Zoom auf 1x ein und suchen Sie dann im Bereich des Schädelfensters nach Mikroglia, die GCaMP6 exprimieren. Suchen Sie in einer Tiefe zwischen 100 μm und 300 μm unter der Gehirnoberfläche.
- Sobald eine geeignete GCaMP6-exprimierende Mikroglia gefunden wurde, maximieren Sie den Zoom, so dass die Ca2+ -Aktivität mit einer Einzelzellauflösung erfasst werden kann.
- Vergewissern Sie sich visuell, dass die Ca2+ -Aktivität eindeutig erfasst wird, und passen Sie bei Bedarf die Laserleistung und die Bildverstärkung an.
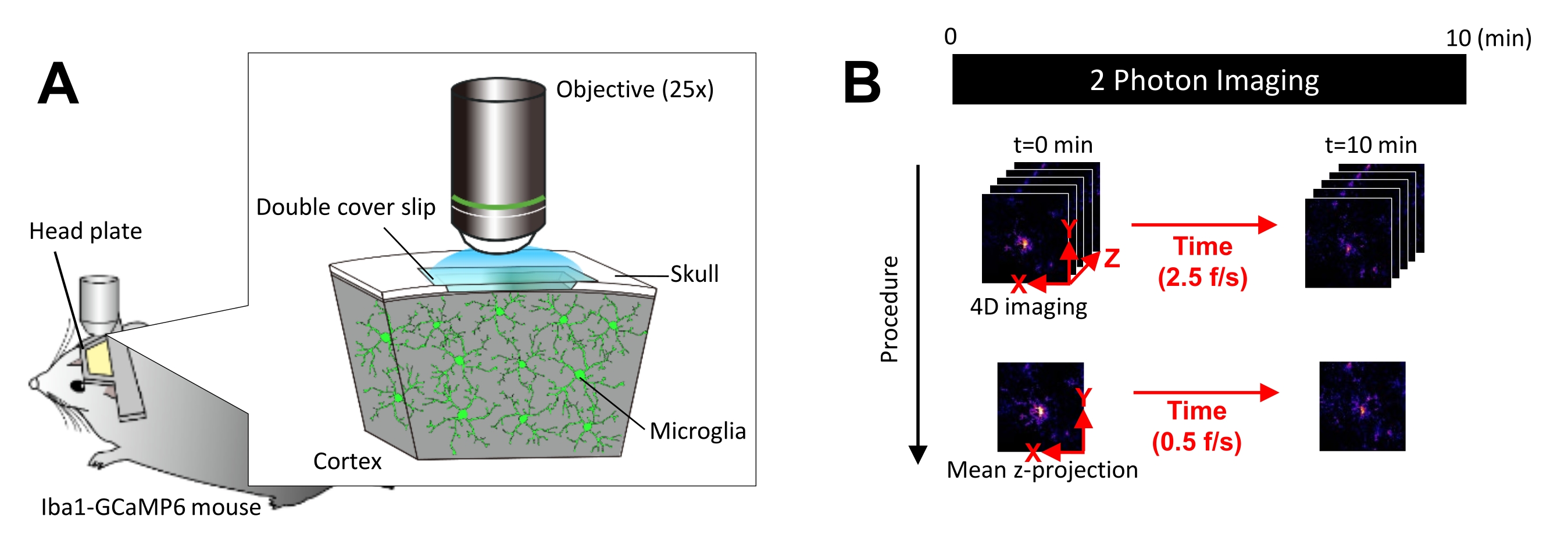
HINWEIS: Die Laserleistung sollte so weit wie möglich minimiert werden, um Photobleiche und Verletzungen der Mikroglia zu vermeiden. - Führen Sie die vierdimensionale (4D) Bilderfassung wie folgt durch (Schritte 3.6.1-3.6.3; Abbildung 1B):
- Stellen Sie die Bildabmessungen auf XY-Bereich = 512 × 512 Pixel, 0,25 μm/Pixel ein; Z-Fläche = fünf Z-Ebenen, 3 μm Z-Schritt (Abbildung 1B).
- Stellen Sie die Erfassungsrate auf 2,5 Bilder/s ein.
HINWEIS: Das Z-Scannen bei dieser Geschwindigkeit wird durch ein Piezo-Nano-Positioniersystem erleichtert. - Erfassen Sie Daten, bis mindestens 10 einzelne Ca2+ -Ereignisse beobachtet werden (in der Regel 10 Minuten).
4. Vorbereitung für die Analyse (Bewegungskorrektur, mittlere/maximale Z-Projektion)
- Bevor Sie mit der Analyse der Ca2+ -Aktivität der Mikroglia fortfahren, korrigieren Sie die 4D-Bilder auf bewegungsbezogene Artefakte (Bildregistrierung) mit dem ECC-Bildausrichtungsalgorithmus22 in der MATLAB-Programmierumgebung (R2020a). Laden Sie den Code herunter unter: https://www.mathworks.com/matlabcentral/fileexchange/27253-ecc-image-alignment-algorithm-image-registration.
- Erstellen Sie ein 3D-Referenzbild, indem Sie alle Bilder innerhalb des ersten Zeitrahmens mit der MATLAB-Operation imregister (Standard Image Processing Toolbox) registrieren.
- Registrieren Sie alle nachfolgenden Bilder und gleichen Sie sie mit der entsprechenden Z-Ebene des Referenz-3D-Bildes mit der Funktion ecc (ECC Image Alignment Algorithm Library) ab.
- Generieren Sie Z-Projektionen der mittleren Intensität aus den registrierten Bildern mit der mittleren MATLAB-Operation. Wenn das Bildsignal zu schwach ist, generieren Sie stattdessen Z-Projektionen mit maximaler Intensität mit der maximalen MATLAB-Operation.
HINWEIS: Das Signal-Rausch-Verhältnis ist schlechter, wenn Z-Projektionen mit maximaler Intensität verwendet werden. - Fahren Sie mit Schritt 5 oder 6 fort. Verwenden Sie die in Schritt 4 generierten Z-Projektionen als Analyseziel, um die raumzeitliche Dynamik der Ca2+ -Aktivität in den Schritten 5 und 6 zu kartieren.
5. ROI-basierte Analyse
- Identifizieren Sie unter Verwendung der in Schritt 4 generierten Z-Projektionen Mikrogliaprozesse, die während des gesamten Bildgebungszeitraums ein stabiles Flächenprofil (stabiler Bereich) für die anschließende Analyse der Ca2+ -Aktivität wie folgt aufrechterhalten (Schritte 5.3-5.6):
- Generieren Sie separate t-Projektionen mit maximaler Intensität der registrierten 4D-Bilder aus 2-minütigen Proben, die zu Beginn und am Ende des Bildgebungszeitraums mit der maximalen MATLAB-Operation aufgenommen wurden.
- Binarisieren Sie die t-Projektionen, um Polygone der Mikroglia-Morphologie zu erzeugen, die dem Beginn und Ende der Bildgebungsperiode entsprechen, indem Sie die imbinarize MATLAB-Operation verwenden. Verwenden Sie den automatisch festgelegten Standardschwellenwert.
HINWEIS: Wenn das Ca2+ -Signal zu Beginn und am Ende des Bildgebungszeitraums schwach ist, kann die Binarisierung Polygone mit fehlenden Rändern erzeugen. Zeichnen Sie in diesem Fall die fehlenden Ränder manuell mit dem Bleistiftwerkzeug in ImageJ. - Überlagern Sie die t-Projektionspolygone mit der MATLAB-Operation imadd . Die überlappenden Bereiche stellen den stabilen Bereich dar (Abbildung 2B).
- Verwenden Sie innerhalb der identifizierten stabilen Bereiche die MATLAB-Operation drawpolygon , um ROIs für jeden der primären Mikrogliaprozesse und alle offensichtlichen Unterzweige zweiter Ordnung manuell zu definieren und zu verfolgen.
- Verfolgen Sie die absoluten Fluoreszenzintensitäten, gemittelt über einen individuellen ROI über alle Zeiträume (Abbildung 2C,D).
- Berechnen Sie aus der Zeitreihe der absoluten Fluoreszenzintensitäten die relative Änderung der Zeitreihe der Fluoreszenzintensität (ΔF/F) gemäß Gleichung (1). Diese Zeitreihe repräsentiert die normierte mikrogliale Ca2+ -Dynamik auf der Ebene der individuellen ROIs.
ΔF/F = (F(t) - F0) / F0 (1)
Dabei ist F(t) die aufgezeichnete Fluoreszenzintensität zu einem bestimmten Zeitpunkt und F0 das 10. Perzentil der Fluoreszenzintensität in allen Zeiträumen (Abbildung 2E). - Identifizieren Sie möglicheCa-2+ -Auslöseereignisse, die in einem bestimmten individuellen ROI auftreten, wie folgt (Schritte 5.5.1-5.5.3):
- Wenden Sie die Tiefpassfilterung vom Typ I mit endlicher Impulsantwort (FIR) auf die ΔF/F-Zeitreihe mit der MATLAB-Operation fir1 an. Stellen Sie die Grenzfrequenz auf den Nyquist-Wert (die Hälfte der Abtastrate) ein.
- Überprüfen Sie die gefilterte Spur visuell, um zu bestätigen, dass die einzelnen Kurvenspitzen den Ausbrüchen der Ca2+ -Aktivität in den registrierten 4D-Bildern entsprechen (Abbildung 2F).
- Identifizieren Sie möglicheCa-2+-Auslöseereignisse als konkave Flexionen in derΔF/F-Zeitreihenkurve. Definieren Sie einen Basisschwellenwert als den Median ΔF/F innerhalb der oberen und unteren Obergrenzen, die die maximalen und minimalen Amplituden von 10 % über alle Zeitrahmen ausschließen. Definieren Sie einen Erkennungsschwellenwert von drei SDs über dem Basisschwellenwert (Abbildung 2F,H).
- Identifizieren Sie echteCa-2+ -Auslöseereignisse von Kandidaten wie folgt (Schritte 5.5.1-5.5.2):
- Berechnen Sie die Steigung jedes Kandidatenereignisses, indem Sie mithilfe der numerischen Gradienten-MATLAB-Operation über die entsprechenden gefilterten ΔF/F-Zeitreihenrahmen differenzieren.
- Identifizieren Sie anschließend echte Ca2+-Ereignisse basierend auf der Anstiegszeit des Profils. Definieren Sie einen Basisschwellenwert als Durchschnitt der Steigungswerte über alle Kandidatenereignisse hinweg. Definieren Sie einen Erkennungsschwellenwert von drei SDs über dem Basisschwellenwert (Abbildung 2G,H).
- Charakterisieren Sie echte Ca2+ -Ereignisse wie folgt (Schritte 5.6.1-5.6.3):
HINWEIS: Im Folgenden finden Sie keine vollständige Liste von Parametern, die zur Charakterisierung von mikroglialen Ca2+ -Ereignissen verwendet werden können. Die interessierenden Parameter hängen vom Zweck der Studie ab.- Leiten Sie die maximale Amplitude eines echten Ca2+ -Auslöseereignisses als ΔF/F-Wert des entsprechenden ΔF/ F-Zeitreihenrahmens ab.
- Leiten Sie die mittlere Amplitude eines echten Ca2+ -Auslöseereignisses als durchschnittlichen ΔF/F-Wert über die gesamte entsprechende ΔF/ F-Zeitreihenteilmenge von Frames ab.
- Leiten Sie die Häufigkeit echter Ca2+ -Ereignisse als Anzahl der Ereignisse geteilt durch die Imaging-Zeit ab.
6. Ereignisbasierte Analyse
- Führen Sie eine ereignisbasierte Analyse der Z-Projektionen aus Schritt 4 mit der AQuA-Bibliothek in der MATLAB-Programmierumgebung durch. Laden Sie den Code herunter von: https://github.com/yu-lab-vt/AQuA.
HINWEIS: Eine exemplarische Vorgehensweise zur allgemeinen Verwendung der AQuA-Bibliothek finden Sie unter: https://drive.google.com/file/d/1a3lhe0dUth-5J1-S2fZlPOCZlPbeuvUr/view. Eine ausführliche Dokumentation zur AQuA-Bibliothek finden Sie unter: https://drive.google.com/file/d/1CckDLbrkw16b7MPlOQdYpZciIz80Snm_/view. - Wechseln Sie nach dem Start von MATLAB mit der cd-Operation vom Standard-Arbeitsverzeichnisordner zum von AQuA angegebenen Arbeitsverzeichnisordner.
- Laden Sie die registrierten 4D-Bilder wie folgt in die AQuA-Analyse-Pipeline (Schritte 6.3.1-6.3.2):
- Starten Sie die AQuA-GUI. Geben Sie aqua_gui in das MATLAB-Befehlsfenster in den von AQuA angegebenen Arbeitsverzeichnisordner ein.
- Klicken Sie in der AQuA-GUI auf Neues Projekt und wählen Sie die registrierten Bilder aus, die analysiert werden sollen. Geben Sie den Datentyp (GCaMPInVivo_cyto_Lck_) und die Bildgebungsparameter an (Zeitauflösung Sekunden pro Frame = 1,993; räumliche Auflösung μm pro Pixel = 0,25; Pixel ausschließen, die kürzer als dieser Abstand zum Rand sind = 5). Klicken Sie auf Öffnen , um die Daten zu laden.
- Definieren Sie Orientierungspunkte für die nachfolgenden Analysepipelines, indem Sie Interessenbereiche nachzeichnen. In der Regel basieren Landmarken auf der Zellgrenze und der Fläche des Zellkörpers.
- Erkennen Sie anschließend möglicheCa-2+ -Ereignisse, indem Sie die automatisierten Analysepipelines für die folgenden Parameter ausführen: aktives Signal, Supervoxel, Ereigniserkennung, saubere Ereignisse und Merge-Ereignisse.
HINWEIS: Eine ausführliche Erläuterung zu jedem dieser Parameter und ihren Ausgaben für die Kandidatenbewertung finden Sie in der exemplarischen Vorgehensweise für AQuA. Kurz gesagt, diese Parameter passen den Schwellenwert für die Ereignisdetektion Ca2+ wie folgt an: aktives Signal = Fluoreszenzamplitude, Supervoxel = Clustering von Fluoreszenz im 3D-Raum, Ereigniserkennung = Anstiegs-/Abklingkinetik, saubere Ereignisse = Signal-Rausch-Verhältnis und Merge-Ereignisse = zeitliche Trennung von Ereignissen. - Visuelle Überprüfung der erkannten Ca2+ -Ereignisse (automatisch überlagert mit den ursprünglich registrierten Bildern). Passen Sie bei Bedarf die Parametereinstellungen der Analysepipelines an, indem Sie die Bildqualität in Bezug auf die oben genannten Parameter berücksichtigen. Wenn die Bildqualität für einen bestimmten Parameter gut ist, kann ein höherer Schwellenwert festgelegt werden und umgekehrt.
- Nachdem alle Ca2+ -Ereignisse ordnungsgemäß erkannt wurden, charakterisieren Sie diese Ereignisse in der MATLAB-Hauptumgebung wie folgt (Schritte 6.8-6.14):
- Exportieren Sie die Ausgabedateien der AQuA-Analyse.
HINWEIS: Eine detaillierte Erläuterung der Ausgabedateien, der extrahierten Features, die zum Definieren von Ca2+ -Ereignissen verwendet werden, und der zugrunde liegenden Parameter dieser extrahierten Features finden Sie in der AQuA-Dokumentation. - (fakultativ) Kategorisieren Sie alle Ca2+ -Ereignisse in zwei Gruppen: 1) Ereignisse, die mit dem Soma beginnen, und 2) Ereignisse, die mit den Prozessen beginnen.
- Greifen Sie auf die Amplitude einzelner Ca2+ -Ereignisse innerhalb der res.dffMat MATLAB-Struktur der AQuA-Analyseausgabedatei (.mat-Datei) zu.
- Leiten Sie die Häufigkeit einzelner Ca2+ -Ereignisse mit der res.fts.loc.x3D-MATLAB-Operation (AQuA-Bibliothek) ab.
- Leiten Sie die Dauer einzelner Ca2+ -Ereignisse mit der MATLAB-Operation res.fts.curve.width11 (AQuA-Bibliothek) ab.
- Leiten Sie den Bereich einzelner Ca2+ -Ereignisse mit der MATLAB-Operation res.fts.basic.area (AQuA-Bibliothek) ab.
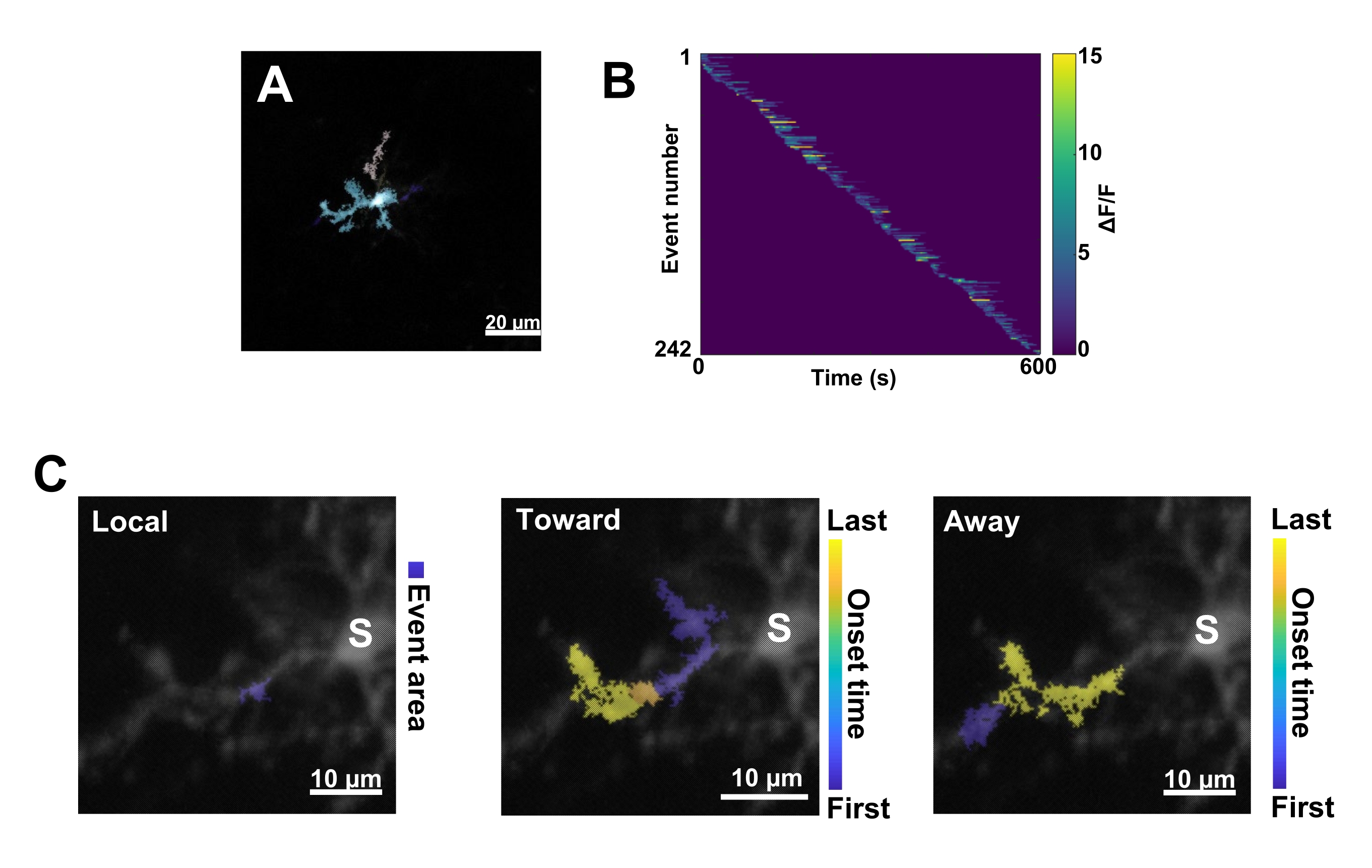
- Kategorisieren Sie alle Ca2+ -Ereignisse als: 1) lokale Ereignisse, 2) fortpflanzungsfähige Ereignisse, die sich auf das Soma zubewegen, und 3) fortlaufende Ereignisse, die sich vom Soma weg bewegen. Verwenden Sie dazu die MATLAB-Operationen res.fts.region.landmarkDir.chgToward und res.fts.region.landmarkDir.chgAway (AQuA-Bibliothek) (Abbildung 3C).
Ergebnisse
Bei transgenen Mäusen, die ausschließlich GCaMP6 (Ca2+-empfindliches fluoreszierendes Protein) in Mikroglia exprimieren, beobachten wir typischerweise unterschiedliche Muster der Mikroglia-Ca2+ -Aktivität (Abbildung 2A). Wichtig ist, dass sich selbst innerhalb einer einzelnen Mikroglia die Muster der Ca2+ -Aktivität zwischen den Prozessen dramatisch unterscheiden können.
Um solche Prozess-zu-Prozess-Unterschiede in der räumlich-zeitlichen Dynamik der mikroglialen Ca2+-Aktivität zu quantifizieren, müssen zunächst stabile Bereiche identifiziert und dann in fein segmentierte ROIs unterteilt werden (Abbildung 2B,C). Für jeden ROI müssen Parameter der Ca2+-Aktivität abgeleitet und quantifiziert werden, wie z. B. Amplitude und Frequenz, indem Merkmale wie lokale Amplituden und Spurensteigungen aus der Zeitreihe der Fluoreszenzintensität extrahiert werden (Abbildung 2D-G).
Als nächstes müssen einzelne Ca2+ -Ereignisse unter Anwendung des genauen Quantifizierungsalgorithmus AQuA untersucht werden (Abbildung 3A). Bei solchen ereignisbasierten Analysen werden in der Regel große Unterschiede in den Eigenschaften von Ursprung, Amplitude, Dauer, Ort und Strömungsrichtung einzelner Ca2+ -Ereignisse beobachtet (Abbildung 3B). Wenn man sich auf die Analyse der Ca2+ -Aktivitätsdynamik in Mikrogliaprozessen konzentriert, ist ein Klassifikationsschema von lokalen Ereignissen, Ereignissen, die sich in Richtung des Somas bewegen, und Ereignissen, die sich vom Soma weg bewegen, informativ (Abbildung 3C).

Abbildung 1: Versuchsaufbau für die in vivo MikrogliaCa 2+ Bildgebung. (A) Versuchsaufbau. Eine Iba1-tTA × tetO-GCaMP6-Maus mit Mikroglia-spezifischer GCaMP6-Expression. Durch das Einsetzen eines Schädelfensters in den Schädel der Maus kann die mikroglialeCa2+ -Aktivität in vivo mit Hilfe der Zwei-Photonen-Mikroskopie beobachtet werden. (B) Versuchsplan und Analyseverfahren. 4D-Bilder werden mindestens 10 Minuten lang als Z-Stapel mit fünf Bildern aufgenommen. Die Bildaufnahmerate beträgt 2,5 Bilder/s. Vor der Analyse der Ca2+ -Aktivität der Mikroglia werden die Fünf-Frame-Z-Stacks in 2D-Z-Projektionen umgewandelt, indem die durchschnittliche (oder maximale) Intensität genommen wird. Die Wiedergaberate der Z-Projektion beträgt 0,5 Bilder/s. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2: ROI-basierte Analyse der mikroglialenCa2+ -Aktivität. (A) Mittlere GCaMP6-Intensitätsprojektion über 10 min für eine einzelne Mikroglia. (B) Stabile Bereiche (weiß) werden durch eine Überlagerung von binarisierten, maximalen GCaMP6-Intensität t-Projektionen definiert, die von 2-minütigen Proben abgeleitet werden, die zu Beginn (magenta) und am Ende (grün) einer Bildgebungsperiode entnommen wurden. (C) Stabile Gebiete werden weiter in regionale ROIs unterteilt. Einzelne Farben zeigen individuelle ROIs an. (D) ΔF/F-Spuren aller einzelnen ROIs in C. Beachten Sie die Unterschiede in den Aktivitätsmustern zwischen den ROIs. (E) Ursprüngliche Spur einer ΔF/ F-Zeitreihe, abgeleitet aus absoluten Intensitätswerten für einen einzelnen ROI. (F) Die gleiche ΔF/F-Zeitreihe nach der Tiefpassfilterung. Kandidaten-Ca-2+ -Ereignisse werden durch einen Amplituden-Cut-off-Schwellenwert (rote Linie) erkannt, der als Baseline + drei SDs definiert ist. Die Basislinie (grüne Linie) ist definiert als der Medianwert über die gesamte ΔF/F-Zeitreihe innerhalb einer oberen und unteren Obergrenze, die die maximalen und minimalen 10 % der ΔF/F-Werte ausschließt. (G) Die Steigungskurve, die aus der gefilterten ΔF/F-Zeitreihe in F abgeleitet wurde. EchteCa-2+ -Ereignisse werden von Kandidaten-Ca-2+ -Ereignissen auf der Grundlage eines Slope-Cut-off-Schwellenwerts (rote Linie) sortiert, der als Baseline + drei SDs definiert ist. Die Basislinie (grüne Linie) ist definiert als der Durchschnittswert über die gesamte Neigungszeitreihe innerhalb einer oberen und unteren Obergrenze, die die maximalen und minimalen 10 % der Neigungswerte ausschließt. (H) Kandidaten-Ca-2+ -Ereignisse, die durch die Amplitudenkriterien in F identifiziert wurden, sind orange gekennzeichnet. EchteCa-2+ -Ereignisse, sortiert von Kandidaten-Ca-2+ -Ereignissen nach Steigungskriterien in G in Rot. Beachten Sie, dass einige Kandidaten fürCa-2+ -Veranstaltungen basierend auf den Neigungskriterien zusammengeführt wurden. Die entsprechende gefilterte ΔF/F-Zeitreihe wird unten als Referenz überlagert. Die schwarze Linie zeigt die Amplitude Null (ΔF/F) an. Die mittlere und maximale Amplitude echter Ca2+ -Ereignisse wird als Mittelwert und Maximum ihrer entsprechenden Peaks in der gefilterten ΔF/F-Zeitreihe abgeleitet. Die Häufigkeit (Ereignisse/min) wird abgeleitet als die Anzahl der echten Ca2+ -Zündereignisse dividiert durch den Bildgebungszeitraum (10 min). Maßstabsleisten = 20 μm (A,B), 10 μm (C). Abkürzung: ROI = Region of Interest. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 3: Ereignisbasierte Analyse der mikroglialenCa2+-Aktivität. (A) Repräsentative Bilder von Ereignissen, die mit dem AQuA-Algorithmus erkannt wurden. Die Farben kennzeichnen einzelne Ereignisbereiche, die zu einem bestimmten Zeitpunkt erkannt wurden. (B) Repräsentative normierte Ca2+-Aktivität (ΔF/F) in einzelnen Ereignissen, sortiert nach der Reihenfolge ihres Auftretens. Der rechte Balken zeigt farblich gekennzeichnetes ΔF/F an. (C) Repräsentative Aktivitäts-Fußabdrücke von Ereignissen, die sich auf das Soma oder lokale Ereignisse hin und von diesem weg ausbreiten. Bei lokalen Ereignissen wird die Ereignisform blau angezeigt. Bei Fortpflanzungsereignissen wird der Zeitpunkt des Auftretens des Ereignisses durch die blau-gelbe Skala angezeigt. Da AQuA Ca2+-Ereignisse zunächst einzeln erkennt, werden später propagative Ereignisse auf der Grundlage der überlappenden räumlichen Orte und Zeitreihen mehrerer einzelner Ca2+-Ereignisse identifiziert. Beachten Sie, dass es sich hierbei um denselben dendritischen Zweig handelt, der für die ROI-Analyse in Abbildung 2E-H verwendet wird. Maßstabsleisten = 20 μm (A), 10 μm (C). Abkürzung: ROI = Region of Interest; S = Soma. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Diskussion
In dieser Arbeit wird ein verbesserter Ansatz für die Abbildung der mikroglialenCa2+ -Aktivität mit hoher räumlich-zeitlicher Auflösung vorgestellt. Diese Methode ist empfindlich genug, um verschiedene Arten von mikroglialerCa2+ -Aktivität auf der Ebene einzelner verzweigter Prozesse zu detektieren und dabei leicht zwischen lokalen und fortpflanzungsfähigen Ereignissen zu unterscheiden.
Bei der allgemeinen Methode zur in vivo Zwei-Photonen-Bildgebung der Mikroglia-Ca2+ -Aktivität muss die folgenden Punkte sorgfältig beachtet werden, um die Bildqualität zu maximieren. Da Mikroglia extrem empfindlich auf Verletzungen reagieren, ist es wichtig, die direkte Berührung der Gehirnoberfläche mit chirurgischen Instrumenten während der Operation zu minimieren. Wichtige Anzeichen dafür, dass die Operation kompetent durchgeführt wurde, sind intakte Blutgefäße und Dura sowie sehr geringe Blutungen während der Operation. Zweitens reduzieren die sichere Befestigung der Kopfplatte am Schädel der Maus und der gute Kontakt zwischen dem doppelten Deckglas und der Gehirnoberfläche bewegungsbedingte Artefakte während der Bildgebung erheblich. Dies ist besonders wichtig bei der Bildgebung mit hoher raumzeitlicher Auflösung und bei vollständig wachen Mäusen. Während die Analysepipeline bewegungsbedingte Artefakte, die sich aus dem Herzschlag, der Atmung und der allgemeinen Drift ergeben, zuverlässig kompensiert, ist sie weniger robust, wenn sie signifikante geometrische Verzerrungen verarbeitet, die durch plötzliche große Bewegungen entstehen.
Die beiden hier beschriebenen Analysemethoden bieten unterschiedliche Vorteile und eignen sich für unterschiedliche Forschungsfragen. Bei der ROI-basierten Analyse definiert der Benutzer den ROI (z. B. einzelne Prozesse) vor, so dass die aggregierte Dynamik der Ca2+ -Aktivität dieses ROI extrahiert werden kann. Daher eignet es sich am besten für Situationen, in denen erwartet wird, dass Phänomene in einem subzellulären Bereich lokalisiert sind, der sowohl gut definierte morphologische Grenzen als auch einen relativ großen Bereich (d.h. einen Prozesszweig) aufweist. Bei der ereignisbasierten Analyse werden einzelne Ereignisse basierend auf der raumzeitlichen Dynamik der Mikrogliaaktivität Ca2+ definiert und müssen dann in den Kontext von benutzerdefinierten Landmarken innerhalb der Mikroglia gestellt werden, damit ihre Funktion interpretiert werden kann. Daher eignet es sich am besten für Situationen, in denen keine Annahmen über die Lokalisierung von Phänomenen getroffen werden können oder in denen der Interessenbereich relativ klein ist (z. B. eine Prozessspitze). Daher bietet die ereignisbasierte Analyse im Vergleich zu früheren Methoden eine verbesserte räumlich-zeitliche Auflösung bei der Charakterisierung der Ca2+ -Aktivität von Mikroglia.
Bei diesen Mäusen ist der einzige fluoreszierende Marker, der von Mikroglia exprimiert wird, der Ca2+-Indikator GCaMP6. In Regionen, in denen die Ca2+-Aktivität gering ist, muss die Mikroglia-Morphologie durch Kombination mehrerer Zeitrahmen extrahiert werden, was die zeitliche Auflösung beeinträchtigen kann. Diese Einschränkung kann jedoch überwunden werden, indem ein separates rotes, stabil fluoreszierendes Protein in Mikroglia exprimiert wird. Insbesondere wurden kürzlich neuartige Adeno-assoziierte Viren beschrieben, die in der Lage sind, Mikroglia zu transfizieren 23,24,25.
Wie dieCa2+ -Aktivität der Mikroglia durch die Umgebung verändert wird, ist ein aufstrebendes Thema. Insbesondere scheint die mikroglialeCa2+ -Aktivität signifikante Korrelationen mit der neuronalen Aktivität zu zeigen, obwohl die funktionelle Bedeutung dieser Aktivität noch nicht vollständig charakterisiert wurde. Die Kombination der Manipulation der neuronalen Aktivität mit den hier vorgestellten bildgebenden und analytischen Methoden für die Ca2+ -Aktivität der Mikroglia sollte daher neue Einblicke in die Physiologie der Mikroglia liefern und unser Verständnis der Rolle, die Mikroglia in physiologischen und pathologischen Zuständen spielen, weiter verbessern.
Offenlegungen
Die Autoren erklären, dass mit diesem Manuskript keine Interessenkonflikte verbunden sind.
Danksagungen
Wir danken Prof. Kenji Tanaka (Keio University, Tokyo, Japan) für die Bereitstellung von Iba1-tTA-Mäusen und tetO-GCaMP6-Mäusen. Diese Arbeit wurde unterstützt durch Grants-in-Aid for Young Scientists (B) [16K19001 (an H.H.)], Grants-in-Aid for Early-Career Scientists [18K14825 (an H.H.)], Grant-in-Aid for Scientific Research (B) [21H03027 (an H.H.)], Grant-in-Aid for Transformative Research Areas (A) [21H05639 (an H.H.)], Grant-in-Aid for Scientific Research (A) [17H01530, 20H00500 (an J.N.)] und JST CREST Grant [JPMJCR1755 (an J.N.)], Japan.
Materialien
| Name | Company | Catalog Number | Comments |
| 2% xylocaine jelly | AstraZeneca, UK | ||
| B6(129S6)-Tg(Aif1-tTA)54Kftnk | RIKEN RBC, Japan | RBRC05769 | Iba1-tTA mice |
| B6;129-Actb(tm3.1(tetO-GCaMP6)Kftnk) | RIKEN RBC, Japan | RBRC09552 | tetO-GCaMP6 mice |
| Forceps | Fine science tools, US | 13008-12 | |
| G-CEM ONE | GC corporation, Japan | ||
| Glass capillary | Narishige, Japan | GDC-1 | |
| ImageJ | NIH, US | ||
| Isofulrane | Pfizer, US | ||
| Ketamin | Daiichi-Sankyo, Japan | ||
| Kwik-sil | World Precision Instruments, US | KWIK-SIL | |
| MATLAB, 2020a | MathWorks, US | ||
| Micro cover glass (2 x 2 mm, No.3) | Matsunami, Japan | custum-made | Bottom glass for cranial window |
| Micro cover glass (3 x 3 mm, No.0) | Matsunami, Japan | custum-made | Upper glass for cranial window |
| N25X-APO-MP | Nikon, Japan | N25X-APO-MP | Objective lens (25x) |
| Norland optical adhesive | Edmund optics, US | 6101 | |
| Piezo nano-positioning system, Nano-Drive | Mao City Labs, US | ||
| Razor blade | Feather, Japan | FA-10 | |
| Scissors | Fine science tools, US | 14060-11 | |
| Steel drill | Minitor, Japan | BS1201 | |
| Stereotaxic instruments | Narishige, Japan | SR-5M-HT | |
| Super-bond (C&B kit) | Sun Medical, Japan | 4560227797382 | |
| Surgical needle hook | Fine science tools, US | 10065-15 | |
| Ti:Sappire laser, MaiTai DeepSee | Spectra Physics, US | Mai Tai eHP DS | |
| Tweezers | Fine science tools, US | 11051-10 | |
| Tweezers | Fine science tools, US | 11255-20 | |
| Two-photon microscope | Nikon, Japan | A1R-MP | |
| UV craft resin | Kiyohara, Japan | UVR | |
| Xylazine | Bayer, Germany |
Referenzen
- Miyamoto, A., et al. Microglia contact induces synapse formation in developing somatosensory cortex. Nature Communications. 7, 12540 (2016).
- Schafer, D. P., et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent. Neuron. 74 (4), 691-705 (2012).
- Davalos, D., et al. ATP mediates rapid microglial response to local brain injury in vivo. Nature Neuroscience. 8 (6), 752-758 (2005).
- Nimmerjahn, A., Kirchhoff, F., Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 308 (5726), 1314-1318 (2005).
- Cserep, C., et al. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science. 367 (6477), 528-537 (2020).
- Kato, G., et al. Microglial contact prevents excess depolarization and rescues neurons from excitotoxicity. eNeuro. 3 (3), (2016).
- Li, Y., Du, X. -. F., Liu, C. -. S., Wen, Z. -. L., Du, J. -. L. Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Developmental Cell. 23 (6), 1189-1202 (2012).
- Tremblay, M. -. E., Lowery, R. L., Majewska, A. K. Microglial interactions with synapses are modulated by visual experience. Plos Biology. 8 (11), 1000527 (2010).
- Wake, H., Moorhouse, A. J., Jinno, S., Kohsaka, S., Nabekura, J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. The Journal of Neuroscience. 29 (13), 3974-3980 (2009).
- Oppenheim, R. W. Cell death during development of the nervous system. Annual Review of Neuroscience. 14 (1), 453-501 (1991).
- Sierra, A., Abiega, O., Shahraz, A., Neumann, H. Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Frontiers in Cellular Neuroscience. 7, 6 (2013).
- Brawek, B., et al. A new approach for ratiometric in vivo calcium imaging of microglia. Scientific Reports. 7 (1), 6030 (2017).
- Eichhoff, G., Brawek, B., Garaschuk, O. Microglial calcium signal acts as a rapid sensor of single neuron damage in vivo. Biochimica et Biophysica Acta. 1813 (5), 1014-1024 (2011).
- Liu, L., et al. Microglial calcium waves during the hyperacute phase of ischemic stroke. Stroke. 52 (1), 274-283 (2021).
- Pozner, A., et al. Intracellular calcium dynamics in cortical microglia responding to focal laser injury in the PC::G5-tdT reporter mouse. Frontiers in Molecular Neuroscience. 8, 12 (2015).
- Umpierre, A. D., et al. Microglial calcium signaling is attuned to neuronal activity in awake mice. Elife. 9, e56502 (2020).
- Ohkura, M., et al. Genetically encoded green fluorescent Ca2+ indicators with improved detectability for neuronal Ca2+ signals. Plos One. 7 (12), e51286 (2012).
- Tanaka, K. F., et al. Expanding the repertoire of optogenetically targeted cells with an enhanced gene expression system. Cell Reports. 2 (2), 397-406 (2012).
- Liu, Y. U., et al. Neuronal network activity controls microglial process surveillance in awake mice via norepinephrine signaling. Nature Neuroscience. 22 (11), 1771-1781 (2019).
- Stowell, R. D., et al. Noradrenergic signaling in the wakeful state inhibits microglial surveillance and synaptic plasticity in the mouse visual cortex. Nature Neuroscience. 22 (11), 1782-1792 (2019).
- Sun, W. L., et al. In vivo two-photon imaging of anesthesia-specific alterations in microglial surveillance and photodamage-directed motility in mouse cortex. Frontiers in Neuroscience. 13, 421 (2019).
- Evangelidis, G. D., Psarakis, E. Z. Parametric image alignment using enhanced correlation coefficient maximization. IEEE Transactions on Pattern Analysis and Machine Intelligence. 30 (10), 1858-1865 (2008).
- Rosario, A. M., et al. Microglia-specific targeting by novel capsid-modified AAV6 vectors. Molecular Therapy. Methods & Clinical Development. 3, 16026 (2016).
- Maes, M. E., Wogenstein, G. M., Colombo, G., Casado-Polanco, R., Siegert, S. Optimizing AAV2/6 microglial targeting identified enhanced efficiency in the photoreceptor degenerative environment. Molecular Therapy. Methods & Clinical Development. 23, 210-224 (2021).
- Lin, R., et al. Directed evolution of adeno-associated virus for efficient gene delivery to microglia. Nature Methods. 19 (8), 976-985 (2022).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten