
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Bestimmung der relativen Zelloberfläche und Total Expression rekombinanter Ionenkanäle Mit Flow Cytometry
In diesem Artikel
Zusammenfassung
Ererbten Arrhythmien werden häufig durch Mutationen verursacht, die die Oberfläche Lieferung eines oder mehrerer Ionenkanäle verändern. Hier passen wir die Durchflusszytometrie-Assays eine Quantifizierung der relativen Gesamt und Zelloberflächenproteinexpression von rekombinanten Ionenkanäle in tsa-201-Zellen exprimiert zu liefern.
Zusammenfassung
Inherited or de novo mutations in cation-selective channels may lead to sudden cardiac death. Alteration in the plasma membrane trafficking of these multi-spanning transmembrane proteins, with or without change in channel gating, is often postulated to contribute significantly in this process. It has thus become critical to develop a method to quantify the change of the relative cell surface expression of cardiac ion channels on a large scale. Herein, a detailed protocol is provided to determine the relative total and cell surface expression of cardiac L-type calcium channels CaV1.2 and membrane-associated subunits in tsA-201 cells using two-color fluorescent cytometry assays. Compared with other microscopy-based or immunoblotting-based qualitative methods, flow cytometry experiments are fast, reproducible, and large-volume assays that deliver quantifiable end-points on large samples of live cells (ranging from 104 to 106 cells) with similar cellular characteristics in a single flow. Constructs were designed to constitutively express mCherry at the intracellular C-terminus (thus allowing a rapid assessment of the total protein expression) and express an extracellular-facing hemagglutinin (HA) epitope to estimate the cell surface expression of membrane proteins using an anti-HA fluorescence conjugated antibody. To avoid false negative, experiments were also conducted in permeabilized cells to confirm the accessibility and proper expression of the HA epitope. The detailed procedure provides: (1) design of tagged DNA (deoxyribonucleic acid) constructs, (2) lipid-mediated transfection of constructs in tsA-201 cells, (3) culture, harvest, and staining of non-permeabilized and permeabilized cells, and (4) acquisition and analysis of fluorescent signals. Additionally, the basic principles of flow cytometry are explained and the experimental design, including the choice of fluorophores, titration of the HA antibody and control experiments, is thoroughly discussed. This specific approach offers objective relative quantification of the total and cell surface expression of ion channels that can be extended to study ion pumps and plasma membrane transporters.
Einleitung
Dieses Dokument bietet einen zuverlässigen Assay, um die relative Zelloberflächenexpression der Membranproteine wie Ionenkanäle exprimiert in rekombinanten Zellen unter Verwendung der bestehenden Technologie Durchflusszytometrie zu melden. Ionenkanäle sind porenbildende Membranproteine, die durch Gating den Fluss von Ionen durch die Zellmembran zum Steuern elektrischer Signale verantwortlich sind. Sie werden durch den Aktivierungsmechanismus, der Natur klassifiziert und Selektivität der Ionenspezies durch die Pore queren, wo sie lokalisiert sind. Auf zellulärer und Gewebespiegel, die makroskopischen Ionenflüsse durch Ionenkanäle sind das Produkt von biophysikalischen (Gating und Permeation), biochemische (Phosphorylierung) und Biogenese (Synthese, Glykosylierung, Menschenhandel und Abbau) Eigenschaften 1. Jedes dieser Verfahren ist einzigartig für jede Art von Ionenkanälen und optimiert wird, um die physiologische Rolle des Ionenkanals zu erfüllen. Folglich Veränderungen in jedem dieser fein abgestimmten Prozesse durch einevererbte oder eine genetische Modifikation, die oft als "Ionenkanal" kann zu Zellhomöostase schädlich sein. Es ist wichtig zu betonen, dass an der Zelloberfläche, die "richtige" Menge an Ionenkanälen liefern zu Zellhomöostase kritisch ist. Selbst kleine Erhöhungen (Gain-of-function) und geringe Abnahmen (loss-of-function) in Ionenkanalaktivität haben das Potenzial, eine ernsthafte Pathologie im Laufe eines Lebens zu verursachen. Defekte in der Zelloberfläche Lieferung von reifen Ionenkanäle ist ein wichtiger Faktor in zahlreichen channelopathies, wie zystischer Fibrose (CFTR Ionenkanal) 2 und Herzrhythmusstörungen des Long - QT - Syndrom Form (kardiale Kaliumkanäle) 3.
Kanalopathien sind mit Herz plötzlichen Tod 4 verbunden. Die aktuelle weltweite Prävalenz aller Herz channelopathies wird angenommen , mindestens 1: 2,000-1: etwa die Hälfte des plötzlichen Herztods arrhythmic ca. 3.000 pro Einzel 5 und sind verantwortlichses 6. Dysfunction in Herzspannungsabhängigen Natrium-, Kalium- und kalzium selektive Ionenkanäle sind dafür bekannt, eine Schlüsselrolle in diesem Prozess spielen. Der L-Typ - Ca V 1.2 spannungsabhängigen Calcium - Kanal erforderlich synchronisiert Herzmuskelkontraktion zu initiieren. Der Herz L-Typ - Ca V 1.2 - Kanal ist ein Multi-Untereinheiten - Proteinkomplex , bestehend aus dem Hauptporenbildenden Ca V α1 - Untereinheit und Ca V SS und V Ca α2δ1 Hilfsuntereinheiten 7-12. Beachten Sie, dass die volle Ergänzung der Hilfsuntereinheiten funktionellen Ca V 1.2 Kanäle an der Plasmamembran und dynamische Wechselwirkungen zu erzeugen erforderlich ist zwischen diese Untereinheiten sind wichtig , die normale elektrische Funktion des Herzens 13 zu unterstützen. Ca V ß fördert die Zelloberflächenexpression von Ca V 1.2 Kanäle , durch eine nicht-kovalente nanomolar hydrophobe Interaktionschromatographie 14. Die Co-Expression des Ca V α2δ1 Untereinheit with Ca V ß-gebundene Ca V α1 stimuliert Spitzenstrom Ausdruck (5 bis 10-fach) und fördert Kanalaktivierung bei negativeren Spannungen. Gain-of-function Mutationen des porenbildende Untereinheit Ca V 1.2 haben mit einer Form von ventrikulären Arrhythmien sogenannte Long - QT - Syndrom 15 , während eine Vielzahl von Punktmutationen in den drei Hauptuntereinheiten bilden , die L-Typ - Ca V 1.2 Kanal zugeordnet worden wurden bei Patienten leiden unter Arrhythmien des Short - QT - Syndrom Form 16,17 identifiziert. Ionenkanäle sind Membranproteine, die aus biochemischer Sicht (Proteinchemie) oder mittels elektro Tools (Stromerzeugungsmaschinen) und häufig diese komplementären Ansätzen untersucht werden können. Electrophysiology, insbesondere Ganzzell - Patch-Clamp ist ein geeigneter Ansatz , um die Funktion von Ionenkanälen 15 zu erläutern , aber nicht lösen können Modifikationen in den Proteintransport durch Veränderungen in ihren biophysikalischenEigenschaften. Proteinchemie hat jedoch häufig den Einsatz aufgrund der relativ geringen Expression großer Membranproteine in Bezug auf kleinere lösliche Proteine beschränkt. Robuste Hochdurchsatzverfahren Fluoreszenzablesung unter Verwendung müssen entwickelt werden, um gezielt Defekte in Proteinbiogenese adressieren Veränderungen in der Zelloberflächenexpression von Ionenkanälen verursacht.
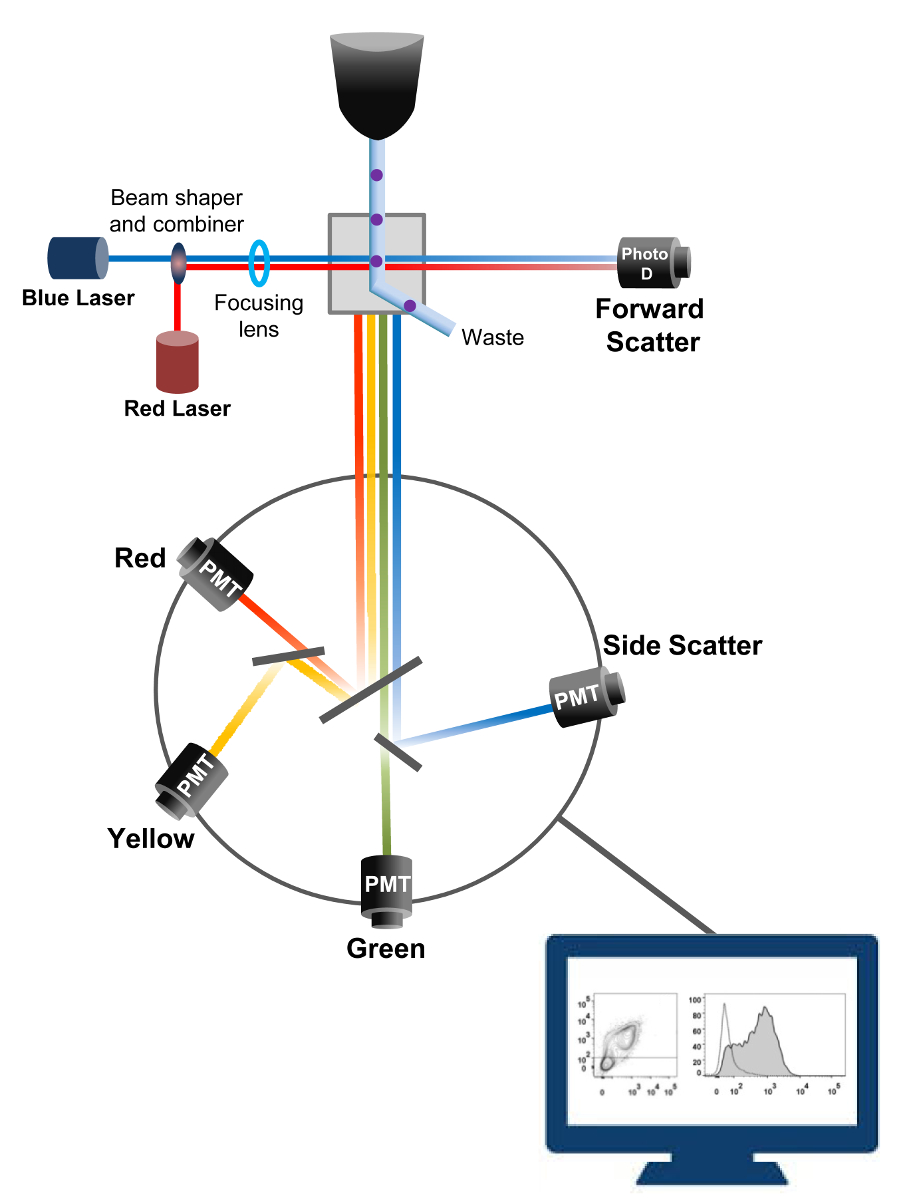
Die Durchflusszytometrie ist ein biophysikalischen Technologie in Zellzählung eingesetzt, das Sortieren, den Nachweis von Biomarkern und Protein - Engineering - 18. Wenn eine Probenlösung von lebenden Zellen oder Partikel in einem Durchflusszytometer eingespritzt wird, werden die Zellen in einen einzigen Strom bestellt, die von der Maschine des Detektionssystems (Figur 1) untersucht werden können. Die erste Durchflusszytometer Instrument 1956 erzeugt 19 detektiert nur einen Parameter , aber moderne Durchflußzytometer weisen mehrere Laser und Fluoreszenzdetektoren, die den Nachweis von mehr als 30 Fluoreszenzparameter 20,21 ermöglichen.Filter und Spiegel (Emissionsoptik) lenken das Licht streuen oder Fluoreszenzlicht von Zellen an ein elektronisches Netzwerk (Photodiode und Detektoren), die das Licht proportional zu seiner Intensität umwandeln. Digitale Daten werden mit spezieller Software analysiert und die primäre Ausgabe wird als Punkt - Diagramm 21 angezeigt.

Abb . 1: Biophysical Prinzipien der Durchflusszytometrie Sortieren einzelner Zellen werden durch eine Düse unter hohem Druck in einem Strom von Hüllflüssigkeit geschoben , der sie bewegt sich über eine oder mehrere Laserabfragepunkten. Der Lichtstrahl wird durch die vorbeifahrenden Zellen umgelenkt und das Licht in der Vorwärtsrichtung gesammelt (Forward Scatter, FCS) auf eine Fotodiode geschickt, die das Licht in ein Signal proportional zur Größe der Zelle umwandelt. Das Licht wird auch in einem 90 ° Winkel zu dem Laserpfad und an Detektoren (auch als Photomultiplier (PMT)) gesammelt.Dieses Licht wird durch den dichroitischen Spiegel geleitet, die die Erkennung des Seitenstreusignal (SSC) zu ermöglichen, die die Granularität innerhalb der Zellen widerspiegelt, und die Fluoreszenzemissionen, wenn erregt Fluorochrome in der Zelle vorhanden sind. Drei Detektoren (grün, gelb und rot) mit unterschiedlichen Wellenlängen-Bandpassfilter dargestellt, so dass die gleichzeitige Detektion von verschiedenen Fluorochromen. Die unterschiedlichen Signale werden von einem externen Computer digitalisiert und in Daten umgewandelt, die die Eigenschaften der Zellen zu quantifizieren analysiert werden. Bitte hier klicken , um eine größere Version dieser Figur zu sehen.
{kind=link}
Die Hochdurchsatzkapazität von Durchflusszytometer wurde die relative Membranexpression von rekombinanten Wildtyp und des Handels-defizienten spannungsabhängigen L-Typ - Ca V 1.2 Kanäle und die damit verbundenen Untereinheiten in lebenden Zellen zu quantifizieren ausgebeutet. cDNA-Konstrukten coding für die Proteine wurden doppelt markiert, um gleichzeitig eine extrazelluläre nichtfluoreszierenden Epitop tragen, die durch eine undurchlässige fluoreszierenden konjugierten Antikörper und einem intrazellulären Fluorophor die konstitutiv fluoresziert detektiert werden kann. Sowohl das extrazelluläre Epitop in einer extrazellulären Schleife des Proteins eingeführt ist, und die intrazelluläre Fluorophor, eingesetzt nach dem C-Terminus mit dem Protein translatiert. In dieser Serie von Experimenten wurde die Ca V α2δ1 proteintechnisch eine extrazelluläre Hämagglutinin (HA) -Epitop (YPYDVPDYA) durch eine undurchlässige FITC (Fluorescein - Isothiocyanat) -konjugiertem anti-HA und mCherry als Intrinsische intrazellulärem Fluorophor detektiert auszudrücken. Um die relative Zelloberflächenexpressionsniveau des mCherry-Ca V α2δ1 HA-markiertes Protein, rekombinante Zellen, die das Fusionsprotein bestimmen , wurden nach der Transfektion geerntet, und gefärbt mit FITC-konjugierten monoklonalen Maus - anti-HA - Epitop - Tag Antibody (Abbildung 2). FITC ist eine organische Fluoreszenzverbindung, die wesentlich kleiner als Enzym-Reporter ist und deshalb nicht so wahrscheinlich mit biologischen Funktion zu stören. mCherry- Ca V α2δ1-HA überexprimiert in Tsa-201cells, erzeugt eine signifikante 3-log Anstieg der FITC - Fluoreszenz und mCherry Fluoreszenz auf zweidimensionalen Plots 22. Da die HA-Epitop in der extrazellulären Teil des Proteins befindet, erhalten die Fluoreszenzintensität für FITC in Gegenwart von intakten Zellen spiegeln die relativen Index der Zelloberflächenexpression von HA-markierten Proteins. Die Zugänglichkeit des HA-Epitops in den Konstrukten wird durch Messung des FITC Signals nach Zellpermeabilisierung systematisch validiert. Auch diese Maßnahme dient der normalisierten Gesamtproteinexpression zu untermauern, da die relativen Fluoreszenzintensitäten für FITC in permeabilisierten Zellen geschätzt sind qualitativ vergleichbar mit den relativen Fluoreszenzwerte for mCherry gemessen unter permeabilisiert und nicht-permeabilisierten Bedingungen 22,23. Es ist wichtig zu beachten, dass die intrinsische Fluoreszenzspektrum zu höheren Werten nach Permeabilisierung verschoben wird, sondern daß der einzige Wert angegeben wird, ist die Änderung in der Fluoreszenzintensität in Bezug auf die Kontrollkonstrukt verglichen. Relative Änderungen in der Fluoreszenzintensität für die Testkonstrukte werden geschätzt die ΔMean Fluoreszenzintensität unter Verwendung von (ΔMFI) Werte für jedes Fluorophor (mCherry oder FITC). Experimente werden die Fluoreszenzintensität des Testkonstrukts relativ zur Fluoreszenzintensität des Kontrollkonstrukts unter den gleichen Bedingungen exprimiert zu messen experimentellen Variationen in der intrinsischen Fluoreszenz des Fluorophors-konjugierten Antikörper zu begrenzen. Zwei Membranproteine wurden erfolgreich mit diesem Test untersucht: das porenbildende Untereinheit des L-Typ spannungsabhängigen Calcium - Kanal - Ca V 1.2 14,22 und in einer anderen Reihe vonExperimente, 22,23 die extrazelluläre Hilfs Ca V α2δ1 Untereinheit. Das folgende Protokoll wurde verwendet , um die Zelloberflächenexpression des Ca V α2δ1 Untereinheit des Herz L-Typ - Ca V 1.2 Kanal unter Kontrollbedingungen und nach Mutationen zu bestimmen , die posttranslationale Modifikation des Ionenkanals beeinflussen. Unter genormten Versuchsbedingungen erhöht die Zelloberfläche Fluoreszenz von FITC quasi-linear mit der Expression der cDNA kodierend für die mCherry-Ca V α2δ1-HA - Proteine (Abbildung 5 aus Lit. 22).

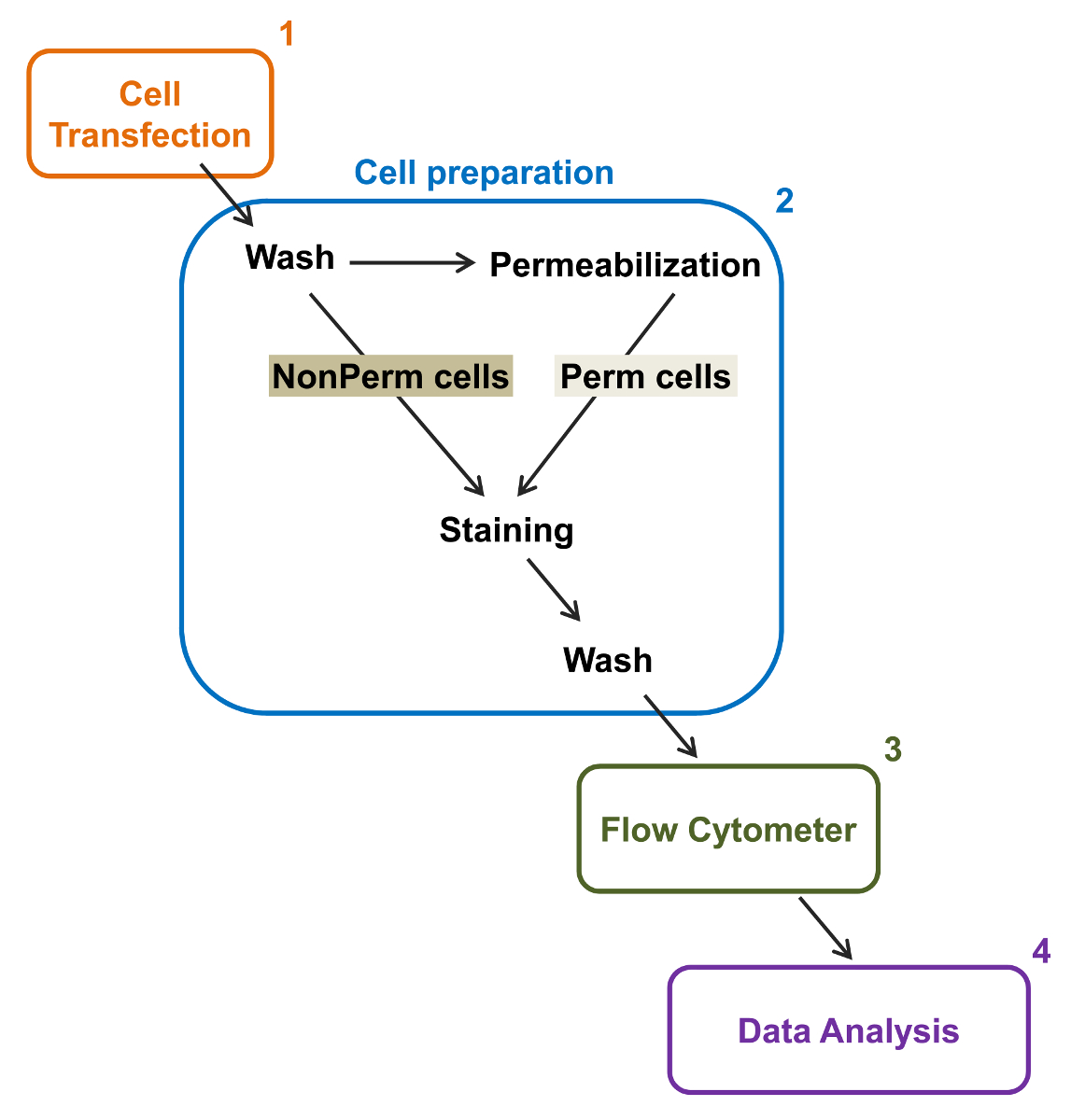
Abbildung 2:. Schematische Darstellung der gesamten und der Membran Kennzeichnung in der Durchflusszytometrie Versuchsprotokoll Das Schema beschreibt einige der wichtigsten Schritte notwendig , um die relativen Gesamt und Zelloberflächenexpression von rekombinanten Ionenkanäle durch fl zu quantifizierenow-Zytometrie. Zellen werden mit dem doppelt markiert Konstruktion mCherry-Ca V α2δ1-HA in TSA-201 - Zellen transfiziert (1) und gefärbt vor oder nach der Permeabilisierung (2). Multi Daten werden in einem Durchflusszytometer (3) für multivariate Analyse (4) übernommen. Bitte hier klicken , um eine größere Version dieser Figur zu sehen.
{kind=link}
Protokoll
1. Doppelt Tagged DNA-Konstrukte
- Legen Sie die HA - Epitop (YPYDVPDYA) in der extrazellulären Linker von Ca V α2δ1 zwischen D676 und R677 durch ortsgerichtete Mutagenese (3B) 20. Verwenden Vorwärtsprimer gccggattatgcgGGAAAACTCCAAACAACC und Reverse-Primer acatcatacggataTCAATAAATTCATTGAAATTTAAAAGAAATTC.
- Subklonierung der cDNA - Sequenz des markierten HA Ca V α2δ1 in den Säuger pmCherry-N1 - Expressionsvektor , der das Protein an den N-Terminus von mCherry zwischen den SacI- und SalI - Stellen (3B) 20 verschmolzen auszudrücken.
HINWEIS: Geeignete Kanalfunktion muss mit dem Kontrollkonstrukt unter Verwendung von Standard - elektrophysiologischen Methoden 24 getestet werden.
2. Liposom-vermittelte transiente Transfektion (30 min, sind alle Schritte ausgeführt unter Laminar Flow Hood)
- Tag 1: Platte, die eine halbe Million von tsa-201-Zellen (oder HEKT) in35 mm Kulturschalen mit 2 ml Dulbeccos hohem Glucoseminimalmedium (DMEM-HG), ergänzt mit 10% fötalem Rinderserum (FBS) und 1% Penicillin-Streptomycin (PS) Kulturmedium. Zählen von Zellen einen Standard Hemacytometer verwenden. Beurteilen, die Lebensfähigkeit der Zellen von einem Bruchteil der Zellprobe unter Verwendung von Trypan Blau. Platte genügend Zellen 90% Konfluenz zum Zeitpunkt der Transfektion zu erreichen.
- Tag 2: Ändern Kulturmedium mit 2 ml frischem vorgewärmten (37 ° C) Kulturmedium ohne PS.
- Für jede Transfektion Probe, bereiten zwei 1,5-ml-Röhrchen. In Rohr 1, verdünne 4 ug DNA in 250 ul reduzierten Serummedium. Im Rohr 2, Mischungs 10 ul der Liposomen-vermittelte Transfektion Reagenzes mit 250 & mgr; l serumreduziertes Kulturmedium. Vorsichtig mischen die Transfektionsreagenz vor dem Gebrauch.
- Inkubieren für 5 min bei Raumtemperatur.
- Kombinieren des Inhalts des Rohres 1 und dem Rohr 2, vorsichtig mischen und Inkubieren mindestens 20 min bei Raumtemperatur.
- Fügen Sie die Liposomen / DNA-Komplexe zu den kultivierten Zellen und sanft die Kulturschale Rock zu mischen.
- Unter 5% CO 2 -Atmosphäre für 24 h bei 37 ° C inkubieren.
3. Anfärben von Zellen für die Durchflusszytometrie (3 Stunden)
- Vorbereiten von Zellproben
- Tag 3: Entfernen Medium aus der Kulturschale vorsichtig und wasche die Zellen mit 400 & mgr; l vorgewärmtes (37 ° C) 0,05% Trypsin-EDTA 1x (Ethylendiamintetraessigsäure).
- Werden 400 & mgr; l Trypsin-EDTA und Inkubation die Schale bei 37 ° C unter 5% CO 2 -Atmosphäre für 5 min Zellen zu ermöglichen , von der Schale zu lösen.
- Stoppen Sie die Enzymverdauung durch Zugabe von 1 ml kaltem Kulturmedium ohne PS und auswaschen alle Zellen von der Oberfläche durch Pipettieren vorsichtig 4-5 mal. Vermeiden Sie Über Verdauung und über Pipettieren Zelltod zu reduzieren.
- Sammeln Zellen in 1,5-ml-Röhrchen und legen Sie sofort auf Eis. Verwenden Sie eiskalten Lösungen und halten Sie die Zellen bei 4 ° C, um die Internalisierung zu verhindernvon Oberflächenantigenen. Verringern Beleuchtung Photobleaching des Fluoreszenzsignals zu begrenzen.
- Zentrifugenröhrchen bei 400 xg für 5 min bei 4 ° C. absaugen und entsorgen Sie vorsichtig den Überstand.
- Resuspendieren des Pellets in 1 ml phosphatgepufferter Kochsalzlösung 1x (PBS), um eine Einzelzellsuspension herzustellen.
- Kurz die Röhrchen Wirbel sehr sanft und wiederholen Sie die Schritte 3.1.5 und 3.1.6 vollständig Kulturmedium zu entfernen.
- Resuspendieren des Pellets in 600 ul 1x PBS und stellen die Zellkonzentration auf ein Minimum von 3 x 10 6 Zellen / ml.
- Teilen Sie die Zellen in zwei neue 1,5-ml-Röhrchen für extra- und intrazellulären Färbung. Fügen Sie die entsprechenden Kontrollen spezifische Färbung von nicht-spezifische Färbung zu unterscheiden.
HINWEIS: Die Isotypkontrollantikörper hilft das Niveau der Hintergrundfärbung zu bewerten und im Idealfall jeder primären Antikörpers Wirtsarten, Isotyp und Fluorophore übereinstimmen sollte. Verwenden Sie Isotypenkontrolle und konjugierte Antikörper mit der gleichenProteinkonzentration.

Tabelle 1: Durchfluss-Zytometrie Experiment Kontrollproben für nicht-permeabilisierten und permeabilisierten Zellen Jedes Experiment benötigt folgende negative Kontrollen enthalten: (1) . Untransfizierten Zellen (ohne Antikörper, mit dem Isotyp oder mit dem konjugierten Antikörper). (2) Die transfizierten Zellen mit dem Protein von Interesse , subkloniert in ein Plasmid ohne konstitutive intrazelluläre Fluoreszenz Fluorochrom (pCMV- Ca V α2δ1-HA) oder mit dem doppelt tagged Konstruktion (pmCherry-Ca V α2δ1 beimpft und ohne Antikörper, mit dem Isotyp oder der konjugierte Antikörper). Einfarbige Kontrollen werden zur Kompensation von Fluorochrom Emissions Überlappung verwendet. Die gleichen Kontrollen laufen für Nicht-permeabilisierten und permeabilisiert Bedingungen in jeder Reihe von Experimenten.
- Zelloberflächenfärbung von Intactlebenden Zellen
- Aliquot 1 x 10 6 Zellen / 100 ul in 1,5 - ml - Röhrchen.
- Fügen Sie die FITC-konjugierten monoklonalen anti-HA-Antikörper mit 5 ug / ml und vortex, bevor die Zellen auf einem Schüttler (200 rpm) im Dunkeln bei 4 ° C für 45 min inkubiert wurde.
HINWEIS: Die optimale Konzentration des Antikörpers in der Vortitration Experimenten bestimmt (Abbildung 4). - Entfernen Sie die Zellen, die aus der Dunkelheit und fügen 900 ul 1x PBS / Rohr. Zentrifuge bei 400 × g für 5 min bei 4 ° C.
- Aspirieren den Überstand und das Pellet in 1 ml 1x PBS, Vortexen und Zentrifugieren bei 400 xg für 5 min bei 4 ° C.
- Wiederholen Sie den Wasch (Schritt 3.2.4) zweimal um alle ungebundenen Antikörper zu entfernen. Wenn ein unkonjugiertes primären Antikörper verwendet wird, mit dem geeigneten sekundären Antikörper inkubiert.
- Nach dem letzten Waschen, um die Zellen in 500 ul 1x PBS resuspendieren und die Einzelzellsuspension in 5 übertragen ml-Zytometrie Rohre fließen. Halten Sie die Zelles im Dunkeln bei 4 ° C, bis die Probe läuft.
- Führen Sie die Proben auf einem Durchflusszytometer. Für die besten Ergebnisse analysieren, um die Zellen auf dem Durchflusszytometer so bald wie möglich und spätestens 24 Stunden nach.
- Intrazelluläre Färbung: Fixierung, Permeabilisierung und Anfärben
- Aliquot 1 x 10 6 Zellen / 100 ul in 1,5 - ml - Röhrchen und zentrifugiert bei 400 g für 5 min bei 4 ° C.
- Überstand verwerfen und resuspendieren Zellen in 100 ul Fixierung-Permeabilisierung Lösung direkt ab Lager.
- im Dunkeln inkubieren 20 min bei 4 ° C.
- Zugabe von 100 ul frisch 1x Permeabilisierung Waschpuffer hergestellt (verdünntes 10x Permeabilisierung-Waschpuffer in destilliertem H 2 O). Zellen Vortex und Sediment einer Tischzentrifuge bei 400 xg für 5 min bei 4 ° C verwendet wird.
- Absaugen und den Überstand verwerfen.
- Wiederholen Sie die Schritte 3.3.4 und 3.3.5.
- Hinzufügen FITC-konjugierten monoklonalen anti-HA-Antikörper bei 5& mgr; g / ml in 100 & mgr; l Waschpuffer Permeabilisierung-1x und vortex bevor die Zellen im Dunkeln bei 4 ° C für 30 min inkubiert wurde.
HINWEIS: Die intrazelluläre Färbung wird nach dem gleichen Verfahren wie die für die Zelloberflächenfärbung verwendet, durchgeführt. Saponin-vermittelte Zellpermeabilisierung ist jedoch ein schnell reversibler Prozess, daher ist es wichtig, 1x PBS mit 1x Perm / Waschpuffer zu ersetzen während die intrazelluläre Färbung der Zellen in der ständigen Gegenwart von Saponin zu halten. - Entfernen Sie die Zellen, die aus der Dunkelheit und fügen 100 ul Permeabilisierung-Waschpuffer. Zentrifuge bei 400 × g für 5 min bei 4 ° C.
- Aspirat vorsichtig der Überstand und das Pellet in 100 & mgr; l Permeabilisierung-Waschpuffer, Vortexen und Zentrifugieren bei 400 xg für 5 min bei 4 ° C.
- Wiederholen Sie die Wäsche (Schritt 3.3.9) noch einmal um alle ungebundenen Antikörper zu entfernen.
- Nach dem letzten Waschen, um die Zellen in 500 ul 1x PBS resuspendieren und die Sünde übertragengle Zellsuspension 5 ml Durchflusszytometrie Rohre. Halten der Zellen in der Dunkelheit bei 4 ° C bis Zytometer die Probe in den Strömungs einzuspritzen.
- Führen Sie die Proben auf einem Durchflusszytometer. Führen Sie die festen Proben auf dem Zytometer so bald wie möglich, jedoch spätestens 1 Woche nach dem Färben. Führen Sie die nicht-permeabilisierten und Zellen am selben Tag permeabilisiert.
4. Durchflusszytometrie
- Durchflusszytometer Cell Sorter Täglich Einrichtung
- Schalten Sie die Durchflusszytometrie-Software. Vor experimentieren, zu kalibrieren und das Setup der Durchflusszytometer Zellsortierer optimale Geräteleistung sicherzustellen (dh Laser und Optik Spezifikation durchführen, die Laser und Zelle fließen richtig ausgerichtet sind ) durch Geräteeinstellung Perlen verwenden.
- Verwenden Sie die 100 & mgr; m Düse mit 20 psi Hülle Druck.
HINWEIS: Die Düse nicht auf einer Bank Durchflusszytometer geändert werden muss. - Stellen Sie die Fördermenge der Zytometer nach der Hersteler Spezifikation. Außerordentlich hohe Strömungsgeschwindigkeiten wird die Empfindlichkeit bei der Erfassung von Variationen in der Fluoreszenz verringern.
- Wählen Sie Blau (488 nm Fluorescein Isothiocayanate oder FITC zu erregen) und gelb-grün (561 nm bis mCherry erregen) Laser. Sammeln FITC und mCherry Fluoreszenzwerte mit einem 530/30 nm und mit einem 610/20 nm Bandpassfilter sind.
- Erwerben Sie die Vorwärtsstreuung (FCS) im Vergleich zu Seitenstreuung (SSC) Dot-Plot für ungefärbte Zellen lineare Skala. Stellen Sie die einzelnen Verstärkung der Detektorzellen in der linken unteren Quadranten des Dot-Plot zu visualisieren.
- Beispiel Lesen von Intact Nicht-permeabilisierten Zellen
- Stellen Sie die P1gate lebende, nicht-permeabilisierten Zellen durch eine freie Form der Abgrenzung um die Zellen ohne Zelltrümmer und Zellaggregate analysiert werden, wodurch das Fluoreszenzsignal zu intakten Zellen zu begrenzen.
Hinweis: Live / Dead-Ausschluss Farbstoffe können verwendet werden, Gate-Platzierung an lebenden Zellen zu erleichtern. Stellen Sie 10.000 Ereignisse aufzeichnenin dem Stopp Gatter P1. Setzen Sie diese auf eine höhere Anzahl von Ereignissen, wenn nötig. - Erwerben Sie mCherry gegen FITC zweiparametrige Konturplot Basisautofluoreszenz von ungefärbten Zellen zu erkennen. Verwenden Sie bi-logarithmischen Skala negative Werte zu zeigen und zu verbessern Auflösung zwischen den Populationen 25. Stellen Sie die einzelnen Spannung des Detektors die ungefärbten negativen Zellen innerhalb des unteren Abschnitts der ersten zehn Einheiten der Log-Fluoreszenzintensität Plots zu setzen.
- Erwerben Sie alle intakten nicht-permeabilisierten Proben etabliert mit den Einstellungen auf 4.1.5 und 4.1.6 und sammeln FSC, SSC und Signale in den Fluoreszenzdetektoren.
- Export und speichern * .fcs Dateien zur Analyse verwendet werden soll unter Verwendung der Durchflusszytometrie-Analyse-Software.
- Stellen Sie die P1gate lebende, nicht-permeabilisierten Zellen durch eine freie Form der Abgrenzung um die Zellen ohne Zelltrümmer und Zellaggregate analysiert werden, wodurch das Fluoreszenzsignal zu intakten Zellen zu begrenzen.
- Beispiel Lesen von permeabilisierten Zellen
- Bewegen Sie den P1-Gate lebenden Zellen in den permeabilisierten Proben wählen und einstellen FSC und SSC-Spannung wie in 4.1.5 und 4.1.6 gezeigt.
- Erwerben Sie alle permeabilisiert Proben und sammeln FSC, SSC einnd Signale in den Fluoreszenzdetektoren.
- Export und speichern * .fcs Dateien zur Analyse verwendet werden soll unter Verwendung der Durchflusszytometrie-Analyse-Software.
- Datenanalyse
- Starten Sie die Durchflusszytometrie-Analyse-Software und Import * .fcs gespeicherten Dateien in 4.2.4 und 4.3.3.
- Klicken Sie auf das erste im Arbeitsbereich Fenster aufgelistet Probe. Ein neues Fenster nach dem Rohr ID-Nummer genannt wird automatisch geöffnet. Starten Sie den Gating-Prozess in der Handlung von SSC gegen FSC. Zeichnen Sie ein Tor (P1) mit dem Symbol Ellipse um lebende Zellen und beseitigen alle Ablagerungen, abgestorbene Zellen oder Aggregate, die unterschiedliche Vorwärtsstreuung und Seitenstreuung als lebende Zellen haben
- Um die Zwei-Parameter-Konturdiagramm der mCherry (y-Achse) gegen FITC (x-Achse) Fluoreszenzintensität der lebenden Zellen zeichnen, klicken Sie zuerst auf der x-Achse und wählen Sie das FITC-A-Kanal und klicken Sie dann auf der y -Achse und die PE-mCherry-A-Kanal wählen. Klicken Sie auf das "Quad" Symbol, um den Quadranten Markierung am Rand eines zu positionierenutofluorescent Zellen in jeder Fluoreszenzkanal.
HINWEIS: Das Tor gesetzt um die FITC und mCherry positiven Zellen ist die P2-Gate. Die Fluoreszenz negativen Zellpopulation wird als P3-Gate bezeichnet. Siehe Abbildung 5 für die repräsentative Gating - Methode in diesem Artikel verwendet. - Wählen Sie P2 und P3 Tore und klicken Sie auf "Hinzufügen Statistics" Symbol im ursprünglichen Arbeitsbereich Fenster. Klicken Sie auf "Count" (Anzahl der positiven Zellen) und klicken Sie auf "Mean" (mittlere Fluoreszenzintensität jedes Fluorochrom) oder "Median" (Median Fluoreszenzintensität jedes Fluorochrom) Statistiken unter der Liste der Optionen. Klicken Sie auf "Hinzufügen Statistics" Symbol wieder. Alle diese Werte werden automatisch auf den ursprünglichen Arbeitsbereich Fenster übertragen.
HINWEIS: Die "mittlere" wird nur verwendet, wenn die Fluoreszenzintensität einer Normalverteilung folgt. In jedem anderen Fall, klicken Sie auf den "Median" aus. so konnte MFI beziehen Fluoreszenz Intensit Meany oder Median der Fluoreszenzintensität.
HINWEIS: Der nächste Schritt ist es, die Tore "Parameter und Statistiken für alle Proben durch das Zytometer sondiert anzuwenden. - Im Arbeitsbereich Fenster, benutzen Sie die Maus, um die Tore und Statistiken Parameter per Drag & Drop auf die Linie alle Proben markiert.
- Generieren Sie einen Batch - Report von zweidimensionalen Konturplots (mCherry vs FITC) und Histogramme (Zelle im Vergleich zu Fluoreszenzintensität zählen) für nicht-permeabilisierten und permeabilisierten Zellen (6A - B).
- Aus den erzeugten Statistiken in Schritt 4.4.4, berechnen die mittlere Fluoreszenzintensität (MFI) für jedes Fluorochrom für die gefärbten Zellen. Aus diesem Wert subtrahieren Sie den MFI-Wert aus ungefärbten Zellen erhalten, die Oberfläche und die Gesamt Expression des Proteins von Interesse zu quantifizieren.
- Melden Sie die ΔMFI Werte für jeden Fluorophor (mCherry und FITC) (6C - D). Normalisieren der ΔMFI für die Ca gemessen V α2δ1 konstruieren Mutanten auf den ΔMFI Wert für FITC und mCherry mit dem WT - Konstrukt erhalten.
HINWEIS: Der Absolutwert der Fluoreszenzintensität kann variieren stark in Abhängigkeit von der Charge von Antikörpern und die technischen Fähigkeiten jedes Laborant, daher die Notwendigkeit, die Fluoreszenzintensität des mutierten Konstrukt mit dem WT-Konstrukt zu normieren.
Ergebnisse
Dieser Artikel beschreibt ein zuverlässiges Protokoll Gesamt- und Zelloberfläche von rekombinanten Ionenkanäle in Tsa-201cells durch eine Zweifarben-Durchflusszytometrie-Assay ausgedrückt zu quantifizieren. Als Beispiel wurde die relative Zelloberfläche und die Gesamtproteinexpression für die Ca V α2δ1subunit quantifiziert. Um wurde die Zweifarben - Durchflusszytometrie - Assay, Ca V α2δ1 doppelt auszuführen getaggt eine extrazelluläre nichtfluoreszieren...
Diskussion
Diese Durchflusszytometrie basierenden Assay wurde zur Messung der relativen Gesamt und Zelloberflächenniveaus von fluoreszenzmarkierten porenbildenden und die zugehörigen Untereinheiten von spannungsabhängigen Calciumkanälen 14,22,26 erfolgreich angewendet. Es wird verwendet, am besten, wenn die Wirkung der genetischen Mutationen zu untersuchen und somit erfordert, daß die intrinsische Fluoreszenzintensität des fluoreszierend markierten markierten Wildtyp-Konstrukt sein, mindestens 10 bis 100-fach grö...
Offenlegungen
The authors declare that they have no competing financial interests.
Danksagungen
We thank Mr. Serge Sénéchal and Dr. Jacques Thibodeau for sharing their expertise and granting us access to their flow cytometry and cell sorting platform. This work was completed with the operating grant 130256 from the Canadian Institutes of Health Research, a grant-in-aid from the Canadian Heart and Stroke Foundation, and support from the "Fondation de l'Institut de Cardiologie de Montréal" to L.P.
Materialien
| Name | Company | Catalog Number | Comments |
| Q5 Site-Directed Mutagenesis Kit | New England Biolabs | E0554S | Can be substitute with QuickChange site-directed mutagenesis Kit (Agilent, #200523). |
| Tubes 1.5 ml | Sarstedt | 72-690-001 | |
| Tubes 15 ml | Sarstedt | 62-554-002 | |
| Disposable graduated Tranfer Pipets | VWR | 160001-192 | |
| 100 mm culture dish | Corning | 430167 | For standard culture of HEKT cells. |
| 35 mm culture dish | Falcon | 353001 | For standard culture of HEKT cells. |
| Serological pipette 1 ml | Sarstedt | 86.1251.001 | |
| Serological pipette 5 ml | Sarstedt | 86.1253.001 | |
| Serological pipette 10 ml | Sarstedt | 86.1254.001 | |
| Serological pipette 25 ml | Sarstedt | 86.1285.001 | |
| Dulbecco's high-glucose medium | Life Technologies | 12100-046 | Warm in 37 °C water bath before use. |
| Fetal Bovine Serum, qualified, heat inactivated, US origin | Life Technologies | 16140-071 | |
| Penicillin-Streptomycin (10,000 U/ml) | Life Technologies | 15140-122 | |
| Lipofectamine 2000 | Life Technologies | 11668-019 | For liposomal transfection. Can be substituted with calcium phosphate transfection. |
| Opti-MEM I Reduced Serum Medium | Life Technologies | 31985-070 | Warm in 37 °C water bath before use. |
| Trypsin-EDTA (1x) 0.05%, phenol red | Life Technologies | 25300-062 | |
| 1.5 ml microtubes | Sarstedt | 72.690.001 | |
| Phosphate Buffered Saline 1x | Fisher | BP661-10 | Can be "home-made". |
| Anti-HA FITC conjugated antibody | Sigma | H7411 | |
| IgG1−FITC Isotype Control antibody | Sigma | F6397 | |
| BD Cytofix/Cytoperm Fixation/Permeabilization Solution Kit | BD Biosciences | 554714 | Fixation/Permeabilization. Permeabilization/Wash solution, store at 4 °C. |
| Hemacytometer | Fisher | 49105161 | |
| Trypan Blue | Fisher | 15250061 | To access cell viability. |
| Refrigerated Microcentrifuge, 5430R | Eppendorf | A14H172200 | |
| Forma Steri-Cycle CO2 Incubator | Fisher | 370 | |
| Laboratory Platform Rocker | Fisher | 545034 | |
| Water Bath | VWR | 89032-216 | |
| BD FACSARIA III | BD Biosciences | 648282 | Flow cytometer. |
| FlowJo Software v10 | FlowJo | FlowJo v10 Dongle | For data analysis. |
Referenzen
- Delisle, B. P., Anson, B. D., Rajamani, S., January, C. T. Biology of Cardiac Arrhythmias: Ion Channel Protein Trafficking. Circ. Res. 94, 1418-1428 (2004).
- Birault, V., Solari, R., Hanrahan, J., Thomas, D. Y. Correctors of the basic trafficking defect of the mutant F508del-CFTR that causes cystic fibrosis. Curr Opin Chem Biol. 17, 353-360 (2013).
- Balijepalli, S. Y., Anderson, C. L., Lin, E. C., January, C. T. Rescue of Mutated Cardiac Ion Channels in Inherited Arrhythmia Syndromes. J. Cardiovas Pharm. 56, 113-122 (2010).
- Gargus, J. J. Unraveling Monogenic Channelopathies and Their Implications for Complex Polygenic Disease. Am. J. Hum. Genet. 72, 785-803 (2003).
- Abriel, H., Zaklyazminskaya, E. V. Cardiac channelopathies: Genetic and molecular mechanisms. Gene. 517, 1-11 (2013).
- Behr, E. R., et al. Sudden arrhythmic death syndrome: familial evaluation identifies inheritable heart disease in the majority of families. Eur Heart J. 29, 1670-1680 (2008).
- Catterall, W. A. Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev.Biol. 16, 521-555 (2000).
- Peterson, B. Z., DeMaria, C. D., Adelman, J. P., Yue, D. T. Calmodulin is the Ca2+ sensor for Ca2+ -dependent inactivation of L- type calcium channels. Neuron. 22, 549-558 (1999).
- Dolphin, A. C. Calcium channel diversity: multiple roles of calcium channel subunits. Curr.Opin.Neurobiol. 19, 237-244 (2009).
- Dai, S., Hall, D. D., Hell, J. W. Supramolecular assemblies and localized regulation of voltage-gated ion channels. Physiol Rev. 89, 411-452 (2009).
- Gao, T., et al. Identification and subcellular localization of the subunits of L-type calcium channels and adenylyl cyclase in cardiac myocytes. J. Biol. Chem. 272, 19401-19407 (1997).
- Carl, S. L., et al. Immunolocalization of sarcolemmal dihydropyridine receptor and sarcoplasmic reticular triadin and ryanodine receptor in rabbit ventricle and atrium. J. Cell Biol. 129, 673-682 (1995).
- Abriel, H., Rougier, J. S., Jalife, J. Ion Channel Macromolecular Complexes in Cardiomyocytes: Roles in Sudden Cardiac Death. Circ. Res. 116, 1971-1988 (2015).
- Bourdin, B., et al. Molecular Determinants of the Cavb-induced Plasma Membrane Targeting of the Cav1.2 Channel. J. Biol. Chem. 285, 22853-22863 (2010).
- Raybaud, A., et al. The Role of the GX9GX3G Motif in the Gating of High Voltage-activated Calcium Channels. J. Biol. Chem. 281, 39424-39436 (2006).
- Burashnikov, E., et al. Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm. 7, 1872-1882 (2010).
- Hennessey, J. A., et al. A CACNA1C Variant Associated with Reduced Voltage-Dependent Inactivation, Increased Cav1.2 Channel Window Current, and Arrhythmogenesis. PLoS ONE. 9, e106982 (2014).
- Adan, A., Alizada, G., Kiraz, Y., Baran, Y., Nalbant, A. Flow cytometry: basic principles and applications. Crit Rev Biotechnol. , 1-14 (2016).
- Graham, M. D. The Coulter Principle: Foundation of an Industry. J. Lab. Autom. 8, 72-81 (2003).
- Baumgarth, N., Roederer, M. A practical approach to multicolor flow cytometry for immunophenotyping. J. Immunol. Methods. 243, 77-97 (2000).
- Rothe, G., Sack, U., Tarnok, A., Rothe, G. . Cellular Diagnostics. Basics, Methods and Clinical Applications of Flow Cytometry. , 53-88 (2009).
- Bourdin, B., et al. Functional Characterization of Cavalpha2delta Mutations Associated with Sudden Cardiac Death. J. Biol. Chem. 290, 2854-2869 (2015).
- Tetreault, M. P., et al. Identification of glycosylation sites essential for surface expression of the Cavalpha2delta1 subunit and modulation of the cardiac Cav1.2 channel activity. J. Biol. Chem. 291, 4826-4843 (2016).
- Senatore, A., Boone, A. N., Spafford, J. D. Optimized Transfection Strategy for Expression and Electrophysiological Recording of Recombinant Voltage-Gated Ion Channels in HEK-293T Cells. J Vis Exp. (47), (2011).
- Herzenberg, L. A., Tung, J., Moore, W. A., Herzenberg, L. A., Parks, D. R. Interpreting flow cytometry data: a guide for the perplexed. Nat.Immunol. 7, 681-685 (2006).
- Shakeri, B., Bourdin, B., Demers-Giroux, P. O., Sauve, R., Parent, L. A quartet of Leucine residues in the Guanylate Kinase domain of Cavbeta determines the plasma membrane density of the Cav2.3 channel. J Biol Chem. 287, 32835-32847 (2012).
- Morton, R. A., Baptista-Hon, D. T., Hales, T. G., Lovinger, D. M. Agonist- and antagonist-induced up-regulation of surface 5-HT3A receptors. Br. J. Pharmacol. 172, 4066-4077 (2015).
- Hoffmann, C., et al. Fluorescent labeling of tetracysteine-tagged proteins in intact cells. Nat. Protocols. 5, 1666-1677 (2010).
- Cockcroft, C. J., Gamper, N. . Ion Channels: Methods and Protocols. , 233-241 (2013).
- Gonzalez-Gutierrez, G., Miranda-Laferte, E., Neely, A., Hidalgo, P. The Src Homology 3 Domain of the beta-Subunit of Voltage-gated Calcium Channels Promotes Endocytosis via Dynamin Interaction. J. Biol.Chem. 282, 2156-2162 (2007).
- Galizzi, J. P., Borsotto, M., Barhanin, J., Fosset, M., Lazdunski, M. Characterization and photoaffinity labeling of receptor sites for the Calcium channel inhibitors d-cis-diltiazem, (+/-)-bepridil, desmethoxyverapamil, and (+)-PN 200-110 in skeletal muscle transverse tubule membranes. J. Biol.Chem. 261, 1393-1397 (1986).
- Bezanilla, F. The voltage sensor in voltage-dependent ion channels. Physiol.Rev. 80, 555-592 (2000).
- Sigworth, F. J. The variance of sodium current fluctuations at the node of Ranvier. J Physiol. 307, 97-129 (1980).
- Bailey, M. A., Grabe, M., Devor, D. C. Characterization of the PCMBS-dependent modification of KCa3.1 channel gating. J. Gen. Physiol. 136, 367-387 (2010).
- Fletcher, P. A., Scriven, D. R., Schulson, M. N., Moore, E. D. Multi-Image Colocalization and Its Statistical Significance. Biophys. J. 99, 1996-2005 (2010).
- Lizotte, E., Tremblay, A., Allen, B. G., Fiset, C. Isolation and characterization of subcellular protein fractions from mouse heart. Anal. Biochem. 345, 47-54 (2005).
- Mattheyses, A. L., Simon, S. M., Rappoport, J. Z. Imaging with total internal reflection fluorescence microscopy for the cell biologist. J. Cell Sci. 123, 3621-3628 (2010).
- Yamamura, H., Suzuki, Y., Imaizumi, Y. New light on ion channel imaging by total internal reflection fluorescence (TIRF) microscopy. J. Pharmacol. Sci. 128, 1-7 (2015).
- Wible, B. A., et al. HERG-Lite-R: A novel comprehensive high-throughput screen for drug-induced hERG risk. J. Pharmacol. Toxicol. Methods. 52, 136-145 (2005).
- Wilde, A. A. M., Brugada, R. Phenotypical Manifestations of Mutations in the Genes Encoding Subunits of the Cardiac Sodium Channel. Circ. Res. 108, 884-887 (2011).
- Milano, A., et al. Sudden Cardiac Arrest and Rare Genetic Variants in the Community. Circ Cardiovasc Genet. , (2016).
- Schnell, U., Dijk, F., Sjollema, K. A., Giepmans, B. N. G. Immunolabeling artifacts and the need for live-cell imaging. Nat. Meth. 9, 152-158 (2012).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten