
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Determinación de la superficie celular y Expresión relativa total de canales de iones recombinante mediante citometría de flujo
En este artículo
Resumen
arritmias cardíacas hereditarias son a menudo causados por mutaciones que alteran la entrega superficie de uno o más canales de iones. Aquí, adaptamos citometría de flujo ensayos para proporcionar una cuantificación de la expresión de la proteína total y la superficie celular relativa de los canales iónicos recombinantes expresadas en células TSA-201.
Resumen
Inherited or de novo mutations in cation-selective channels may lead to sudden cardiac death. Alteration in the plasma membrane trafficking of these multi-spanning transmembrane proteins, with or without change in channel gating, is often postulated to contribute significantly in this process. It has thus become critical to develop a method to quantify the change of the relative cell surface expression of cardiac ion channels on a large scale. Herein, a detailed protocol is provided to determine the relative total and cell surface expression of cardiac L-type calcium channels CaV1.2 and membrane-associated subunits in tsA-201 cells using two-color fluorescent cytometry assays. Compared with other microscopy-based or immunoblotting-based qualitative methods, flow cytometry experiments are fast, reproducible, and large-volume assays that deliver quantifiable end-points on large samples of live cells (ranging from 104 to 106 cells) with similar cellular characteristics in a single flow. Constructs were designed to constitutively express mCherry at the intracellular C-terminus (thus allowing a rapid assessment of the total protein expression) and express an extracellular-facing hemagglutinin (HA) epitope to estimate the cell surface expression of membrane proteins using an anti-HA fluorescence conjugated antibody. To avoid false negative, experiments were also conducted in permeabilized cells to confirm the accessibility and proper expression of the HA epitope. The detailed procedure provides: (1) design of tagged DNA (deoxyribonucleic acid) constructs, (2) lipid-mediated transfection of constructs in tsA-201 cells, (3) culture, harvest, and staining of non-permeabilized and permeabilized cells, and (4) acquisition and analysis of fluorescent signals. Additionally, the basic principles of flow cytometry are explained and the experimental design, including the choice of fluorophores, titration of the HA antibody and control experiments, is thoroughly discussed. This specific approach offers objective relative quantification of the total and cell surface expression of ion channels that can be extended to study ion pumps and plasma membrane transporters.
Introducción
Este documento proporciona un ensayo fiable para informar de la superficie celular de expresión relativa de las proteínas de membrana como canales iónicos expresados en las células recombinantes utilizando la tecnología de citometría de flujo existente. Los canales iónicos son proteínas de membrana formadores de poros que son responsables del control de señales eléctricas por gating el flujo de iones a través de la membrana celular. Se clasifican por el mecanismo de activación, la naturaleza, y la selectividad de las especies de iones que transitan a través del poro donde se localizan. En los niveles celulares y tisulares, los flujos de iones a través de los canales iónicos macroscópicas son el producto de las propiedades biofísicas 1 (gating y la permeabilidad), bioquímica (fosforilación), y la biogénesis (síntesis, glicosilación, el tráfico y la degradación). Cada uno de estos procesos es única para cada tipo de canales de iones y está optimizado para cumplir el papel fisiológico del canal de iones. En consecuencia, las alteraciones en cualquiera de estos procesos-finas sintonizado a través de unaheredada o una modificación genética, a menudo referido como "canalopatía", puede ser perjudicial para la homeostasis celular. Es importante hacer hincapié en que la entrega de la cantidad "correcta" de los canales iónicos en la superficie celular es crítica para la homeostasis celular. Incluso pequeños incrementos (ganancia de función) y disminuciones pequeñas (pérdida de la función) en la actividad de los canales iónicos tienen el potencial de causar una patología grave durante toda la vida. Los defectos en la entrega de la superficie celular de los canales iónicos maduro es un determinante importante en numerosos canalopatías, como la fibrosis quística (CFTR canal iónico) 2 y arritmias cardiacas de forma síndrome de QT largo (canales de potasio cardíacos) 3.
Canalopatías se asocian con la muerte súbita cardiaca 4. La prevalencia actual en todo el mundo de todas las canalopatías cardiacas se piensa que es al menos 1: 2,000-1: 3,000 por persona 5 y son responsables de cerca de la mitad de la súbita arrítmica ca muerte cardíacases 6. Disfunción cardíaca en sodio dependientes de voltaje, potasio, calcio y los canales iónicos selectivos se sabe que juegan un papel clave en este proceso. Se requiere que el 1.2 canales de calcio dependientes de voltaje tipo L Ca V para iniciar la contracción del músculo cardíaco sincronizado. La cardiaca de tipo L Ca V 1.2 canales es un complejo de proteínas de múltiples subunidades compuesto por el principal Ca subunidad α1 V de formación de poros y Ca V SS y Ca V α2δ1 subunidades auxiliares 7-12. Tenga en cuenta que se requiere el complemento completo de subunidades auxiliares para producir funcionales Ca V 1.2 canales en la membrana plasmática y las interacciones dinámicas entre estas subunidades son esenciales para apoyar la función eléctrica normal del corazón 13. Ca V ß promueve la expresión de la superficie celular de Ca V 1.2 canales a través de un nanomolar de interacción hidrofóbica no covalente 14. La co-expresión de la subunidad V wi α2δ1 Caunido a la ß TH Ca V V α1 Ca estimula la expresión corriente de pico (de 5 a 10 veces) y promueve la activación del canal a voltajes más negativos. Ganancia de función de las mutaciones de la subunidad formadora de poros Ca V 1.2 se han asociado con una forma de arritmias ventriculares llamado el síndrome de QT largo 15 mientras que una serie de mutaciones puntuales en las tres subunidades principales que forman el tipo L Ca V 1.2 canales han sido identificados en sujetos que sufren de arritmias de forma síndrome de QT corto 16,17. Los canales iónicos son proteínas de membrana que pueden ser investigados desde una perspectiva bioquímica (la química de proteínas) o utilizando herramientas electrofisiológicos (máquinas generadoras de corriente) y, a menudo utilizando estos enfoques complementarios. Electrofisiología, en particular de células enteras patch-sujeción, es un enfoque adecuado para dilucidar la función de los canales iónicos 15 pero no puede resolver modificaciones en el tráfico de proteínas a partir de los cambios en su biofísicopropiedades. química de proteínas ha, sin embargo, a menudo limitado el uso debido a la relativamente baja expresión de grandes proteínas de la membrana en relación con las proteínas solubles más pequeños. Robustos métodos de alto rendimiento utilizando la lectura de fluorescencia se deben desarrollar con el fin de abordar específicamente los defectos de la biogénesis de proteínas que causan cambios en la expresión de la superficie celular de los canales iónicos.
La citometría de flujo es una tecnología biofísico empleada en el recuento de células, la clasificación, la detección de biomarcadores y la ingeniería de proteínas 18. Cuando se inyecta una solución de muestra de células vivas o partículas en un citómetro de flujo, las células se ordenan en una única corriente que se puede probar por el sistema de detección de la máquina (Figura 1). La primera citómetro de flujo instrumento producido en 1956 19 detectado sólo un parámetro pero citómetros de flujo modernos tienen múltiples láseres y detectores de fluorescencia que permiten la detección de más de 30 parámetros fluorescentes 20,21.Filtros y espejos (óptica de emisión) dirigen la dispersión de la luz o luz fluorescente de las células a una red electrónica (fotodiodo y detectores) que convierten la luz en proporción a su intensidad. Los datos digitales se analizan usando un software especializado y la salida principal se muestra como un gráfico de puntos 21.

Figura 1:. Principios biofísicos de citometría de flujo clasificar las células individuales son empujados a través de una boquilla a alta presión dentro de una corriente de fluido de funda que les mueve a través de uno o más puntos de interrogación láser. El haz de luz es desviado por las células que pasan y la luz recogida en la dirección hacia adelante (Forward Scatter, FCS) se envía a un fotodiodo que convierte la luz en una señal proporcional al tamaño de la célula. La luz también se recoge en un ángulo de 90 ° a la trayectoria de láser y se envía a detectores (también llamados fotomultiplicadores (PMT)).Esta luz se dirige a través de espejos dicroicos que permiten la detección de la señal de dispersión lateral (SSC), que refleja la granularidad dentro de las células, y las emisiones fluorescentes si fluorocromos excitados están presentes en la célula. Tres detectores (verde, amarillo y rojo) se representan con diferentes filtros de paso de banda de longitud de onda, lo que permite la detección simultánea de diferentes fluorocromos. Las diferentes señales son digitalizadas por un ordenador externo y se convierten en datos que serán analizados para cuantificar las características de las células. Por favor, haga clic aquí para ver una versión más grande de esta figura.
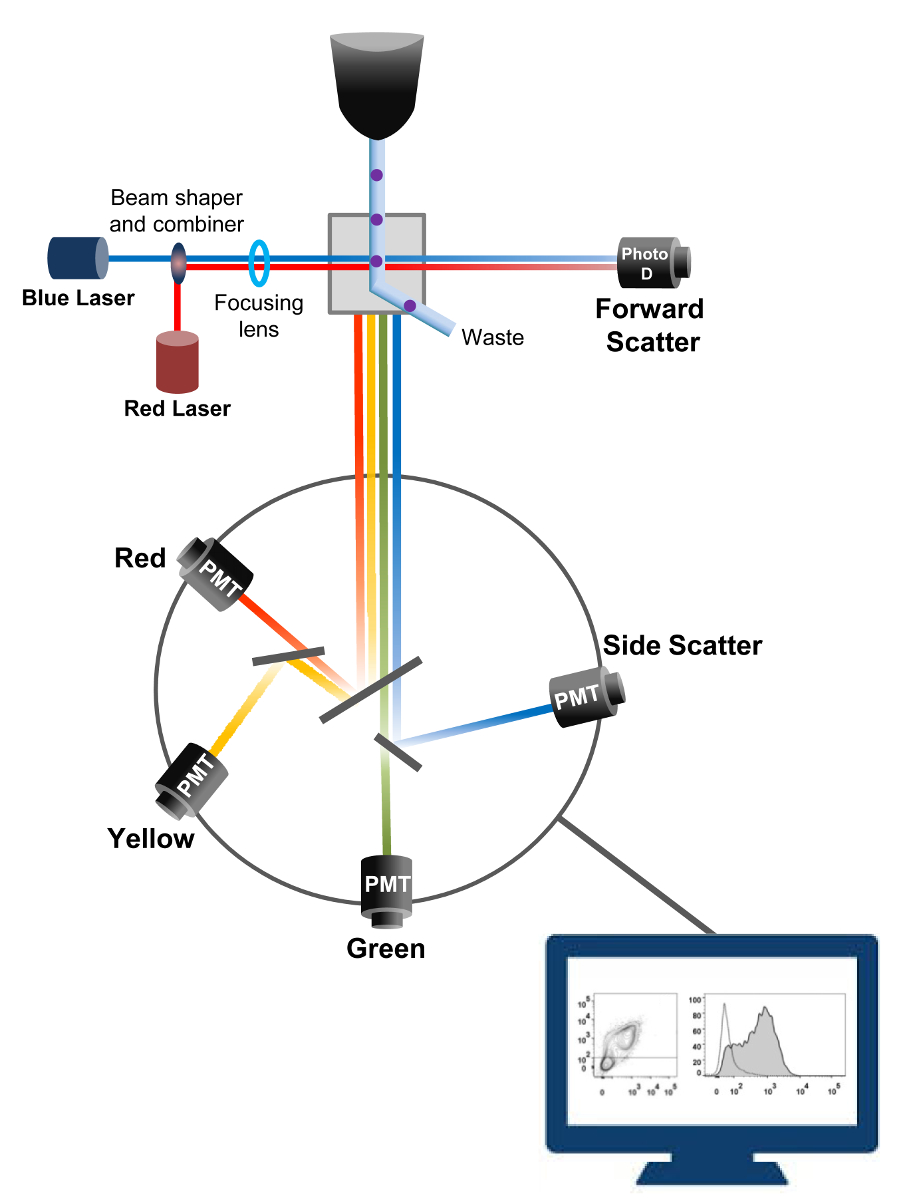{kind=link}
La capacidad de alto rendimiento de citómetros de flujo fue explotada para cuantificar la expresión en la membrana relativa de tipo salvaje recombinante y la trata con deficiencia de voltaje de tipo L Ca V 1.2 canales y subunidades asociadas en las células vivas. construcciones de ADNc coding para las proteínas se marcaron doblemente para llevar simultáneamente un epítopo no fluorescente extracelular que puede ser detectado por un anticuerpo conjugado fluorescente impermeable y un fluoróforo intracelular que es constitutivamente fluorescente. Tanto el epítopo extracelular, insertado en un bucle extracelular de la proteína, y el fluoróforo intracelular, insertado después de la C-terminal, se traducen con la proteína. En esta serie de experimentos, la proteína V α2δ1 Ca fue diseñado para expresar una hemaglutinina extracelular (HA) epítopo (YPYDVPDYA) detectada por un impermeable FITC (isotiocianato de fluoresceína) conjugada anti-HA y mCherry como el fluoróforo intracelular intrínseco. Para determinar el nivel de expresión de la superficie celular relativa de la V α2δ1 mCherry-Ca proteína etiquetada con HA, se recogieron las células recombinantes que expresan la proteína de fusión después de la transfección, y se tiñeron con el anticuerpo monoclonal de ratón anti-HA antibod etiqueta de epítopo conjugado con FITCy (Figura 2). FITC es un compuesto fluorescente orgánico que es considerablemente más pequeño que los reporteros de la enzima y por lo tanto no es tan probable que interfiera con la función biológica. mCherry- Ca V α2δ1-HA sobreexpresa en TSA-201cells, produce un aumento de 3 registro significativo de la fluorescencia FITC y la fluorescencia mCherry en gráficos bidimensionales 22. Dado que el epítopo HA se encuentra en la porción extracelular de la proteína, la intensidad de fluorescencia de FITC obtiene en presencia de células intactas reflejan el índice relativo de la expresión de la superficie celular de la proteína etiquetada con HA. La accesibilidad del epítopo HA en las construcciones se valida sistemáticamente mediante la medición de la señal de FITC después de la permeabilización celular. Esta medida también sirve para corroborar la expresión de la proteína total normalizada ya que las intensidades relativas de fluorescencia de FITC estimaron en células permeabilizadas son cualitativamente comparables con los valores de fluorescencia relativa FOr mCherry mide bajo permeabilizadas y no permeabilizadas condiciones 22,23. Es importante tener en cuenta que el espectro de fluorescencia intrínseca se desplaza hacia valores más altos después de la permeabilización, pero que el único valor que se informa es el cambio en la intensidad de fluorescencia en comparación con la construcción de control. Los cambios relativos en la intensidad de fluorescencia de las construcciones de ensayo se calculan utilizando la intensidad de fluorescencia ΔMean (ΔMFI) los valores para cada fluoróforo (mCherry o FITC). Los experimentos están diseñados para medir la intensidad de fluorescencia de la construcción de ensayo con respecto a la intensidad de fluorescencia de la construcción de control expresado en las mismas condiciones para limitar las variaciones experimentales en la fluorescencia intrínseca del anticuerpo fluoróforo conjugado. Dos proteínas de la membrana fueron estudiados con éxito utilizando este ensayo: la subunidad formadora de poros del canal de calcio dependiente de voltaje de tipo L Ca V 1.2 14,22 y en una serie diferente deexperimentos, el auxiliar extracelular Ca V α2δ1 subunidad 22,23. Se usó el siguiente protocolo para determinar la expresión de la superficie celular de la Ca V α2δ1 subunidad de la cardíaco L-tipo Ca V 1.2 canal en condiciones de control y después de las mutaciones que afectan a la modificación postraduccional del canal iónico. En condiciones experimentales estandarizadas, la fluorescencia de la superficie celular de FITC aumenta casi linealmente con la expresión de ADNc que codifica para las proteínas α2δ1-HA mCherry-Ca V (Figura 5 de la referencia 22).

Figura 2:. Representación esquemática de etiquetado total y membrana en la citometría de flujo protocolo experimental El esquema se describen algunos de los principales pasos necesarios para cuantificar la expresión total y superficie celular relativa de los canales iónicos recombinantes por flflujo citometría. Las células se transfectaron con la construcción de doble etiquetado mCherry-Ca V α2δ1-HA en TSA-201 células (1) y se tiñeron antes o después de la permeabilización (2). Multiparamétricos datos se adquieren en un citómetro de flujo (3) para el análisis multivariado (4). Haga clic aquí para ver una versión más grande de esta figura.
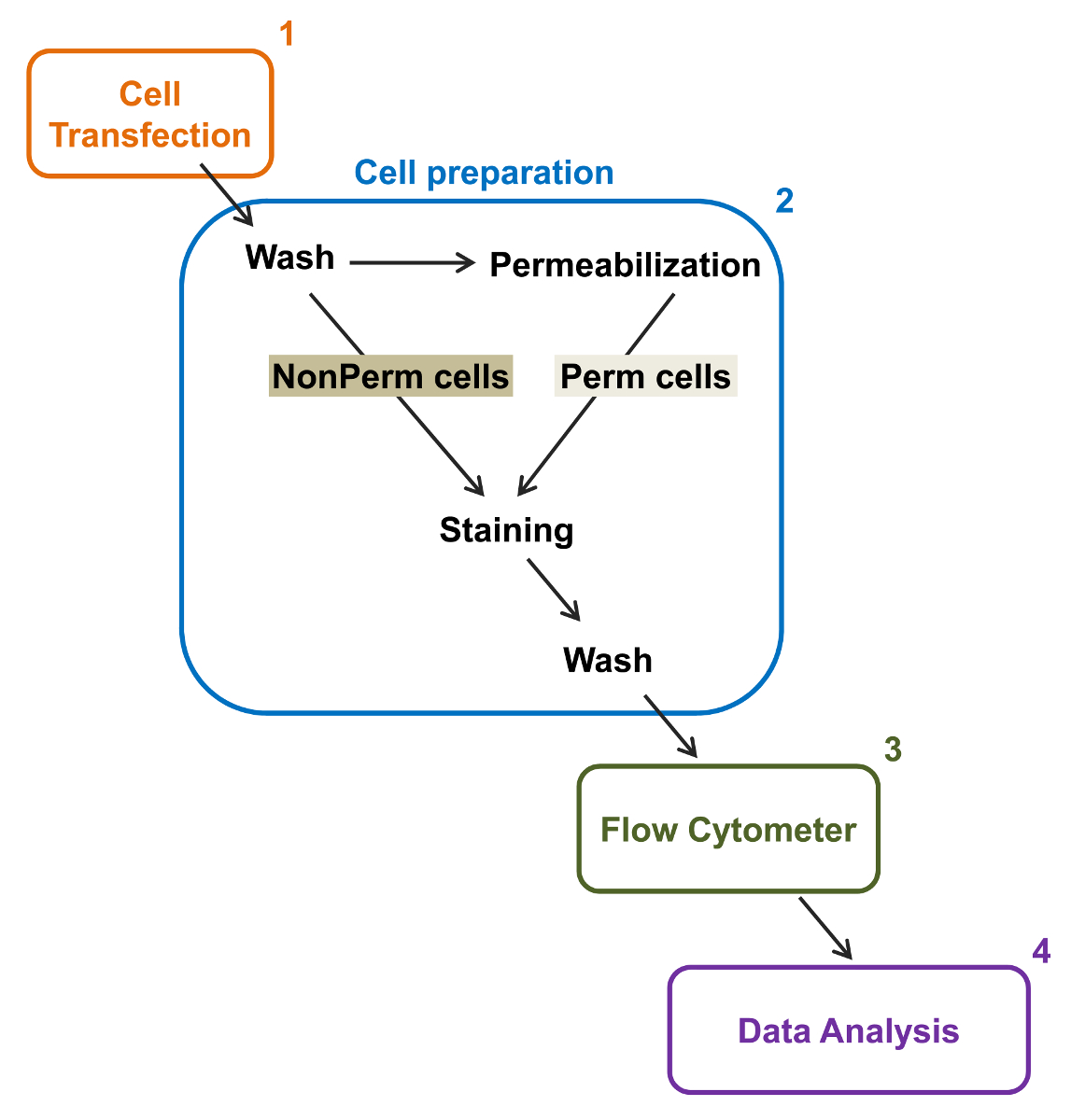{kind=link}
Protocolo
1. construcciones de ADN Tagged Doblemente
- Insertar el epítopo HA (YPYDVPDYA) en el enlazador extracelular de Ca V α2δ1 entre D676 y R677 por mutagénesis dirigida al sitio (Figura 3B) 20. Utilice gccggattatgcgGGAAAACTCCAAACAACC Cebador inverso y acatcatacggataTCAATAAATTCATTGAAATTTAAAAGAAATTC imprimación.
- Subclonar la secuencia de ADNc del V α2δ1 HA Ca etiquetado en el vector de expresión de mamífero pmCherry-N1 diseñado para expresar la proteína fusionada al N-terminal de mCherry entre los sitios SacI y SalI (Figura 3B) 20.
NOTA: la función del canal apropiado necesita ser probado con la construcción de control usando métodos electrofisiológicos estándar 24.
2. La transfección transitoria mediada por liposomas (30 min, todos los pasos se realizan bajo flujo laminar Hood)
- Día 1: Placa de medio millón de TSA-201 células (o) en HEKT35 placas de cultivo mm con 2 ml de Dulbecco medio esencial mínimo de alta glucosa (DMEM-HG) complementado con 10% de suero bovino fetal (FBS) y medio (PS) cultura de penicilina-estreptomicina al 1%. Recuento de células utilizando un hemocitómetro estándar. Evaluar la viabilidad celular a partir de una fracción de la muestra de células usando azul tripán. suficientes células de la placa para alcanzar 90% de confluencia en el momento de la transfección.
- Día 2: Cambio medio de cultivo con 2 ml de (37 ° C) medio de cultivo fresco precalentado sin PS.
- Para cada muestra de transfección, preparar dos tubos de 1,5 ml. En el tubo 1, diluir 4 g de ADN en 250 l de medio de suero reducido. En el tubo 2, la mezcla 10 l de reactivo de transfección mediada por liposomas con 250 l de medio de cultivo reducido suero. Mezclar suavemente el reactivo de transfección antes de su uso.
- Incubar durante 5 min a temperatura ambiente.
- Combinar los contenidos de tubo 1 y el tubo 2, se mezcla suavemente y se incuba al menos 20 min a temperatura ambiente.
- Añadir los liposomas / DNA los complejos a las células cultivadas y agitándolo suavemente la placa de cultivo para mezclar.
- Se incuba a 37 ° C en 5% de CO2 durante 24 horas.
3. La tinción de células para citometría de flujo (3 hr)
- Preparación de muestras de células
- Día 3: Eliminar medio de la placa de cultivo cuidadosamente y se lavan las células con 400 l de pre-calentado (37 ° C) 0,05% de tripsina-EDTA 1x (ácido etilendiaminotetraacético).
- Añadir 400 l de tripsina-EDTA y se incuba la placa a 37 ° C bajo 5% de CO2 durante 5 min para permitir que las células se desprenden de la placa.
- Detener la digestión con enzimas mediante la adición de 1 ml de medio de cultivo frío sin PS y lavar todas las células de la superficie pipeteando suavemente 4-5 veces. Evitar el exceso de la digestión y el exceso de pipeteado para reducir la muerte celular.
- Recoger las células en tubos de 1,5 ml y colocar inmediatamente en hielo. Utilice soluciones frías de hielo y mantener las células a 4 ° C para evitar la internalizaciónantígenos de la superficie. Disminución de iluminación para limitar photobleaching de la señal fluorescente.
- tubos de centrifugación a 400 xg durante 5 min a 4 ° C. aspirar con cuidado y descartar el sobrenadante.
- Vuelva a suspender el sedimento en 1 ml de 1x solución salina tamponada con fosfato (PBS) para preparar una suspensión de células individuales.
- En resumen vórtice los tubos suavemente y repita los pasos 3.1.5 y 3.1.6 para eliminar por completo el medio de cultivo.
- Vuelva a suspender el precipitado en 600 l de PBS 1x y ajustar la concentración de células a un mínimo de 3 x 10 6 células / ml.
- Dividir las células en dos nuevos tubos de 1,5 ml para la tinción extracelular e intracelular. Incluir los controles adecuados para discriminar la tinción específica de tinción no específica.
NOTA: El anticuerpo de control de isotipo ayuda a evaluar el nivel de la tinción de fondo e idealmente debe coincidir con las especies huéspedes de cada anticuerpo primario, isotipo y fluoróforo. Utilice el control de isotipo y el anticuerpo conjugado al mismola concentración de proteínas.

Tabla 1: citometría de flujo de muestras de control para el experimento no se permeabilizaron y se permeabilizaron células Cada experimento tiene que incluir los siguientes controles negativos:. (1) células no transfectadas (sin anticuerpo, con el isotipo o con el anticuerpo conjugado). (2) Las células transfectadas con la proteína de interés se subclona en un plásmido sin constitutiva fluorocromo intracelular fluorescente (pCMV-Ca V α2δ1-HA) o con la construcción doblemente etiquetada (pmCherry-Ca V α2δ1 y se incubaron sin anticuerpo, con el isotipo o con el anticuerpo conjugado). controles individuales de color se utilizan para la compensación de superposición de emisión fluorocromo. Los mismos controles se ejecutan para no permeabilized y se permeabilizaron las condiciones de cada serie de experimentos.
- La tinción de superficie celular de IntactLas células vivas
- Alícuota de 1 x 10 6 células / 100 l en tubos de 1,5 ml.
- Añadir el anticuerpo monoclonal conjugado con FITC anti-HA en 5 mg / ml y, vórtice antes de incubar las células en una plataforma de balancín (200 rpm) en la oscuridad a 4 ° C durante 45 min.
NOTA: Se determinó la concentración óptima de anticuerpo en los experimentos de titulación preliminar (Figura 4). - Eliminar las células de la oscuridad y añadir 900 l de PBS 1x / tubo. Centrifugar a 400 xg durante 5 min a 4 ° C.
- Aspirar el sobrenadante y resuspender el precipitado en 1 ml de PBS 1x, vortex y centrifugar a 400 g durante 5 min a 4 ° C.
- Repetir el lavado (etapa 3.2.4) dos veces para eliminar cualquier anticuerpo no unido. Si se utiliza un anticuerpo primario sin conjugar, se incuban con el anticuerpo secundario apropiado.
- Después del lavado final, resuspender las células en 500 l de 1x PBS y transferir la suspensión de células individuales en 5 ml de citometría de flujo tubos. Mantenga la células en la oscuridad a 4 ° C hasta que se ejecuta la muestra.
- Ejecutar las muestras en un citómetro de flujo. Para obtener los mejores resultados, analizar las células en el citómetro de flujo tan pronto como sea posible y no más tarde de 24 horas después.
- La tinción intracelular: La fijación, permeabilización y tinción
- Alícuota 1 x 10 6 células / 100 l en tubos de 1,5 ml y centrifugar a 400 xg durante 5 min a 4 ° C.
- Desechar las células sobrenadante y resuspender en 100 l de solución de fijación-permeabilización directamente desde el almacén.
- Incubar en la oscuridad a 4 ° C durante 20 minutos.
- Añadir 100 l de 1 x tampón de permeabilización lavado recién preparada (diluida 10x tampón de permeabilización de lavado en agua destilada H 2 O). células de vórtice y sedimento utilizando una centrífuga de mesa a 400 xg durante 5 min a 4 ° C.
- Aspirar y descartar el sobrenadante.
- Repita los pasos 3.3.4 y 3.3.5.
- Añadir anticuerpo anti-HA monoclonal conjugado con FITC a 5mg / ml en 100 l de 1 x tampón de permeabilización y el lavado, vórtice antes de incubar las células en la oscuridad a 4 ° C durante 30 minutos.
NOTA: La tinción intracelular se lleva a cabo siguiendo el mismo procedimiento que el utilizado para la tinción de la superficie celular. permeabilización celular mediada saponina es sin embargo, un proceso reversible de forma rápida, por lo tanto, es importante reemplazar 1x PBS con tampón 1x Perm / Wash para mantener las células en la presencia constante de la saponina durante la tinción intracelular. - Eliminar las células de la oscuridad y añadir 100 l de tampón de permeabilización-lavado. Centrifugar a 400 xg durante 5 min a 4 ° C.
- Aspirar cuidadosamente el sobrenadante y resuspender el precipitado en 100 l de tampón de permeabilización de lavado, vórtice y se centrifuga a 400 g durante 5 min a 4 ° C.
- Repetir el lavado (paso 3.3.9) una vez más para eliminar cualquier anticuerpo no unido.
- Después del lavado final, resuspender las células en 500 l de 1x PBS y transferir el pecadosuspensión de células GLE a 5 ml de citometría de flujo tubos. Mantener las células en la oscuridad a 4 ° C hasta que la inyección de la muestra en el citómetro de flujo.
- Ejecutar las muestras en un citómetro de flujo. Ejecutar las muestras fijadas en el citómetro tan pronto como sea posible pero no más tarde de 1 semana después de la tinción. Ejecutar las células no permeabilizadas y permeabilizadas en el mismo día.
4. Citometría de Flujo
- Citómetro de flujo célula de establecimiento Clasificador diario
- Encienda el software de citometría de flujo. Antes del experimento, calibrar y configurar el citómetro de flujo separador celular para garantizar un rendimiento óptimo del instrumento (es decir, láser y la óptica están funcionando según las especificaciones, el láser y la celda de flujo están alineados correctamente) mediante el uso de cuentas de configuración del instrumento.
- Utilice la boquilla 100 micras con presión de funda 20 psi.
NOTA: La boquilla no tiene que ser cambiado en un banco citómetro de flujo. - Ajuste del caudal del citómetro de acuerdo con la fabriespecificación er. Excesivamente altas velocidades de flujo disminuirá la sensibilidad en la detección de variaciones en la fluorescencia.
- Seleccione azul (488 nm para excitar la fluoresceína Isothiocayanate o FITC) y amarillo-verde (561 nm para excitar mCherry) láser. Recoger los niveles de fluorescencia FITC y mCherry con una nm 530/30 y con un filtro de paso de banda 610/20 nm respectivamente.
- Adquirir la dispersión frontal (FCS) frente a dispersión lateral (SSC) diagrama de puntos de células no teñidas utilizando la escala lineal. Ajustar la amplificación de cada detector para visualizar las células en el cuadrante inferior izquierdo del gráfico de puntos.
- Lectura muestra de células intactas para no permeabilizadas-
- Ajuste el P1gate para células no permeabilizadas en vivo por la delimitación de una forma libre alrededor de las células a analizar con exclusión de los desechos celulares y agregados de células, lo que limita la señal de fluorescencia de las células intactas.
NOTA: en vivo colorantes de exclusión / muertas se pueden utilizar para facilitar la colocación de compuerta en células vivas. Conjunto 10.000 eventos para grabaren la puerta de parada P1. Establecer esto a un mayor número de eventos si es necesario. - Adquirir mCherry frente FITC gráfico de contorno de dos parámetros de línea de base para detectar autofluorescencia de las células teñidas. Utilice la escala bi-logarítmica para mostrar los valores negativos y mejorar la resolución entre las poblaciones 25. Ajustar el voltaje de cada detector para establecer las células negativas no teñidas dentro de la parte inferior de los primeros diez unidades de las parcelas de intensidad de fluorescencia de registro.
- Adquirir todas las muestras intactas no permeabilizadas utilizando los ajustes establecidos en 4.1.5 y 4.1.6 y recoger FSC, SSC y señales de los detectores de fluorescencia.
- Exportar y guardar archivos * .fcs que se utilizarán para el análisis mediante citometría de flujo de software de análisis.
- Ajuste el P1gate para células no permeabilizadas en vivo por la delimitación de una forma libre alrededor de las células a analizar con exclusión de los desechos celulares y agregados de células, lo que limita la señal de fluorescencia de las células intactas.
- Lectura de la muestra de células permeabilizadas
- Mover la puerta P1 para seleccionar células vivas en las muestras permeabilizadas y ajustar el FSC y SSC tensión como se muestra en 4.1.5 y 4.1.6.
- Adquirir todas las muestras permeabilizadas y recoger FSC, SSC unaseñales de ND en los detectores de fluorescencia.
- Exportar y guardar archivos * .fcs que se utilizarán para el análisis mediante citometría de flujo de software de análisis.
- Análisis de los datos
- Poner en marcha el análisis de citometría de flujo de software e importación * .fcs archivos guardados en 4.2.4 y 4.3.3.
- Haga clic en el primer ejemplo que aparece en la ventana del espacio de trabajo. Una nueva ventana llamada después de que el número de identificación de tubo se abre automáticamente. Iniciar el proceso de compuerta en la trama de SSC frente a FSC. Dibuje una puerta (P1) mediante el icono de la elipse alrededor de las células vivas y eliminar cualquier desecho, células muertas, o agregados que tienen diferentes dispersión frontal y dispersión lateral de células vivas
- Para dibujar el gráfico de contorno de dos parámetros de la mCherry (eje y) frente a FITC (eje x) la intensidad de fluorescencia de las células vivas, primero haga clic en el eje x y elegir el FITC-Un canal y, a continuación, haga clic en la ordenada y elegir el eje x-PE-A mCherry canal. Haga clic en el icono "Quad" para colocar el marcador cuadrante en el borde de unautofluorescent células en cada canal de fluorescencia.
NOTA: La puerta en torno a las células FITC y mCherry positivos es la puerta P2. La población de células negativo de fluorescencia se conoce como la puerta P3. Vea la Figura 5 para el método de compuerta representativo utilizado en este artículo. - Seleccionar P2 y P3 puertas y hacer clic en el icono "Agregar Estadísticas" en la ventana del área de trabajo original. Haga clic en "Count" (número de células positivas) y hacer clic en "Mean" (intensidad media de fluorescencia de cada fluorocromo) o "mediana" (Mediana Intensidad de fluorescencia de cada fluorocromo) estadísticas entre la lista de opciones. Haga clic en la opción "Agregar Estadísticas" de nuevo. Todos estos valores se transfieren automáticamente a la ventana de área de trabajo original.
NOTA: El "medio" se utiliza sólo si la intensidad de la fluorescencia sigue una distribución normal. En cualquier otro caso, haga clic en la pestaña "mediana". IMF por lo tanto puede referirse a fluorescencia media intensitLa intensidad de la fluorescencia y o mediana.
NOTA: El siguiente paso es aplicar los parámetros y estadísticas de Gates a todas las muestras sondeadas por el citómetro. - En la ventana de área de trabajo, utilizar el ratón para arrastrar y soltar los parámetros puertas y estadísticas sobre la línea marcada todas las muestras.
- Generar un informe lote de gráficos bidimensionales de nivel, (mCherry vs FITC) y los histogramas (recuento de células frente a la intensidad de fluorescencia) para las células no permeabilizadas y permeabilizadas (Figuras 6A - B).
- De las estadísticas generadas en el paso 4.4.4, calcular la intensidad media de fluorescencia (MFI) para cada fluorocromo para las células teñidas. A partir de este valor, restar el valor de MFI obtenido a partir de células no teñidas de cuantificar la superficie y la expresión total de la proteína de interés.
- Reportar los valores ΔMFI para cada fluoróforo (mCherry y FITC) (Figura 6 C - D). Normalizar la ΔMFI medido para el Ca V α2δ1 construcción de mutantes con el valor obtenido para ΔMFI FITC y mCherry con el constructo WT.
NOTA: El valor absoluto de la intensidad de fluorescencia puede variar mucho dependiendo del lote de anticuerpos y las capacidades técnicas de cada trabajador de laboratorio, de ahí la necesidad de normalizar la intensidad de fluorescencia de la construcción de mutantes utilizando la construcción WT.
Resultados
En este artículo se describe un protocolo fiable para cuantificar la superficie total y celular de los canales iónicos recombinantes expresadas en TSA-201cells por un flujo de dos colores citometría de ensayo. A modo de ejemplo, la superficie de la célula y la expresión relativa de proteína total se cuantificó para el V α2δ1subunit Ca. Para llevar a cabo el flujo de dos colores ensayo de citometría, Ca V α2δ1 fue etiquetado doblemente para expresar un ep...
Discusión
Este ensayo basado en citometría de flujo se aplicó con éxito a la medición de los niveles totales y de la superficie celular relativas de las subunidades marcado fluorescentemente formadores de poros y asociado de los canales de calcio dependientes de voltaje 14,22,26. Se utiliza mejor en la investigación de los efectos de mutaciones genéticas y por lo tanto requiere que la intensidad de la fluorescencia intrínseca de la construcción de tipo salvaje marcado fluorescentemente etiquetado sea al menos 1...
Divulgaciones
The authors declare that they have no competing financial interests.
Agradecimientos
We thank Mr. Serge Sénéchal and Dr. Jacques Thibodeau for sharing their expertise and granting us access to their flow cytometry and cell sorting platform. This work was completed with the operating grant 130256 from the Canadian Institutes of Health Research, a grant-in-aid from the Canadian Heart and Stroke Foundation, and support from the "Fondation de l'Institut de Cardiologie de Montréal" to L.P.
Materiales
| Name | Company | Catalog Number | Comments |
| Q5 Site-Directed Mutagenesis Kit | New England Biolabs | E0554S | Can be substitute with QuickChange site-directed mutagenesis Kit (Agilent, #200523). |
| Tubes 1.5 ml | Sarstedt | 72-690-001 | |
| Tubes 15 ml | Sarstedt | 62-554-002 | |
| Disposable graduated Tranfer Pipets | VWR | 160001-192 | |
| 100 mm culture dish | Corning | 430167 | For standard culture of HEKT cells. |
| 35 mm culture dish | Falcon | 353001 | For standard culture of HEKT cells. |
| Serological pipette 1 ml | Sarstedt | 86.1251.001 | |
| Serological pipette 5 ml | Sarstedt | 86.1253.001 | |
| Serological pipette 10 ml | Sarstedt | 86.1254.001 | |
| Serological pipette 25 ml | Sarstedt | 86.1285.001 | |
| Dulbecco's high-glucose medium | Life Technologies | 12100-046 | Warm in 37 °C water bath before use. |
| Fetal Bovine Serum, qualified, heat inactivated, US origin | Life Technologies | 16140-071 | |
| Penicillin-Streptomycin (10,000 U/ml) | Life Technologies | 15140-122 | |
| Lipofectamine 2000 | Life Technologies | 11668-019 | For liposomal transfection. Can be substituted with calcium phosphate transfection. |
| Opti-MEM I Reduced Serum Medium | Life Technologies | 31985-070 | Warm in 37 °C water bath before use. |
| Trypsin-EDTA (1x) 0.05%, phenol red | Life Technologies | 25300-062 | |
| 1.5 ml microtubes | Sarstedt | 72.690.001 | |
| Phosphate Buffered Saline 1x | Fisher | BP661-10 | Can be "home-made". |
| Anti-HA FITC conjugated antibody | Sigma | H7411 | |
| IgG1−FITC Isotype Control antibody | Sigma | F6397 | |
| BD Cytofix/Cytoperm Fixation/Permeabilization Solution Kit | BD Biosciences | 554714 | Fixation/Permeabilization. Permeabilization/Wash solution, store at 4 °C. |
| Hemacytometer | Fisher | 49105161 | |
| Trypan Blue | Fisher | 15250061 | To access cell viability. |
| Refrigerated Microcentrifuge, 5430R | Eppendorf | A14H172200 | |
| Forma Steri-Cycle CO2 Incubator | Fisher | 370 | |
| Laboratory Platform Rocker | Fisher | 545034 | |
| Water Bath | VWR | 89032-216 | |
| BD FACSARIA III | BD Biosciences | 648282 | Flow cytometer. |
| FlowJo Software v10 | FlowJo | FlowJo v10 Dongle | For data analysis. |
Referencias
- Delisle, B. P., Anson, B. D., Rajamani, S., January, C. T. Biology of Cardiac Arrhythmias: Ion Channel Protein Trafficking. Circ. Res. 94, 1418-1428 (2004).
- Birault, V., Solari, R., Hanrahan, J., Thomas, D. Y. Correctors of the basic trafficking defect of the mutant F508del-CFTR that causes cystic fibrosis. Curr Opin Chem Biol. 17, 353-360 (2013).
- Balijepalli, S. Y., Anderson, C. L., Lin, E. C., January, C. T. Rescue of Mutated Cardiac Ion Channels in Inherited Arrhythmia Syndromes. J. Cardiovas Pharm. 56, 113-122 (2010).
- Gargus, J. J. Unraveling Monogenic Channelopathies and Their Implications for Complex Polygenic Disease. Am. J. Hum. Genet. 72, 785-803 (2003).
- Abriel, H., Zaklyazminskaya, E. V. Cardiac channelopathies: Genetic and molecular mechanisms. Gene. 517, 1-11 (2013).
- Behr, E. R., et al. Sudden arrhythmic death syndrome: familial evaluation identifies inheritable heart disease in the majority of families. Eur Heart J. 29, 1670-1680 (2008).
- Catterall, W. A. Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev.Biol. 16, 521-555 (2000).
- Peterson, B. Z., DeMaria, C. D., Adelman, J. P., Yue, D. T. Calmodulin is the Ca2+ sensor for Ca2+ -dependent inactivation of L- type calcium channels. Neuron. 22, 549-558 (1999).
- Dolphin, A. C. Calcium channel diversity: multiple roles of calcium channel subunits. Curr.Opin.Neurobiol. 19, 237-244 (2009).
- Dai, S., Hall, D. D., Hell, J. W. Supramolecular assemblies and localized regulation of voltage-gated ion channels. Physiol Rev. 89, 411-452 (2009).
- Gao, T., et al. Identification and subcellular localization of the subunits of L-type calcium channels and adenylyl cyclase in cardiac myocytes. J. Biol. Chem. 272, 19401-19407 (1997).
- Carl, S. L., et al. Immunolocalization of sarcolemmal dihydropyridine receptor and sarcoplasmic reticular triadin and ryanodine receptor in rabbit ventricle and atrium. J. Cell Biol. 129, 673-682 (1995).
- Abriel, H., Rougier, J. S., Jalife, J. Ion Channel Macromolecular Complexes in Cardiomyocytes: Roles in Sudden Cardiac Death. Circ. Res. 116, 1971-1988 (2015).
- Bourdin, B., et al. Molecular Determinants of the Cavb-induced Plasma Membrane Targeting of the Cav1.2 Channel. J. Biol. Chem. 285, 22853-22863 (2010).
- Raybaud, A., et al. The Role of the GX9GX3G Motif in the Gating of High Voltage-activated Calcium Channels. J. Biol. Chem. 281, 39424-39436 (2006).
- Burashnikov, E., et al. Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm. 7, 1872-1882 (2010).
- Hennessey, J. A., et al. A CACNA1C Variant Associated with Reduced Voltage-Dependent Inactivation, Increased Cav1.2 Channel Window Current, and Arrhythmogenesis. PLoS ONE. 9, e106982 (2014).
- Adan, A., Alizada, G., Kiraz, Y., Baran, Y., Nalbant, A. Flow cytometry: basic principles and applications. Crit Rev Biotechnol. , 1-14 (2016).
- Graham, M. D. The Coulter Principle: Foundation of an Industry. J. Lab. Autom. 8, 72-81 (2003).
- Baumgarth, N., Roederer, M. A practical approach to multicolor flow cytometry for immunophenotyping. J. Immunol. Methods. 243, 77-97 (2000).
- Rothe, G., Sack, U., Tarnok, A., Rothe, G. . Cellular Diagnostics. Basics, Methods and Clinical Applications of Flow Cytometry. , 53-88 (2009).
- Bourdin, B., et al. Functional Characterization of Cavalpha2delta Mutations Associated with Sudden Cardiac Death. J. Biol. Chem. 290, 2854-2869 (2015).
- Tetreault, M. P., et al. Identification of glycosylation sites essential for surface expression of the Cavalpha2delta1 subunit and modulation of the cardiac Cav1.2 channel activity. J. Biol. Chem. 291, 4826-4843 (2016).
- Senatore, A., Boone, A. N., Spafford, J. D. Optimized Transfection Strategy for Expression and Electrophysiological Recording of Recombinant Voltage-Gated Ion Channels in HEK-293T Cells. J Vis Exp. (47), (2011).
- Herzenberg, L. A., Tung, J., Moore, W. A., Herzenberg, L. A., Parks, D. R. Interpreting flow cytometry data: a guide for the perplexed. Nat.Immunol. 7, 681-685 (2006).
- Shakeri, B., Bourdin, B., Demers-Giroux, P. O., Sauve, R., Parent, L. A quartet of Leucine residues in the Guanylate Kinase domain of Cavbeta determines the plasma membrane density of the Cav2.3 channel. J Biol Chem. 287, 32835-32847 (2012).
- Morton, R. A., Baptista-Hon, D. T., Hales, T. G., Lovinger, D. M. Agonist- and antagonist-induced up-regulation of surface 5-HT3A receptors. Br. J. Pharmacol. 172, 4066-4077 (2015).
- Hoffmann, C., et al. Fluorescent labeling of tetracysteine-tagged proteins in intact cells. Nat. Protocols. 5, 1666-1677 (2010).
- Cockcroft, C. J., Gamper, N. . Ion Channels: Methods and Protocols. , 233-241 (2013).
- Gonzalez-Gutierrez, G., Miranda-Laferte, E., Neely, A., Hidalgo, P. The Src Homology 3 Domain of the beta-Subunit of Voltage-gated Calcium Channels Promotes Endocytosis via Dynamin Interaction. J. Biol.Chem. 282, 2156-2162 (2007).
- Galizzi, J. P., Borsotto, M., Barhanin, J., Fosset, M., Lazdunski, M. Characterization and photoaffinity labeling of receptor sites for the Calcium channel inhibitors d-cis-diltiazem, (+/-)-bepridil, desmethoxyverapamil, and (+)-PN 200-110 in skeletal muscle transverse tubule membranes. J. Biol.Chem. 261, 1393-1397 (1986).
- Bezanilla, F. The voltage sensor in voltage-dependent ion channels. Physiol.Rev. 80, 555-592 (2000).
- Sigworth, F. J. The variance of sodium current fluctuations at the node of Ranvier. J Physiol. 307, 97-129 (1980).
- Bailey, M. A., Grabe, M., Devor, D. C. Characterization of the PCMBS-dependent modification of KCa3.1 channel gating. J. Gen. Physiol. 136, 367-387 (2010).
- Fletcher, P. A., Scriven, D. R., Schulson, M. N., Moore, E. D. Multi-Image Colocalization and Its Statistical Significance. Biophys. J. 99, 1996-2005 (2010).
- Lizotte, E., Tremblay, A., Allen, B. G., Fiset, C. Isolation and characterization of subcellular protein fractions from mouse heart. Anal. Biochem. 345, 47-54 (2005).
- Mattheyses, A. L., Simon, S. M., Rappoport, J. Z. Imaging with total internal reflection fluorescence microscopy for the cell biologist. J. Cell Sci. 123, 3621-3628 (2010).
- Yamamura, H., Suzuki, Y., Imaizumi, Y. New light on ion channel imaging by total internal reflection fluorescence (TIRF) microscopy. J. Pharmacol. Sci. 128, 1-7 (2015).
- Wible, B. A., et al. HERG-Lite-R: A novel comprehensive high-throughput screen for drug-induced hERG risk. J. Pharmacol. Toxicol. Methods. 52, 136-145 (2005).
- Wilde, A. A. M., Brugada, R. Phenotypical Manifestations of Mutations in the Genes Encoding Subunits of the Cardiac Sodium Channel. Circ. Res. 108, 884-887 (2011).
- Milano, A., et al. Sudden Cardiac Arrest and Rare Genetic Variants in the Community. Circ Cardiovasc Genet. , (2016).
- Schnell, U., Dijk, F., Sjollema, K. A., Giepmans, B. N. G. Immunolabeling artifacts and the need for live-cell imaging. Nat. Meth. 9, 152-158 (2012).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados