
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Détermination de la surface de la cellule relative et d'expression totale de Recombinant Canaux ioniques par cytométrie en flux
Dans cet article
Résumé
les arythmies cardiaques héréditaires sont souvent provoquées par des mutations qui modifient la distribution de surface d'un ou plusieurs canaux ioniques. Ici, on adapte les essais de cytométrie en flux pour fournir une quantification de l'expression de la protéine totale et de la surface cellulaire relative des canaux ioniques recombinants exprimés dans des cellules 201 TSA.
Résumé
Inherited or de novo mutations in cation-selective channels may lead to sudden cardiac death. Alteration in the plasma membrane trafficking of these multi-spanning transmembrane proteins, with or without change in channel gating, is often postulated to contribute significantly in this process. It has thus become critical to develop a method to quantify the change of the relative cell surface expression of cardiac ion channels on a large scale. Herein, a detailed protocol is provided to determine the relative total and cell surface expression of cardiac L-type calcium channels CaV1.2 and membrane-associated subunits in tsA-201 cells using two-color fluorescent cytometry assays. Compared with other microscopy-based or immunoblotting-based qualitative methods, flow cytometry experiments are fast, reproducible, and large-volume assays that deliver quantifiable end-points on large samples of live cells (ranging from 104 to 106 cells) with similar cellular characteristics in a single flow. Constructs were designed to constitutively express mCherry at the intracellular C-terminus (thus allowing a rapid assessment of the total protein expression) and express an extracellular-facing hemagglutinin (HA) epitope to estimate the cell surface expression of membrane proteins using an anti-HA fluorescence conjugated antibody. To avoid false negative, experiments were also conducted in permeabilized cells to confirm the accessibility and proper expression of the HA epitope. The detailed procedure provides: (1) design of tagged DNA (deoxyribonucleic acid) constructs, (2) lipid-mediated transfection of constructs in tsA-201 cells, (3) culture, harvest, and staining of non-permeabilized and permeabilized cells, and (4) acquisition and analysis of fluorescent signals. Additionally, the basic principles of flow cytometry are explained and the experimental design, including the choice of fluorophores, titration of the HA antibody and control experiments, is thoroughly discussed. This specific approach offers objective relative quantification of the total and cell surface expression of ion channels that can be extended to study ion pumps and plasma membrane transporters.
Introduction
Ce document fournit un test fiable pour signaler l'expression relative de la surface cellulaire des protéines membranaires telles que des canaux ioniques exprimés dans les cellules recombinantes en utilisant la technologie de cytométrie de flux existant. Les canaux ioniques sont des protéines membranaires porogènes qui sont responsables de la commande des signaux électriques par gating le flux d'ions à travers la membrane cellulaire. Ils sont classés par le mécanisme d'activation, la nature et la sélectivité des espèces d'ions qui transitent à travers le pore où ils sont localisés. Au niveau cellulaire et tissulaire, les flux ioniques macroscopiques à travers les canaux ioniques sont le produit de biophysique propriétés 1 (gating et perméation), biochimique (phosphorylation), et la biogenèse (synthèse, glycosylation, le trafic et la dégradation). Chacun de ces processus est unique pour tous les types de canaux ioniques et elle est optimisée pour remplir le rôle physiologique du canal ionique. Par conséquent, des modifications dans l'un de ces processus affiné grâce à unhéréditaire ou une modification génétique, souvent appelée "canalopathie", peut être préjudiciable à l'homéostasie cellulaire. Il est important de souligner que la prestation du «droit» quantité de canaux ioniques à la surface cellulaire est essentielle pour l'homéostasie cellulaire. Même de petites augmentations (gain de fonction) et de petites diminutions (perte de fonction) dans l'activité des canaux ioniques ont le potentiel de provoquer une pathologie grave au cours d'une vie. Défauts dans la livraison de la surface cellulaire des canaux ioniques matures est un déterminant important dans de nombreux channelopathies, comme la fibrose kystique (canal ionique CFTR) 2 et les arythmies cardiaques de la forme longue de syndrome du QT (canaux potassiques cardiaques) 3.
Channelopathies sont associés à la mort subite cardiaque 4. La prévalence mondiale actuelle de tous les channelopathies cardiaques est considéré comme au moins 1: 2,000-1: 3.000 par personne 5 et sont responsables d'environ la moitié d' un coup arythmique cardiaque mort caSES 6. Dysfonction cardiaque en voltage-dépendants de sodium, de potassium et de calcium canaux ioniques sélectifs sont connus pour jouer un rôle clé dans ce processus. Le 1.2 canaux calciques voltage-dépendants de type L Ca V est nécessaire pour engager synchronisé coeur la contraction musculaire. L cardiaque de type Ca 1,2 V de canal est un complexe protéique sous-unités multiples composée de la sous - unité principale Ca porogène V α1 et Ca Vss et Ca V α2δ1 sous - unités auxiliaires 7-12. Notez que la liste complète des sous - unités auxiliaires est nécessaire pour produire fonctionnels Ca V 1.2 canaux à la membrane plasmique et les interactions dynamiques entre ces sous - unités sont essentiels pour soutenir la fonction électrique normale du cœur 13. Ca Vss favorise l'expression de surface cellulaire de Ca 1,2 V canaux à travers une nanomolaire interaction hydrophobe non covalente 14. Co-expression de la Ca V sous - unité α2δ1 wiCa th Ca V ß lié V α1 stimule l' expression du courant de crête (5 à 10 fois) et favorise l' activation du canal à des tensions négatives. Gain de fonction des mutations de la sous - unité de formation de pores Ca V 1.2 ont été associés à une forme d'arythmie ventriculaire appelé le syndrome du QT long 15 tandis qu'une foule de mutations ponctuelles dans les trois principales sous - unités formant le type L Ca V 1.2 canaux ont été identifiés chez des sujets souffrant d'arythmies du QT court formulaire de syndrome 16,17. Les canaux ioniques sont des protéines membranaires qui peuvent être étudiés du point de vue biochimique (chimie des protéines) ou à l'aide d'outils électrophysiologiques (machines génératrices de courant) et souvent l'aide de ces approches complémentaires. Électrophysiologie, notamment la cellule entière patch-serrage, est une approche appropriée pour élucider la fonction des canaux ioniques 15 mais ne peut pas résoudre des modifications dans le trafic de protéines à partir des changements dans leur biophysiquePropriétés. la chimie des protéines a cependant souvent un usage limité en raison de la relativement faible expression de grandes protéines de la membrane par rapport aux protéines solubles plus petits. méthodes à haut débit robustes en utilisant la fluorescence de lecture doivent être développées afin de traiter spécifiquement les défauts de la protéine biogenèse provoquant des changements dans l'expression de la surface cellulaire des canaux ioniques.
La cytométrie en flux est une technologie biophysique employée dans le comptage des cellules, le tri, la détection de biomarqueurs et l' ingénierie des protéines 18. Lorsqu'une solution de cellules vivantes ou des particules de l' échantillon est injecté dans un cytomètre de flux, les cellules sont ordonnées en un flux unique qui peut être sondé par le système de détection de la machine (figure 1). Le premier instrument cytomètre produit en 1956 19 un seul paramètre détecté , mais cytomètres de flux modernes ont de multiples lasers et les détecteurs de fluorescence permettant la détection de plus de 30 paramètres fluorescents 20,21.Filtres et miroirs (optique d'émission) dirigent la diffusion de la lumière ou de la lumière fluorescente de cellules à un réseau électronique (photodiode et détecteurs) qui convertissent la lumière proportionnellement à son intensité. Les données numériques sont analysées à l' aide de logiciels spécialisés et la sortie primaire est affiché comme un complot point 21.

Figure 1:. Principes biophysiques des flux de tri cytométrie Les cellules individuelles sont poussées à travers une buse sous haute pression dans un courant de fluide de gaine qui les déplace à travers un ou plusieurs points d'interrogation laser. Le faisceau lumineux est dévié par les cellules qui passent et la lumière collectée dans le sens avant (Forward Scatter, FCS) est envoyé à une photodiode qui convertit la lumière en un signal proportionnel à la taille de la cellule. La lumière est également recueillie à un angle de la trajectoire du laser 90 ° et envoyé aux détecteurs (également appelés photomultiplicateurs (PMT)).Cette lumière est acheminée à travers des miroirs dichroïques qui permettent la détection du signal de diffusion latérale (SSC), qui reflète la granularité dans les cellules, et les émissions fluorescentes si fluorochromes excités sont présents dans la cellule. Trois détecteurs (vert, jaune et rouge) sont représentés avec différents filtres de longueur d'onde de bande passante, ce qui permet la détection simultanée de différents fluorochromes. Les différents signaux sont numérisés par un ordinateur externe et converties en données qui seront analysées afin de quantifier les caractéristiques des cellules. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
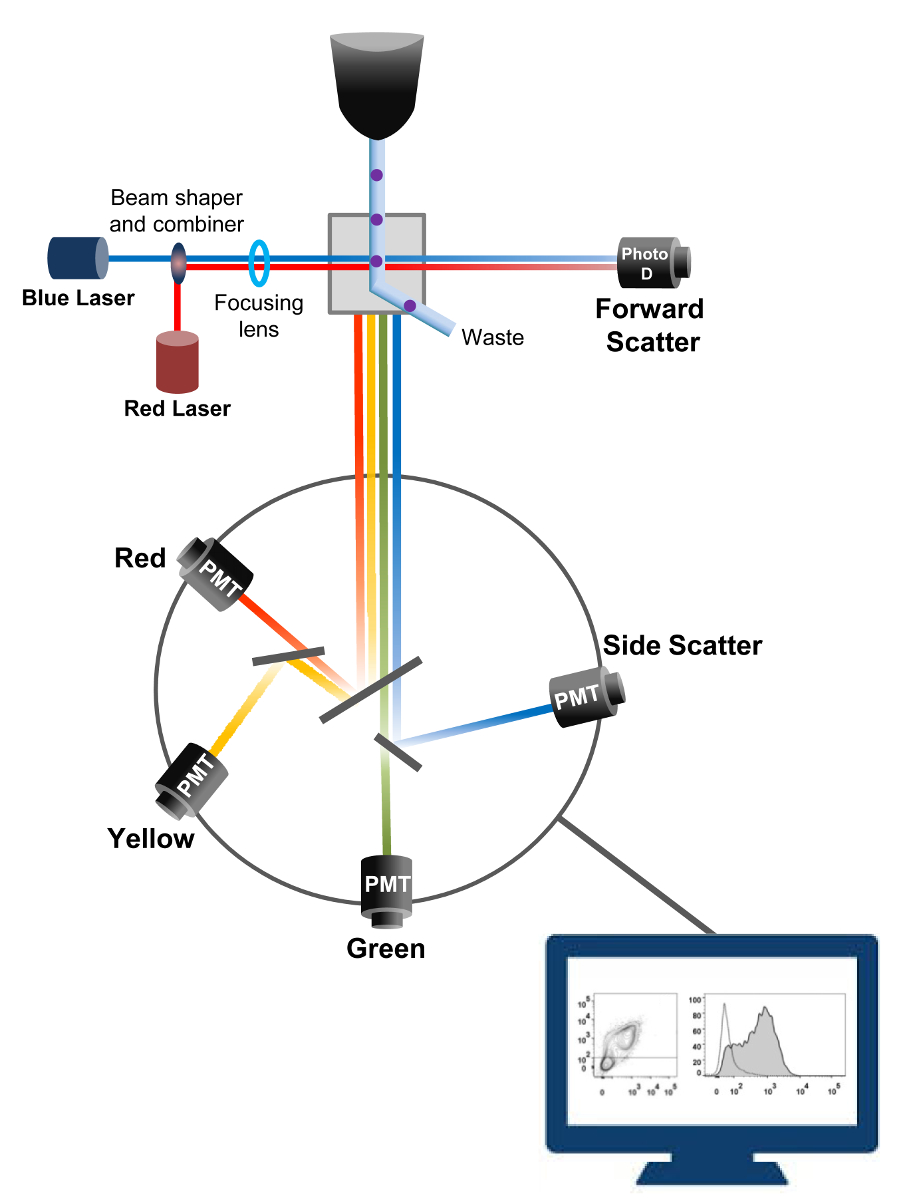{kind=link}
La capacité à haut débit de cytomètres de flux a été exploité pour quantifier l'expression membranaire relative de type sauvage recombinant et de type L voltage-dépendants trafic déficient Ca V 1.2 canaux et sous - unités associées à des cellules vivantes. constructions d'ADNc de coding pour les protéines ont été doublement marquées pour transporter simultanément un épitope extracellulaire non fluorescent qui peut être détecté par un anticorps conjugué fluorescent imperméable et un fluorophore intracellulaire qui est constitutivement fluorescente. À la fois l'épitope extracellulaire, inséré dans une boucle extracellulaire de la protéine, et le fluorophore intracellulaire, après avoir inséré l'extrémité C-terminale, sont convertis avec la protéine. Dans cette série d'expériences, la protéine V α2δ1 Ca a été conçu pour exprimer une hémagglutinine extracellulaire (HA) épitope (YPYDVPDYA) détectée par un matériau imperméable au FITC (isothiocyanate de fluorescéine) conjugué à anti-HA et mCherry comme fluorophore intrinsèque intracellulaire. Pour déterminer le niveau d'expression de surface cellulaire relative du V α2δ1 mCherry-Ca protéine HA étiquetée, des cellules recombinées exprimant la protéine de fusion ont été récoltées après la transfection, et colorées avec l'anticorps monoclonal de souris anti-HA étiquette d' épitope Antibod conjugué au FITCy (Figure 2). FITC est un composé organique fluorescent qui est considérablement plus petite que les journalistes enzymes et donc moins susceptibles d'interférer avec la fonction biologique. mCherry- Ca V α2δ1-HA surexprimé dans Tsa-201cells, produit une augmentation de 3 log significative de la fluorescence FITC et mCherry fluorescence sur des parcelles en deux dimensions 22. Etant donné que l'épitope HA est situé dans la partie extracellulaire de la protéine, l'intensité de fluorescence FITC obtenue en présence de cellules intactes reflètent l'indice relatif de l'expression de surface cellulaire de la protéine HA étiquetée. L'accessibilité de l'épitope HA dans les constructions est systématiquement validée en mesurant le signal FITC après perméabilisation cellulaire. Cette mesure permet aussi de confirmer l'expression de la protéine totale normalisée depuis les intensités de fluorescence relatives pour FITC estimées dans les cellules perméabilisées sont qualitativement comparables aux valeurs de fluorescence relative for mCherry mesurée sous perméabilisées et non perméabilisées conditions 22,23. Il est important de noter que le spectre de fluorescence intrinsèque est déplacée vers des valeurs plus élevées après perméabilisation, mais que la seule valeur étant rapportée est la variation de l'intensité de la fluorescence par comparaison avec la construction témoin. Les changements relatifs à l'intensité de fluorescence pour les constructions d'essai sont estimées en utilisant l'intensité ΔMean Fluorescence (de ΔMFI) des valeurs pour chaque fluorophore (mCherry ou FITC). Les expériences sont conçues pour mesurer l'intensité de la fluorescence du produit d'assemblage de test par rapport à l'intensité de fluorescence du produit d'assemblage de commande exprimée dans les mêmes conditions expérimentales pour limiter les variations de la fluorescence intrinsèque de l'anticorps conjugué à un fluorophore. Deux protéines de la membrane ont été étudiés avec succès en utilisant ce dosage: la sous - unité formant des pores du canal calcium de type L voltage-dépendants Ca 1,2 V 14,22 et dans une autre série d'expériences, l'auxiliaire extracellulaire Ca V α2δ1 sous - unité 22,23. Le protocole suivant a été utilisé pour déterminer l'expression de surface cellulaire de la sous - unité V α2δ1 Ca du type L cardiaque Ca V 1.2 canal dans des conditions de contrôle et d' après des mutations affectant la modification post - traductionnelle du canal ionique. Dans des conditions expérimentales normalisées, la fluorescence à la surface cellulaire de FITC augmente quasi linéairement avec l'expression de l' ADNc codant pour les mCherry-Ca V protéines α2δ1-HA (figure 5 de la référence 22).

Figure 2:. Représentation schématique de l' étiquetage totale et de la membrane dans la cytométrie protocole expérimental Le schéma décrit quelques - unes des principales mesures nécessaires pour quantifier l'expression relative totale et surface cellulaire des canaux ioniques recombinantes par flow cytométrie. Les cellules sont transfectées avec la construction double marquage mCherry-Ca V α2δ1-HA dans Tsa-201 cellules (1) et colorées avant ou après perméabilisation (2). Les données multiparamètres sont acquises dans un cytomètre de flux (3) pour l' analyse multivariée (4). S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
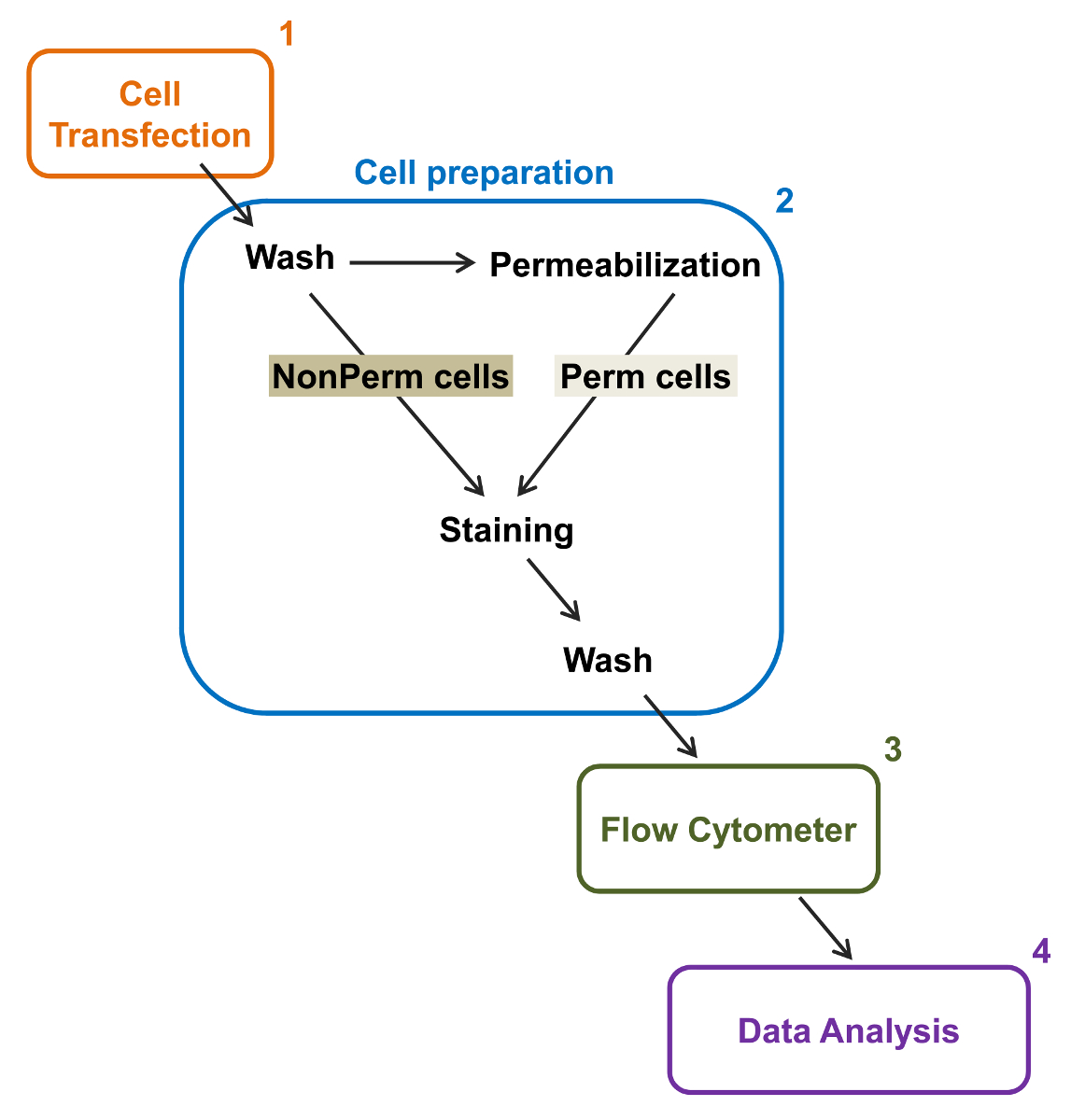{kind=link}
Protocole
1. constructions Doublement ADN Tagged
- Insérer l'épitope HA (YPYDVPDYA) dans le segment de liaison extracellulaire du Ca V α2δ1 entre D676 et R677 par mutagénèse dirigée (figure 3B) 20. Utilisez gccggattatgcgGGAAAACTCCAAACAACC Amorce et Reverse acatcatacggataTCAATAAATTCATTGAAATTTAAAAGAAATTC primer.
- Sous - cloner la séquence d'ADNc de HA V Ca α2δ1 étiqueté dans le vecteur d'expression de mammifère pmCherry-N1 conçu pour exprimer la protéine fusionnée à l' extrémité N-terminale de mCherry entre les sites Sacl et Sali (figure 3B) 20.
REMARQUE: La fonction de canal approprié doit être testé avec la construction de commande en utilisant des méthodes standards électrophysiologiques 24.
2. liposome Transfection transitoire (30 min, toutes les étapes sont réalisées sous Hotte à flux laminaire)
- Jour 1: Plaque d'un demi-million de Tsa-201 cellules (ou Hekt) dans35 mm des boîtes de culture avec du milieu essentiel minimum élevé de glucose de 2 ml de Dulbecco (DMEM-HG) supplémenté avec 10% de sérum bovin fœtal (FBS) et 1% de milieu (PS) culture de la pénicilline-streptomycine. Compter les cellules en utilisant un hématimètre standard. Évaluer la viabilité des cellules à partir d'une fraction de l'échantillon de cellules en utilisant du bleu Trypan. suffisamment de cellules de plaques pour atteindre 90% de confluence au moment de la transfection.
- Jour 2: Changement milieu de culture avec 2 ml de pré-chauffé (37 ° C) milieu de culture frais sans PS.
- Pour chaque échantillon de transfection, on prépare deux tubes de 1,5 ml. Dans le tube 1, diluer 4 pg d'ADN dans 250 ul de milieu sérique réduit. Dans le tube 2, mélanger 10 ul du réactif de transfection médiée par des liposomes avec 250 ul de milieu de culture sérum réduit. Mélanger délicatement le réactif de transfection avant utilisation.
- Incuber pendant 5 min à température ambiante.
- Combiner le contenu du tube 1 et le tube 2, mélanger doucement et incuber au moins 20 min à température ambiante.
- Ajouter les liposomes / DNA complexes aux cellules en culture et en douceur roche la boîte de culture pour mélanger.
- Incuber à 37 ° C sous 5% de CO 2 , l' atmosphère pendant 24 heures.
3. Coloration des cellules pour cytométrie de flux (3 h)
- Préparation des échantillons de cellules
- Jour 3: Supprimer moyen de la boîte de culture avec soin et laver les cellules avec 400 pi de préchauffé (37 ° C) 0,05% de trypsine-EDTA 1x (acide éthylènediaminetétraacétique).
- Ajouter 400 pi de trypsine-EDTA et incuber le plat à 37 ° C sous 5% de CO 2 atmosphère pendant 5 min pour permettre aux cellules de se détacher de l'antenne.
- Mettre un terme à la digestion enzymatique par addition de 1 ml de milieu de culture froid sans PS et laver toutes les cellules de la surface par un léger pipetage 4-5 fois. Évitez de trop digestion et over-pipetage pour réduire la mort cellulaire.
- Recueillir les cellules dans 1,5 ml tubes et placer immédiatement sur la glace. Utiliser des solutions froides de glace et de maintenir les cellules à 4 ° C pour empêcher l'intériorisationdes antigènes de surface. Diminution de l'éclairage pour limiter photoblanchiment du signal fluorescent.
- Tubes à centrifuger à 400 g pendant 5 min à 4 ° C. Aspirer soigneusement et jeter le surnageant.
- Remettre en suspension le culot dans 1 ml de 1 x solution saline tamponnée au phosphate (PBS) pour préparer une suspension cellulaire unique.
- vortex brièvement les tubes très doucement et répétez les étapes 3.1.5 et 3.1.6 pour supprimer complètement le milieu de culture.
- Remettre en suspension le culot dans 600 pl de PBS 1x et ajuster la concentration cellulaire à un minimum de 3 x 10 6 cellules / ml.
- Diviser les cellules dans deux nouveaux tubes de 1,5 ml pour la coloration extracellulaire et intracellulaire. Inclure des contrôles appropriés pour discriminer une coloration spécifique de la coloration non spécifique.
NOTE: L'anticorps de contrôle d'isotype permet d'évaluer le niveau de coloration de fond et doit correspondre idéalement les espèces hôtes, isotype et fluorophore de chaque anticorps primaire. Utilisez le contrôle isotypique et de l'anticorps conjugué à la mêmeconcentration en protéines.

Tableau 1: cytométrie de flux d' échantillons de contrôle de test pour la non-perméabilisées et perméabilisées cellules Chaque expérience doit inclure les témoins négatifs suivants:. (1) des cellules non transfectées (sans anticorps, avec l'isotype ou avec l'anticorps conjugué). (2) Les cellules transfectées avec la protéine d'intérêt sous - cloné dans un plasmide constitutif sans fluorochrome intracellulaire fluorescent (V pCMV Ca α2δ1-HA) ou avec la construction doublement étiquetée (V pmCherry-Ca α2δ1 et mis à incuber sans anticorps, avec l'isotype ou l'anticorps conjugué). Simples contrôles de couleur sont utilisés pour la compensation de chevauchement des émissions fluorochrome. Les mêmes contrôles sont exécutés pour les non perméabilisées et perméabilisées conditions dans chaque série d'expériences.
- Coloration de surface cellulaire d'Intactcellules vivantes
- Aliquote de 1 x 10 6 cellules / 100 ul dans des tubes de 1,5 ml.
- Ajouter l'anticorps monoclonal conjugué à FITC anti-HA à 5 pg / ml et, vortex avant l'incubation des cellules sur une plate-forme de bascule (200 tpm) dans l'obscurité à 4 ° C pendant 45 min.
REMARQUE: La concentration optimale de l' anticorps a été déterminée dans des expériences préliminaires de titrage (Figure 4). - Retirer les cellules de l'obscurité et ajouter 900 ul de 1 x PBS / tube. Centrifuger à 400 g pendant 5 min à 4 ° C.
- Aspirer le surnageant et resuspendre le culot dans 1 ml de PBS 1x, vortex et centrifuger à 400 g pendant 5 min à 4 ° C.
- Répétez le lavage (étape 3.2.4) deux fois pour éliminer tout anticorps non lié. Si un anticorps primaire non conjugué est utilisée, laisser incuber avec l'anticorps secondaire approprié.
- Après le lavage final, la remise en suspension des cellules dans 500 ul de 1 x PBS et transférer la suspension monocellulaire dans 5 ml cytométrie de flux des tubes. Gardez la cellules dans l'obscurité à 4 ° C jusqu'à ce que l'exécution de l'échantillon.
- Exécutez les échantillons sur un cytomètre de flux. Pour de meilleurs résultats, analyser les cellules sur la cytométrie de flux le plus tôt possible et au plus tard 24 heures après.
- Coloration intracellulaires: Fixation, perméabilisation et Coloration
- Aliquoter 1 x 10 6 cellules / 100 ul dans 1,5 ml tubes et centrifuger à 400 g pendant 5 min à 4 ° C.
- Jeter les cellules surnageant et remettre en suspension dans 100 pi de solution de fixation-perméabilisation directement du stock.
- Incuber dans l'obscurité à 4 ° C pendant 20 min.
- Ajouter 100 ul de 1 x tampon de perméabilisation-linge fraîchement préparé (diluée 10 x tampon de perméabilisation-lavage à l' eau distillée 2 O). cellules Vortex et de sédiments à l'aide d'une centrifugeuse de table à 400 g pendant 5 min à 4 ° C.
- Aspirer et jeter le surnageant.
- Répétez les étapes 3.3.4 et 3.3.5.
- Ajouter anticorps FITC-conjugué monoclonal anti-HA à 5pg / ml dans 100 pi de 1 x tampon de lavage et perméabilisation-vortex, avant l'incubation des cellules dans l'obscurité à 4 ° C pendant 30 min.
REMARQUE: La coloration intracellulaire est réalisée en suivant la même procédure que celle utilisée pour la coloration de la surface cellulaire. cellule perméabilisation saponine médiation est cependant un processus rapidement réversible, il est donc important de remplacer 1x PBS avec un tampon 1x Perm / Wash pour maintenir les cellules en la présence constante de saponine pendant la coloration intracellulaire. - Retirez les cellules de l'obscurité et d'ajouter un tampon perméabilisation-lavage 100 ul. Centrifuger à 400 g pendant 5 min à 4 ° C.
- Aspirer soigneusement le surnageant et remettre le culot dans 100 ul de tampon perméabilisation-lavage, vortex et centrifuger à 400 g pendant 5 min à 4 ° C.
- Répétez le lavage (étape 3.3.9) une fois de plus pour éliminer tout anticorps non lié.
- Après le lavage final, remettre en suspension les cellules dans 500 ul de PBS 1x et transférer le péchégle suspension cellulaire à 5 mL flux cytométrie tubes. Maintenir les cellules dans l'obscurité à 4 ° C jusqu'à l'injection de l'échantillon dans le cytomètre de flux.
- Exécutez les échantillons sur un cytomètre de flux. Exécutez les échantillons fixés sur le cytomètre dès que possible mais au plus tard 1 semaine après la coloration. Exécuter les cellules non perméabilisées et perméabilisées le même jour.
4. cytométrie de flux
- Cytomètre de flux cellulaire Setup Sorter Daily
- Allumez le logiciel de cytométrie de flux. Avant d'expérimenter, calibrer et configurer le cytomètre de flux trieur de cellules pour assurer une performance optimale de l' instrument (c. -à- laser et de l' optique sont performants à la spécification, le laser et le débit cellulaire sont correctement alignés) en utilisant des perles de configuration de l' appareil.
- Utilisez la buse de 100 um avec la pression de la gaine 20 psi.
NOTE: La buse ne doit pas être modifiée sur un banc cytomètre de flux. - Réglez le débit du cytomètre selon la fabrispécification er. Très hauts débits diminuent la sensibilité de la détection des variations de fluorescence.
- Sélectionnez bleu (488 nm pour exciter la fluorescéine Isothiocayanate ou FITC) et jaune-vert (561 nm pour exciter mCherry) lasers. Collecter les niveaux de fluorescence FITC et mCherry avec un nm 530/30 et avec respectivement un 610/20 nm filtre passe-bande.
- Acquérir la diffusion vers l'avant (FCS) par rapport à la diffusion latérale (SSC) tracé de points pour les cellules non colorées à l'aide de l'échelle linéaire. Ajustez l'amplification de chaque détecteur pour visualiser les cellules dans le quadrant inférieur gauche du tracé de points.
- Lecture des échantillons de cellules intactes non-perméabilisées
- Définir la P1gate pour les cellules vivantes non perméabilisées en définissant une forme libre autour des cellules à analyser l'exclusion des débris cellulaires et des agrégats de cellules, limitant ainsi le signal de fluorescence pour des cellules intactes.
NOTE: direct / morts colorants d'exclusion peuvent être utilisés pour faciliter le placement de grille sur les cellules vivantes. Définir 10.000 événements pour enregistrerdans le portillon d'arrêt P1. Réglez ce paramètre sur un nombre plus élevé d'événements si besoin est. - Acquérir mCherry contre FITC à deux paramètres tracé de contour pour détecter autofluorescence base de cellules non colorées. Utilisez l' échelle bi-logarithmique pour afficher les valeurs négatives et améliorer la résolution entre les populations 25. Régler la tension de chaque détecteur pour définir les cellules négatives non colorées dans la partie inférieure des dix premières unités des parcelles d'intensité de fluorescence du journal.
- Acquérir tous les échantillons intacts non perméabilisées en utilisant les paramètres établis dans 4.1.5 et 4.1.6 et de recueillir FSC, SSC et des signaux dans les détecteurs de fluorescence.
- Exporter et enregistrer des fichiers * .fcs à utiliser pour l'analyse à l'aide de cytométrie de flux logiciel d'analyse.
- Définir la P1gate pour les cellules vivantes non perméabilisées en définissant une forme libre autour des cellules à analyser l'exclusion des débris cellulaires et des agrégats de cellules, limitant ainsi le signal de fluorescence pour des cellules intactes.
- Lecture des échantillons de cellules perméabilisées
- Déplacer la porte P1 pour sélectionner des cellules vivantes dans les échantillons perméabilisées et ajuster FSC et de la tension SSC comme indiqué dans 4.1.5 et 4.1.6.
- Acquérir tous les échantillons perméabilisées et recueillir FSC, SSC asignaux nd dans les détecteurs de fluorescence.
- Exporter et enregistrer des fichiers * .fcs à utiliser pour l'analyse à l'aide de cytométrie de flux logiciel d'analyse.
- L'analyse des données
- Lancer le flux cytométrie fichiers logiciels d'analyse et de l'importation * .fcs enregistrés dans 4.2.4 et 4.3.3.
- Cliquez sur le premier échantillon répertorié dans la fenêtre d'espace de travail. Une nouvelle fenêtre nommée d'après le nombre tube ID ouvre automatiquement. Démarrer le processus de déclenchement dans la parcelle de SSC par rapport FSC. Dessinez une porte (P1) en utilisant l'icône Ellipse autour des cellules vivantes et d'éliminer tous les débris, les cellules mortes, ou des agrégats qui ont différentes diffusion vers l'avant et la diffusion latérale que les cellules vivantes
- Pour dessiner le contour tracé à deux paramètres de l'mCherry (y de l'axe) par rapport à FITC (axe-x) l'intensité de fluorescence des cellules vivantes, cliquez d'abord sur l'axe des x et choisissez-A FITC canal, puis cliquez sur le y -axis et choisissez le-mCherry-A PE canal. Cliquez sur l'icône "Quad" pour positionner le marqueur quadrant au bord d'unutofluorescent cellules dans chaque canal de fluorescence.
NOTE: La porte fixé autour des cellules positives FITC et mCherry est la porte P2. Négative population de cellules à fluorescence est appelée la porte P3. Voir la figure 5 pour la méthode de déclenchement représentatif utilisé dans cet article. - Sélectionnez P2 et P3 portes et cliquez sur l'icône "Ajouter Statistiques" dans la fenêtre de l'espace de travail d'origine. Cliquez sur "Count" (nombre de cellules positives) et cliquez sur "Moyenne" (Mean Fluorescence Intensity de chaque fluorochrome) ou "médian" (médian Fluorescence Intensity de chaque fluorochrome) statistiques parmi la liste des options. Cliquez sur l'icône "Ajouter Statistiques" à nouveau. Toutes ces valeurs sont transférées automatiquement dans la fenêtre de l'espace de travail d'origine.
NOTE: La «moyenne» est utilisé seulement si l'intensité de fluorescence suit une distribution normale. Dans tous les autres cas, cliquez sur l'onglet "médian". MFI pourrait donc se référer à Mean Fluorescence Intensity ou médian intensité de fluorescence.
REMARQUE: L'étape suivante consiste à appliquer les paramètres et statistiques des portes à tous les échantillons testés par le cytomètre. - Dans la fenêtre d'espace de travail, utilisez la souris pour faire glisser et déposer les portes et les statistiques des paramètres sur la ligne marquée tous les échantillons.
- Générer un rapport de lot de parcelles à deux dimensions contour (mCherry vs FITC) et histogrammes (nombre de cellules par rapport à l' intensité de fluorescence) pour les cellules non perméabilisées et perméabilisées (figures 6A - B).
- D'après les statistiques générées à l'étape 4.4.4, calculer l'intensité de fluorescence moyenne (MFI) pour chaque fluorochrome pour les cellules colorées. A partir de cette valeur, on soustrait la valeur MFI obtenu à partir de cellules non colorées pour quantifier la surface et l'expression totale de la protéine d'intérêt.
- Signaler les valeurs ΔMFI pour chaque fluorophore (mCherry et FITC) (Figure 6C - D). Normaliser l'ΔMFI mesurée pour le Ca V α2δ1 construire des mutants à la valeur obtenue pour ΔMFI FITC et mCherry avec la construction WT.
NOTE: La valeur absolue de l'intensité de fluorescence peut varier fortement en fonction du lot d'anticorps et les capacités techniques de chaque travailleur de laboratoire, d'où la nécessité de normaliser l'intensité de fluorescence de la construction mutant en utilisant la construction de WT.
Résultats
Cet article décrit un protocole fiable pour quantifier la surface totale et cellulaire des canaux ioniques recombinantes exprimées dans Tsa-201cells par un flux à deux couleurs cytométrie dosage. A titre d'exemple, la surface de la cellule par rapport et une expression de protéine totale a été quantifiée pour le V Ca α2δ1subunit. Afin d'effectuer le flux en deux couleurs cytométrie test, Ca V α2δ1 a été doublement marqué à exprimer une HA é...
Discussion
Cet essai à base de cytométrie de flux a été appliqué avec succès à la mesure des niveaux relatifs totale et la surface des cellules des sous - unités marquées par fluorescence porogène et associé à des canaux calciques voltage-dépendants 14,22,26. Il est préférable d'utiliser lors d'enquêtes sur l'impact des mutations génétiques et exige donc que l'intensité de fluorescence intrinsèque du type sauvage construction marqué par fluorescence marqué soit au moins 10 à 100 fo...
Déclarations de divulgation
The authors declare that they have no competing financial interests.
Remerciements
We thank Mr. Serge Sénéchal and Dr. Jacques Thibodeau for sharing their expertise and granting us access to their flow cytometry and cell sorting platform. This work was completed with the operating grant 130256 from the Canadian Institutes of Health Research, a grant-in-aid from the Canadian Heart and Stroke Foundation, and support from the "Fondation de l'Institut de Cardiologie de Montréal" to L.P.
matériels
| Name | Company | Catalog Number | Comments |
| Q5 Site-Directed Mutagenesis Kit | New England Biolabs | E0554S | Can be substitute with QuickChange site-directed mutagenesis Kit (Agilent, #200523). |
| Tubes 1.5 ml | Sarstedt | 72-690-001 | |
| Tubes 15 ml | Sarstedt | 62-554-002 | |
| Disposable graduated Tranfer Pipets | VWR | 160001-192 | |
| 100 mm culture dish | Corning | 430167 | For standard culture of HEKT cells. |
| 35 mm culture dish | Falcon | 353001 | For standard culture of HEKT cells. |
| Serological pipette 1 ml | Sarstedt | 86.1251.001 | |
| Serological pipette 5 ml | Sarstedt | 86.1253.001 | |
| Serological pipette 10 ml | Sarstedt | 86.1254.001 | |
| Serological pipette 25 ml | Sarstedt | 86.1285.001 | |
| Dulbecco's high-glucose medium | Life Technologies | 12100-046 | Warm in 37 °C water bath before use. |
| Fetal Bovine Serum, qualified, heat inactivated, US origin | Life Technologies | 16140-071 | |
| Penicillin-Streptomycin (10,000 U/ml) | Life Technologies | 15140-122 | |
| Lipofectamine 2000 | Life Technologies | 11668-019 | For liposomal transfection. Can be substituted with calcium phosphate transfection. |
| Opti-MEM I Reduced Serum Medium | Life Technologies | 31985-070 | Warm in 37 °C water bath before use. |
| Trypsin-EDTA (1x) 0.05%, phenol red | Life Technologies | 25300-062 | |
| 1.5 ml microtubes | Sarstedt | 72.690.001 | |
| Phosphate Buffered Saline 1x | Fisher | BP661-10 | Can be "home-made". |
| Anti-HA FITC conjugated antibody | Sigma | H7411 | |
| IgG1−FITC Isotype Control antibody | Sigma | F6397 | |
| BD Cytofix/Cytoperm Fixation/Permeabilization Solution Kit | BD Biosciences | 554714 | Fixation/Permeabilization. Permeabilization/Wash solution, store at 4 °C. |
| Hemacytometer | Fisher | 49105161 | |
| Trypan Blue | Fisher | 15250061 | To access cell viability. |
| Refrigerated Microcentrifuge, 5430R | Eppendorf | A14H172200 | |
| Forma Steri-Cycle CO2 Incubator | Fisher | 370 | |
| Laboratory Platform Rocker | Fisher | 545034 | |
| Water Bath | VWR | 89032-216 | |
| BD FACSARIA III | BD Biosciences | 648282 | Flow cytometer. |
| FlowJo Software v10 | FlowJo | FlowJo v10 Dongle | For data analysis. |
Références
- Delisle, B. P., Anson, B. D., Rajamani, S., January, C. T. Biology of Cardiac Arrhythmias: Ion Channel Protein Trafficking. Circ. Res. 94, 1418-1428 (2004).
- Birault, V., Solari, R., Hanrahan, J., Thomas, D. Y. Correctors of the basic trafficking defect of the mutant F508del-CFTR that causes cystic fibrosis. Curr Opin Chem Biol. 17, 353-360 (2013).
- Balijepalli, S. Y., Anderson, C. L., Lin, E. C., January, C. T. Rescue of Mutated Cardiac Ion Channels in Inherited Arrhythmia Syndromes. J. Cardiovas Pharm. 56, 113-122 (2010).
- Gargus, J. J. Unraveling Monogenic Channelopathies and Their Implications for Complex Polygenic Disease. Am. J. Hum. Genet. 72, 785-803 (2003).
- Abriel, H., Zaklyazminskaya, E. V. Cardiac channelopathies: Genetic and molecular mechanisms. Gene. 517, 1-11 (2013).
- Behr, E. R., et al. Sudden arrhythmic death syndrome: familial evaluation identifies inheritable heart disease in the majority of families. Eur Heart J. 29, 1670-1680 (2008).
- Catterall, W. A. Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev.Biol. 16, 521-555 (2000).
- Peterson, B. Z., DeMaria, C. D., Adelman, J. P., Yue, D. T. Calmodulin is the Ca2+ sensor for Ca2+ -dependent inactivation of L- type calcium channels. Neuron. 22, 549-558 (1999).
- Dolphin, A. C. Calcium channel diversity: multiple roles of calcium channel subunits. Curr.Opin.Neurobiol. 19, 237-244 (2009).
- Dai, S., Hall, D. D., Hell, J. W. Supramolecular assemblies and localized regulation of voltage-gated ion channels. Physiol Rev. 89, 411-452 (2009).
- Gao, T., et al. Identification and subcellular localization of the subunits of L-type calcium channels and adenylyl cyclase in cardiac myocytes. J. Biol. Chem. 272, 19401-19407 (1997).
- Carl, S. L., et al. Immunolocalization of sarcolemmal dihydropyridine receptor and sarcoplasmic reticular triadin and ryanodine receptor in rabbit ventricle and atrium. J. Cell Biol. 129, 673-682 (1995).
- Abriel, H., Rougier, J. S., Jalife, J. Ion Channel Macromolecular Complexes in Cardiomyocytes: Roles in Sudden Cardiac Death. Circ. Res. 116, 1971-1988 (2015).
- Bourdin, B., et al. Molecular Determinants of the Cavb-induced Plasma Membrane Targeting of the Cav1.2 Channel. J. Biol. Chem. 285, 22853-22863 (2010).
- Raybaud, A., et al. The Role of the GX9GX3G Motif in the Gating of High Voltage-activated Calcium Channels. J. Biol. Chem. 281, 39424-39436 (2006).
- Burashnikov, E., et al. Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm. 7, 1872-1882 (2010).
- Hennessey, J. A., et al. A CACNA1C Variant Associated with Reduced Voltage-Dependent Inactivation, Increased Cav1.2 Channel Window Current, and Arrhythmogenesis. PLoS ONE. 9, e106982 (2014).
- Adan, A., Alizada, G., Kiraz, Y., Baran, Y., Nalbant, A. Flow cytometry: basic principles and applications. Crit Rev Biotechnol. , 1-14 (2016).
- Graham, M. D. The Coulter Principle: Foundation of an Industry. J. Lab. Autom. 8, 72-81 (2003).
- Baumgarth, N., Roederer, M. A practical approach to multicolor flow cytometry for immunophenotyping. J. Immunol. Methods. 243, 77-97 (2000).
- Rothe, G., Sack, U., Tarnok, A., Rothe, G. . Cellular Diagnostics. Basics, Methods and Clinical Applications of Flow Cytometry. , 53-88 (2009).
- Bourdin, B., et al. Functional Characterization of Cavalpha2delta Mutations Associated with Sudden Cardiac Death. J. Biol. Chem. 290, 2854-2869 (2015).
- Tetreault, M. P., et al. Identification of glycosylation sites essential for surface expression of the Cavalpha2delta1 subunit and modulation of the cardiac Cav1.2 channel activity. J. Biol. Chem. 291, 4826-4843 (2016).
- Senatore, A., Boone, A. N., Spafford, J. D. Optimized Transfection Strategy for Expression and Electrophysiological Recording of Recombinant Voltage-Gated Ion Channels in HEK-293T Cells. J Vis Exp. (47), (2011).
- Herzenberg, L. A., Tung, J., Moore, W. A., Herzenberg, L. A., Parks, D. R. Interpreting flow cytometry data: a guide for the perplexed. Nat.Immunol. 7, 681-685 (2006).
- Shakeri, B., Bourdin, B., Demers-Giroux, P. O., Sauve, R., Parent, L. A quartet of Leucine residues in the Guanylate Kinase domain of Cavbeta determines the plasma membrane density of the Cav2.3 channel. J Biol Chem. 287, 32835-32847 (2012).
- Morton, R. A., Baptista-Hon, D. T., Hales, T. G., Lovinger, D. M. Agonist- and antagonist-induced up-regulation of surface 5-HT3A receptors. Br. J. Pharmacol. 172, 4066-4077 (2015).
- Hoffmann, C., et al. Fluorescent labeling of tetracysteine-tagged proteins in intact cells. Nat. Protocols. 5, 1666-1677 (2010).
- Cockcroft, C. J., Gamper, N. . Ion Channels: Methods and Protocols. , 233-241 (2013).
- Gonzalez-Gutierrez, G., Miranda-Laferte, E., Neely, A., Hidalgo, P. The Src Homology 3 Domain of the beta-Subunit of Voltage-gated Calcium Channels Promotes Endocytosis via Dynamin Interaction. J. Biol.Chem. 282, 2156-2162 (2007).
- Galizzi, J. P., Borsotto, M., Barhanin, J., Fosset, M., Lazdunski, M. Characterization and photoaffinity labeling of receptor sites for the Calcium channel inhibitors d-cis-diltiazem, (+/-)-bepridil, desmethoxyverapamil, and (+)-PN 200-110 in skeletal muscle transverse tubule membranes. J. Biol.Chem. 261, 1393-1397 (1986).
- Bezanilla, F. The voltage sensor in voltage-dependent ion channels. Physiol.Rev. 80, 555-592 (2000).
- Sigworth, F. J. The variance of sodium current fluctuations at the node of Ranvier. J Physiol. 307, 97-129 (1980).
- Bailey, M. A., Grabe, M., Devor, D. C. Characterization of the PCMBS-dependent modification of KCa3.1 channel gating. J. Gen. Physiol. 136, 367-387 (2010).
- Fletcher, P. A., Scriven, D. R., Schulson, M. N., Moore, E. D. Multi-Image Colocalization and Its Statistical Significance. Biophys. J. 99, 1996-2005 (2010).
- Lizotte, E., Tremblay, A., Allen, B. G., Fiset, C. Isolation and characterization of subcellular protein fractions from mouse heart. Anal. Biochem. 345, 47-54 (2005).
- Mattheyses, A. L., Simon, S. M., Rappoport, J. Z. Imaging with total internal reflection fluorescence microscopy for the cell biologist. J. Cell Sci. 123, 3621-3628 (2010).
- Yamamura, H., Suzuki, Y., Imaizumi, Y. New light on ion channel imaging by total internal reflection fluorescence (TIRF) microscopy. J. Pharmacol. Sci. 128, 1-7 (2015).
- Wible, B. A., et al. HERG-Lite-R: A novel comprehensive high-throughput screen for drug-induced hERG risk. J. Pharmacol. Toxicol. Methods. 52, 136-145 (2005).
- Wilde, A. A. M., Brugada, R. Phenotypical Manifestations of Mutations in the Genes Encoding Subunits of the Cardiac Sodium Channel. Circ. Res. 108, 884-887 (2011).
- Milano, A., et al. Sudden Cardiac Arrest and Rare Genetic Variants in the Community. Circ Cardiovasc Genet. , (2016).
- Schnell, U., Dijk, F., Sjollema, K. A., Giepmans, B. N. G. Immunolabeling artifacts and the need for live-cell imaging. Nat. Meth. 9, 152-158 (2012).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.