
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Determinação da superfície celular Relativa e Expressão total de canais recombinante de iões utilizando Citometria de Fluxo
Neste Artigo
Resumo
arritmias cardíacas hereditárias são geralmente causadas por mutações que alteram a entrega de superfície de um ou mais canais de íons. Aqui, nós adaptar ensaios de citometria de fluxo para proporcionar uma quantificação da expressão de proteína total e a superfície celular relativa dos canais de iões recombinantes expressas em células TSA-201.
Resumo
Inherited or de novo mutations in cation-selective channels may lead to sudden cardiac death. Alteration in the plasma membrane trafficking of these multi-spanning transmembrane proteins, with or without change in channel gating, is often postulated to contribute significantly in this process. It has thus become critical to develop a method to quantify the change of the relative cell surface expression of cardiac ion channels on a large scale. Herein, a detailed protocol is provided to determine the relative total and cell surface expression of cardiac L-type calcium channels CaV1.2 and membrane-associated subunits in tsA-201 cells using two-color fluorescent cytometry assays. Compared with other microscopy-based or immunoblotting-based qualitative methods, flow cytometry experiments are fast, reproducible, and large-volume assays that deliver quantifiable end-points on large samples of live cells (ranging from 104 to 106 cells) with similar cellular characteristics in a single flow. Constructs were designed to constitutively express mCherry at the intracellular C-terminus (thus allowing a rapid assessment of the total protein expression) and express an extracellular-facing hemagglutinin (HA) epitope to estimate the cell surface expression of membrane proteins using an anti-HA fluorescence conjugated antibody. To avoid false negative, experiments were also conducted in permeabilized cells to confirm the accessibility and proper expression of the HA epitope. The detailed procedure provides: (1) design of tagged DNA (deoxyribonucleic acid) constructs, (2) lipid-mediated transfection of constructs in tsA-201 cells, (3) culture, harvest, and staining of non-permeabilized and permeabilized cells, and (4) acquisition and analysis of fluorescent signals. Additionally, the basic principles of flow cytometry are explained and the experimental design, including the choice of fluorophores, titration of the HA antibody and control experiments, is thoroughly discussed. This specific approach offers objective relative quantification of the total and cell surface expression of ion channels that can be extended to study ion pumps and plasma membrane transporters.
Introdução
Este documento proporciona um ensaio fiável para relatar a expressão na superfície celular relativa das proteínas de membrana, tais como canais de iões expressos em células recombinantes que utilizam a tecnologia de citometria de fluxo existente. canais de iões são proteínas de membrana de poros de formação que são responsáveis por controlar os sinais eléctricos por gating o fluxo de iões através da membrana celular. Eles são classificados pelo mecanismo de ativação, natureza, e seletividade de espécies de íon de trânsito através do poro onde eles estão localizados. Nos níveis celulares e teciduais, os fluxos de iões macroscópicas através de canais iónicos são o produto de imóveis 1 biofísico (gating e permeação), bioquímicos (fosforilação) e biogênese (síntese, glicosilação, tráfico e degradação). Cada um destes processos é único para cada tipo de canais iónicos e é optimizado para desempenhar o papel fisiológico do canal de iões. Consequentemente, as alterações em qualquer um destes processos afinadas através de umherdada ou uma modificação genética, muitas vezes referida como "canalopatia", pode ser prejudicial para a homeostase celular. É importante salientar que entregar a quantidade "certa" dos canais de íons na superfície da célula é fundamental para a homeostase celular. Mesmo pequenos aumentos (ganho de função) e pequenas diminuições (perda de função) em actividade de canal iónico tem o potencial de causar uma patologia grave ao longo da vida. Os defeitos na superfície da célula de entrega dos canais de iões maduros é um determinante importante em numerosos canalopatias, tais como a fibrose cística (CFTR canal de iões) 2 e arritmias cardíacas de forma a síndrome de QT longo (canais de potássio cardíacos) 3.
Canalopatias estão associadas à morte súbita cardíaca 4. A prevalência mundial atual de todos os canalopatias cardíacas é pensado para ser, pelo menos, 1: 2,000-1: 3.000 por pessoa 5 e são responsáveis por cerca de metade da súbita por arritmia ca morte cardíacases 6. Disfunção cardíaca na voltagem-sódio, potássio, cálcio e canais iônicos seletivos são conhecidos por desempenhar um papel fundamental neste processo. O 1.2 canais de cálcio voltagem-L-type Ca V é necessária para iniciar a contração do músculo do coração sincronizados. A cardíaca L-type Ca V 1.2 canal é um complexo de proteínas multi-subunidade composta de o principal formador de poros Ca subunidade V α1 e Ca V ß e Ca V α2δ1 subunidades auxiliares 7-12. Note-se que o complemento total de subunidades auxiliares é necessário para produzir funcionais Ca V 1.2 canais na membrana plasmática e interacções dinâmicas entre as subunidades são essenciais para suportar a função eléctrica normal do coração 13. Ca V SS promove a expressão na superfície celular de Ca V 1.2 canais através de um não-covalente nanomolar de interacção hidrófoba 14. A co-expressão do Ca V Wi subunidade α2δ1th Ca V ß-bound Ca V α1 estimula a expressão corrente de pico (de 5 a 10 vezes) e promove a ativação do canal em voltagens mais negativas. Ganho de função mutações da subunidade formadora de poros Ca V 1.2 têm sido associados com uma forma de arritmias ventriculares chamado síndroma do QT longo de 15 enquanto que uma série de mutações pontuais nas três subunidades principais que formam o tipo L-Ca V canal 1.2 foram identificados em indivíduos que sofrem de arritmias de forma a síndroma QT curto 16,17. canais iônicos são proteínas de membrana que podem ser investigados a partir de um ponto de vista bioquímico (química de proteínas), ou usando ferramentas eletrofisiológicos (máquinas geradoras de corrente) e muitas vezes usando estas abordagens complementares. Eletrofisiologia, em particular de células inteiras patch-fixação, é uma abordagem adequada para elucidar a função de canais iônicos 15, mas não pode resolver modificações no tráfego de proteínas a partir de mudanças em seu biofísicopropriedades. Protein Chemistry tem, no entanto, muitas vezes limitado uso, devido à relativamente baixa expressão de proteínas de membrana grande em relação às proteínas solúveis menores. métodos de alto rendimento robustos usando leitura de fluorescência precisam ser desenvolvidos, a fim de abordar especificamente defeitos de biogênese de proteínas que causam alterações na expressão na superfície celular de canais iônicos.
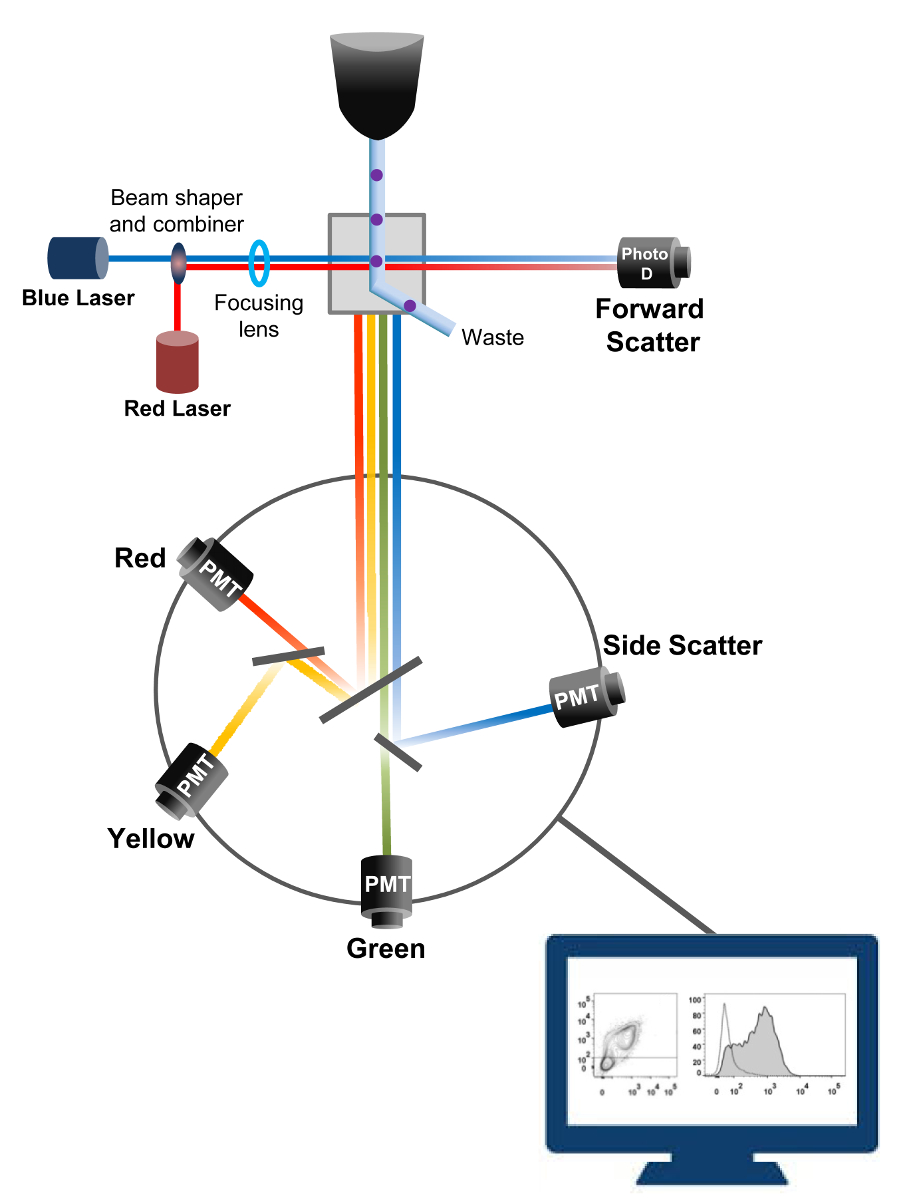
A citometria de fluxo é uma tecnologia biofísica empregada na contagem de células, a triagem, detecção de biomarcadores e engenharia de proteínas 18. Quando uma solução de amostra de células vivas ou em partículas é injectado num citómetro de fluxo, as células são ordenados numa única corrente que pode ser sondado por sistema de detecção da máquina (Figura 1). O primeiro instrumento de citometria de fluxo produzido em 1956 19 detectada apenas um parâmetro, mas citómetros de fluxo modernos têm múltiplos lasers e detectores de fluorescência que permitem a detecção de mais do que 30 parâmetros fluorescentes 20,21.Filtros e espelhos (óptica de emissão) dirigir a difusão da luz ou luz fluorescente de células a uma rede electrónica (fotodiodo e detectores) que convertem a luz proporcionalmente à sua intensidade. Os dados digitais são analisados usando software especializado e a saída primária é exibido como um gráfico de pontos 21.

Figura 1:. Biophysical princípios de citometria de fluxo de triagem células individuais são empurradas através de um bico sob alta pressão dentro de um fluxo de fluido de revestimento que as move entre um ou mais pontos de interrogação laser. O feixe de luz é desviada por as células que passam e a luz recolhida na direcção para a frente (Forward Scatter, FCS) é enviada para um fotodíodo que converte a luz em um sinal proporcional ao tamanho da célula. A luz também é recolhida em um ângulo de 90 ° para o caminho do laser e enviado para detectores (também chamados de fotomultiplicadores (PMT)).Esta luz é encaminhado através de espelhos dicróicos que permitem a detecção do sinal de dispersão lateral (SSC), que reflecte a granularidade dentro das células, e as emissões fluorescentes excitadas se fluorocromos estão presentes na célula. Três detectores (verde, amarelo e vermelho) são representados com diferentes filtros de banda de comprimento de onda, permitindo a detecção simultânea de diferentes fluorocromos. Os diferentes sinais são digitalizados por um computador externo e convertidos em dados que serão analisados para quantificar as características das células. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
A capacidade de alto rendimento de citómetros de fluxo foi explorada para quantificar a expressão relativa de membrana do tipo selvagem recombinante e do tipo L dependentes da voltagem tráfico deficiente em Ca V 1.2 canais e subunidades associadas em células vivas. co construções de ADNcDing para as proteínas foram duplamente marcado para realizar simultaneamente um epitopo extracelular não fluorescente que pode ser detectado por um anticorpo conjugado fluorescente impermeável e um fluoróforo intracelular que é constitutivamente fluorescente. Tanto o epitopo extracelular, inserido num loop extracelular da proteína, e o fluoróforo intracelular, inserido depois do terminal C, são convertidas com a proteína. Nesta série de experiências, a proteína V α2δ1 Ca foi manipulada para expressar um epitopo extracelular de hemaglutinina (HA) (YPYDVPDYA) detectado por um revestimento impermeável com FITC (isotiocianato de fluoresceína) -conjugated anti-HA e mCherry como o fluororo intracelular intrínseca. Para determinar o nível de expressão na superfície celular relativa do mCherry-Ca V α2δ1 proteína marcada com HA, as células recombinantes que expressam a proteína de fusão foram colhidas após a transfecção, e coradas com FITC-conjugado monoclonal de murganho anti-HA tag de epitopo AntibodY (Figura 2). FITC é um composto fluorescente orgânico que é consideravelmente menor do que os repórteres de enzimas e, portanto, não como susceptível de interferir com a função biológica. mCherry- Ca V α2δ1-HA sobre-expresso em TSA-201cells, produz um aumento de 3 log significativa na fluorescência FITC e mCherry fluorescência em parcelas bidimensionais 22. Uma vez que o epitopo de HA situa-se na porção extracelular da proteína, a intensidade de fluorescência obtida para FITC na presença de células intactas reflectir o índice relativo da expressão na superfície celular de proteína marcada com HA. A acessibilidade do epitopo de HA nas construções é sistematicamente validada medindo o sinal de FITC após permeabilização celular. Esta medida serve também para confirmar a expressão de proteína total normalizado desde as intensidades de fluorescência relativas para FITC estimado em células permeabilizadas são qualitativamente comparáveis com os valores de fluorescência relativa for mCherry medidos sob condições permeabilizadas e não permeabilizadas 22,23. É importante notar que o espectro de fluorescência intrínseca é deslocada para valores mais elevados após permeabilização, mas que o único valor a ser reportada é a alteração na intensidade de fluorescência em comparação com a construção de controlo. mudanças relativas na intensidade de fluorescência para as construções de teste são estimadas utilizando a intensidade de fluorescência ΔMean (ΔMFI) valores para cada fluoróforo (mCherry ou FITC). As experiências são concebidos para medir a intensidade de fluorescência do construto de teste em relação à intensidade de fluorescência da construção de controlo expresso sob as mesmas condições experimentais para limitar as variações na fluorescência intrínseca do anticorpo fluoróforos. Duas proteínas de membrana foram estudados com êxito com este ensaio: a subunidade formadora de poros do tipo L, canal de cálcio dependentes da voltagem V Ca 1,2 e 14,22 de uma série diferente deexperimentos, o auxiliar extracelular Ca V α2δ1 subunidade 22,23. O protocolo seguinte foi utilizado para determinar a expressão na superfície celular do Ca V α2δ1 subunidade do tipo L cardíaca Ca V 1.2 canal sob condições de controlo e após a mutações que afectam a modificação pós-tradução do canal de iões. Sob condições experimentais padronizadas, a fluorescência da superfície celular de FITC aumenta quase linearmente com a expressão de ADNc que codifica para os mCherry-Ca V proteínas α2δ1-HA (Figura 5 de referência 22).

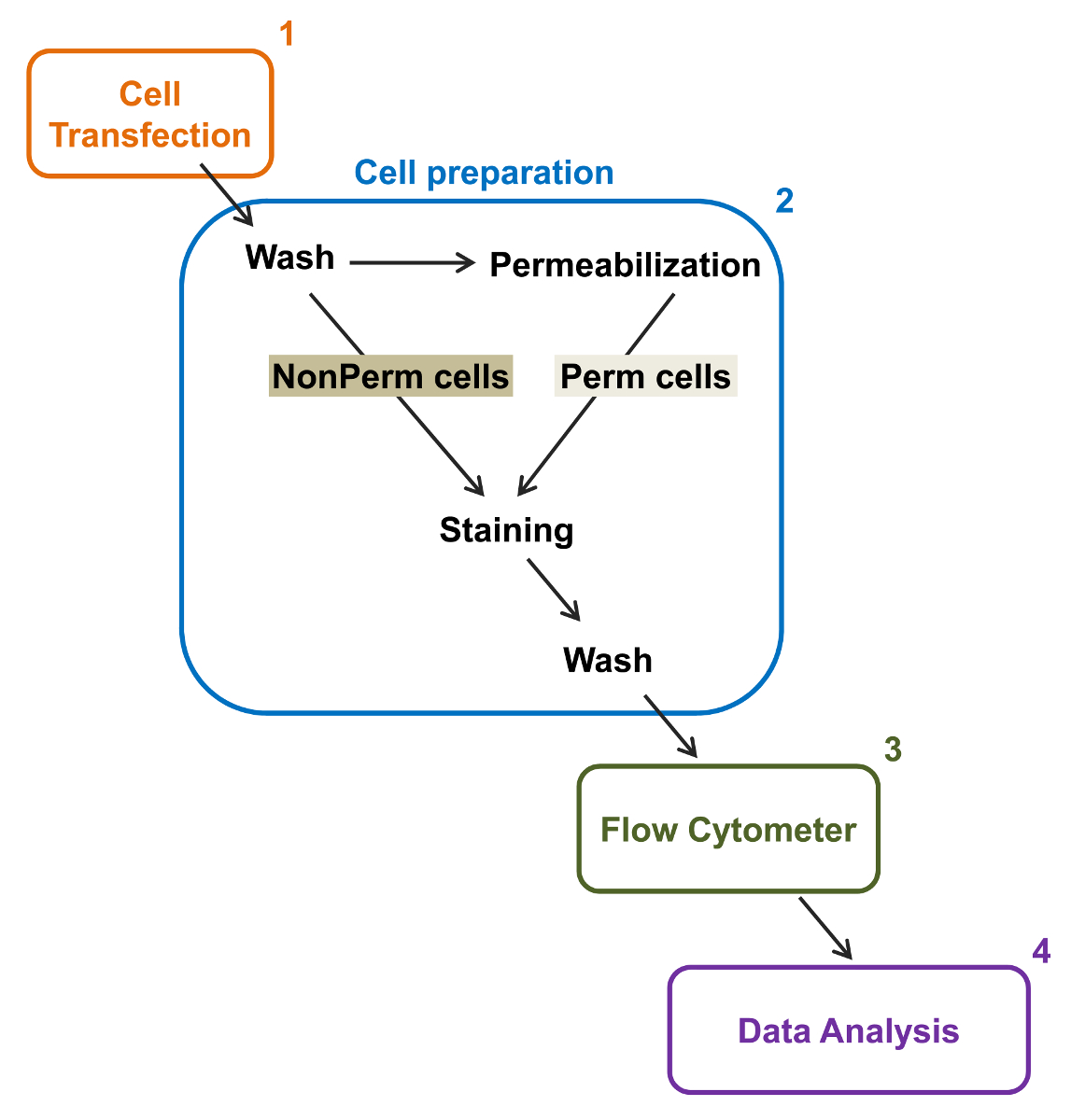
Figura 2:. Representação esquemática da rotulagem total e membrana na citometria de fluxo protocolo experimental O esquema descreve algumas das principais medidas necessárias para quantificar a expressão total e da superfície celular relativa de canais de iões recombinantes por flow citometria. As células são transfectadas com a construção de marcação dupla mCherry-Ca V α2δ1-HA em células (1) TSA-201 e coradas antes ou após permeabilização (2). Dados multiparâmetros são adquiridos em um citômetro de fluxo (3) para a análise multivariada (4). Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
Protocolo
1. construções Duplamente Tagged DNA
- Inserir o epitopo de HA (YPYDVPDYA) no ligante extracelular de Ca V α2δ1 entre D676 e R677 por mutagénese dirigida ao local (Figura 3B) 20. Use gccggattatgcgGGAAAACTCCAAACAACC Atacante primer e Reverso acatcatacggataTCAATAAATTCATTGAAATTTAAAAGAAATTC primer.
- Subclone da sequência de ADNc da HA etiquetado Ca V α2δ1 no vector de expressão de mamífero pmCherry-N1 concebido para expressar a proteína fundida com o terminal N de mCherry entre os locais SacI e SalI (Figura 3B) 20.
NOTA: A função de canal apropriado precisa ser testado com a construção controle usando métodos eletrofisiológicos padrão 24.
2. mediada por lipossomas transfecção transiente (30 min, todas as etapas são realizadas sob Fluxo Laminar Hood)
- Dia 1: Placa de meio milhão de células TSA-201 (ou Hekt) emPlacas de cultura de 35 mm com meio essencial 2 mL de Dulbecco de alta glucose mínimo (DMEM-HG), suplementado com 10% de Soro Fetal Bovino (FBS) e médio (PS) cultura de penicilina-estreptomicina a 1%. Contagem de células utilizando um hemacitómetro padrão. Avaliar a viabilidade celular de uma fracção da amostra de células utilizando azul de tripano. Placa células suficientes para alcançar 90% de confluência no momento da transfecção.
- Dia 2: Mudança de meio de cultura com 2 ml de meio de cultura fresco pré-aquecido (37 ° C) sem PS.
- Para cada amostra de transfeco, preparar dois tubos de 1,5 ml. No tubo 1, dilui-se 4 mg de DNA em 250 ul de meio de soro reduzido. No tubo 2, misturar 10 mL de reagente de transfecção mediada por lipossomas com 250 de meio de cultura em soro reduzido ul. Misture suavemente o reagente de transfecção antes do uso.
- Incubar durante 5 min à temperatura ambiente.
- Combinar o conteúdo de um tubo e o tubo 2, misturar suavemente e incuba-se, pelo menos, 20 min à temperatura ambiente.
- Adicione os lipossomas / DNA complexos para as células cultivadas e agite levemente o prato de cultura para misturar.
- Incubar a 37 ° C sob 5% de atmosfera de CO2 durante 24 h.
3. Coloração de células para citometria de fluxo (3 h)
- Preparando amostras de células
- Dia 3: Remover o meio a partir do prato de cultura com cuidado e lavar as células com 400 ul de pré-aquecido (37 ° C) de 0,05% de tripsina-EDTA 1x (ácido etilenodiaminotetracético).
- Adicionar 400 ul de tripsina-EDTA e incubar o prato a 37 ° C sob 5% de CO2 durante 5 min para permitir que as células a separar a partir do prato.
- Parar a digestão enzimática por adição de 1 ml de meio de cultura frio sem PS e lavar todas as células da superfície pipetando suavemente 4-5 vezes. Evitar o excesso de digestão e excesso de pipetagem para reduzir a morte celular.
- Recolher as células em tubos de 1,5 ml e coloque imediatamente em gelo. Utilizar soluções gelada e manter as células a 4 ° C para evitar a internalizaçãode antígenos de superfície. Diminuir iluminação para limitar o fotobranqueamento do sinal fluorescente.
- tubos de centrifugação a 400 xg durante 5 min a 4 ° C. Aspirar cuidadosamente e descartar o sobrenadante.
- Re-suspender o sedimento em 1 ml de solução salina tamponada 1x com fosfato (PBS) para preparar uma suspensão de célula única.
- Resumidamente vortex os tubos muito suavemente e repita os passos 3.1.5 e 3.1.6 para remover completamente o meio de cultura.
- Re-suspender o sedimento em 600 ul de 1x PBS e ajustar a concentração de células a um mínimo de 3 X 10 6 células / ml.
- Dividir as células em dois novos tubos de 1,5 ml para coloração intracelular e extracelular. Incluir controlos adequados para discriminar coloração específica de coloração não específica.
NOTA: O anticorpo de controlo de isotipo ajuda a avaliar o nível de coloração de fundo e idealmente deve corresponder de cada anticorpo primário espécies hospedeiras, isótipo e fluoróforo. Use de controlo de isotipo e o anticorpo conjugado ao mesmoconcentração de proteína.

Tabela 1: citometria de fluxo amostras de controlo experimento para não-permeabilizadas e permeabilizadas células Cada experiência deve incluir os seguintes controlos negativos:. (1) células não transfectadas (sem anticorpo, com o isotipo ou com o anticorpo conjugado). (2) As células transf ectadas com a proteína de interesse subclonado num plasmídeo sem constitutiva fluorocromo intracelular fluorescente (pCMV- Ca V α2δ1-HA) ou com a construção duplamente etiquetado (pmCherry-Ca V α2δ1 e incubados sem anticorpo, com o isotipo ou com o anticorpo conjugado). controles de cores individuais são usados para a compensação de sobreposição de emissão de fluorocromo. Os mesmos controles são executados por não permeabilizadas e permeabilizadas condições em cada série de experimentos.
- A coloração da superfície celular da IntactAs células vivas
- Alíquota 1 X 10 6 células / 100 uL em tubos de 1,5 ml.
- Adicionar o anticorpo monoclonal conjugado com FITC anti-HA a 5 ug / ml e, de vórtice antes da incubação das células sobre uma plataforma oscilante (200 rpm) no escuro a 4 ° C durante 45 min.
NOTA: A concentração óptima de anticorpo foi determinada em experiências preliminares de titulação (Figura 4). - Remover as células a partir do escuro e adicionar 900 ul de 1x PBS / tubo. Centrifuga-se a 400 xg durante 5 min a 4 ° C.
- Aspirar o sobrenadante e ressuspender o sedimento em 1 ml de PBS 1x, vortex e centrifugar a 400 xg durante 5 min a 4 ° C.
- Repetir a lavagem (passo 3.2.4) duas vezes para remover qualquer anticorpo não ligado. Se for utilizado um anticorpo primário não conjugado, incubar com o anticorpo secundário adequado.
- Após a última lavagem, ressuspender as células em 500 ul de 1x PBS e transferir a suspensão de célula única em 5 ml de tubos de citometria de fluxo. Mantenha o celulars no escuro a 4 ° C até à execução da amostra.
- Execute as amostras em um citômetro de fluxo. Para melhores resultados, analisar as células no citômetro de fluxo, logo que possível eo mais tardar 24 horas depois.
- A coloração intracelular: Fixação, permeabilização, e coloração
- Alíquota 1 X 10 6 células / 100 uL em tubos de 1,5 ml e centrifugar a 400 xg durante 5 min a 4 ° C.
- Descarte as células sobrenadante e ressuspender em 100 ul de solução de fixação-permeabilização diretamente do estoque.
- Incubar no escuro a 4 ° C durante 20 min.
- Adicionar 100 ul de tampão de permeabilização de lavagem 1x preparado de fresco (diluído 10x tampão de permeabilização de lavagem em H2O destilada). células de vórtice e de sedimentos, utilizando uma centrífuga de mesa a 400 xg durante 5 min a 4 ° C.
- Aspirar e desprezar o sobrenadante.
- Repita os passos 3.3.4 e 3.3.5.
- Adicionar anticorpos anti-HA monoclonal conjugado com FITC em 5ug / ml em 100 ul de 1x tampão de permeabilização de lavagem e, de vórtice antes de incubar as células no escuro a 4 ° C durante 30 min.
NOTA: A coloração intracelular é realizado seguindo o mesmo procedimento que o utilizado para a coloração da superfície celular. permeabilização celular mediada por A saponina é, no entanto, um processo rapidamente reversíveis, por isso, é importante substituir 1x PBS com tampão 1x Perm / Wash para manter as células na presença constante de saponina durante a coloração intracelular. - Remover as células a partir do escuro e adiciona-se 100 ul de tampão de permeabilização de lavagem. Centrifuga-se a 400 xg durante 5 min a 4 ° C.
- Aspirar cuidadosamente o sobrenadante e ressuspender o sedimento em 100 ul de tampão de permeabilização de lavagem, vortex e centrifugar a 400 xg durante 5 min a 4 ° C.
- Repetir a lavagem (passo 3.3.9) mais uma vez para remover qualquer anticorpo não ligado.
- Após a última lavagem, ressuspender as células em 500 ul de 1x PBS e transferir o pecadosuspensão de células para GLE 5 mL fluxo citometria de tubos. Manter as células no escuro a 4 ° C até à injecção de amostra num citómetro de fluxo.
- Execute as amostras em um citômetro de fluxo. Execute as amostras fixadas no citômetro o mais rapidamente possível, mas o mais tardar uma semana após a coloração. Executar as células não-permeabilizadas e permeabilizadas no mesmo dia.
4. Citometria de Fluxo
- Citômetro de fluxo celular Sorter diário Setup
- Ligue o software citometria de fluxo. Antes de experimentar, calibrar e configurar o citômetro de fluxo classificador de células para garantir o melhor desempenho do instrumento (ou seja, laser e óptica estão realizando a especificação, o laser e célula de fluxo estão bem alinhados) usando esferas de configuração do instrumento.
- Utilizar o bocal 100 um com bainha pressão de 20 psi.
NOTA: O bico não tem de ser alterada em um banco de citometria de fluxo. - Ajustar a taxa de fluxo do citômetro de acordo com o FABRICAÇÃOespecificação er. Excessivamente altas taxas de fluxo vai diminuir a sensibilidade na detecção de variações na fluorescência.
- Escolha azul (488 nm para excitar Fluoresceína Isothiocayanate ou FITC) e amarelo-verde (561 nm para excitar mCherry) lasers. Recolhe níveis de fluorescência FITC e com uma mCherry 530/30 nm e 610/20 nm com um filtro de passagem de banda, respectivamente.
- Adquirir a dispersão para a frente (FCS) versus dispersão lateral (SSC) gráfico de pontos para as células não coradas utilizando escala linear. Ajustar a amplificação de cada detector para visualizar as células no quadrante inferior esquerdo do gráfico de pontos.
- Reading amostra de células intactas não-permeabilizadas
- Definir a P1gate para células não-permeabilizadas vivo delineando uma forma livre em torno das células a serem analisados excluindo os detritos celulares e agregados de células, limitando assim o sinal de fluorescência para células intactas.
NOTA: Live / corantes de exclusão mortos pode ser usado para facilitar a colocação portão em células vivas. Definir 10.000 eventos para gravarna P1 portão de parada. Defina esta opção para um maior número de eventos se for necessário. - Adquirir mCherry contra o traçado de contorno de dois parâmetros FITC para detectar autofluorescência de linha de base de células não coradas. Use escala bi-logarítmica para mostrar valores negativos e melhorar a resolução entre as populações 25. Ajustar a tensão de cada detector para definir as células não coradas negativas dentro da porção inferior das primeiras dez unidades de log as parcelas de intensidade de fluorescência.
- Adquirir todas as amostras não permeabilizadas intactas usando as configurações estabelecidas em 4.1.5 e 4.1.6 e recolher FSC, SSC e sinais nos detectores de fluorescência.
- Exportação e salvar arquivos * .fcs a ser utilizado para análise de citometria de fluxo de software de análise.
- Definir a P1gate para células não-permeabilizadas vivo delineando uma forma livre em torno das células a serem analisados excluindo os detritos celulares e agregados de células, limitando assim o sinal de fluorescência para células intactas.
- Reading amostra de células permeabilizadas
- Mova o portão P1 para selecionar células vivas nas amostras permeabilizadas e ajustar FSC e tensão SSC como mostrado na 4.1.5 e 4.1.6.
- Adquirir todas as amostras permeabilizadas e recolher FSC, SSC and sinais em detectores de fluorescência.
- Exportação e salvar arquivos * .fcs a ser utilizado para análise de citometria de fluxo de software de análise.
- Análise de dados
- Lançar a citometria de fluxo arquivos de software de análise e importação * .fcs salvos em 4.2.4 e 4.3.3.
- Clique no primeiro exemplo listado na janela do espaço de trabalho. Uma nova janela com o nome do tubo número ID é aberto automaticamente. Inicie o processo gating na trama de SSC contra FSC. Desenhe um portão (P1), utilizando o ícone Elipse em torno de células vivas e eliminar quaisquer detritos, células mortas, ou agregados que têm dispersão para a frente diferente e dispersão lateral do que as células vivas
- Para desenhar o gráfico de contorno de dois parâmetros do mCherry (eixo y) versus FITC (eixo-x) a intensidade de fluorescência das células vivas, clique primeiro no eixo-x e escolher o FITC-A do canal e, em seguida, clique no y -axis e escolher o PE-mCherry-a do canal. Clique no ícone "Quad" para posicionar o marcador quadrante na extremidade de umcélulas utofluorescent em cada canal de fluorescência.
NOTA: A porta definido em torno das células FITC e mCherry positivos é a porta P2. A população de células negativa fluorescência é referido como o portão P3. Ver Figura 5 para o método de propagação representativa usada neste artigo. - Selecione P2 e P3 portões e clique no ícone "Adicionar Estatísticas" na janela área de trabalho original. Clique em "Conde" (número de células positivas) e clique em "média" (Média de Fluorescência intensidade de cada fluorocromo) ou "Median" (mediana da intensidade da fluorescência de cada fluorocromo) estatísticas entre a lista de opções. Clique no ícone "Adicionar Statistics" novamente. Todos estes valores são automaticamente transferidos para a janela área de trabalho original.
NOTA: O "significam" só é usado se a intensidade de fluorescência segue uma distribuição normal. Em qualquer outro caso, clique na aba "Median". MFI, portanto, poderia referir-se a média de fluorescência IntensitY ou mediana da intensidade de fluorescência.
NOTA: O próximo passo é a aplicação de parâmetros e estatísticas dos portões para todas as amostras testados pelo citômetro. - Na janela do espaço de trabalho, use o mouse para arrastar e soltar os parâmetros portões e estatísticas sobre a linha marcada todas as amostras.
- Gerar um relatório lote de lotes bidimensionais de contorno (mCherry vs FITC) e histogramas (contagem de células versus intensidade de fluorescência) para células não permeabilizadas e permeabilizadas (Figuras 6A - B).
- A partir das estatísticas geradas no passo 4.4.4, calcular a intensidade de fluorescência média (IFM) de cada fluorocromo para as células coradas. A partir deste valor, subtrair o valor MFI obtido a partir de células não coradas para quantificar a expressão de superfície e total da proteína de interesse.
- Informar os valores ΔMFI para cada fluoróforo (mCherry e ITCF) (Figura 6C - D). Normalizar o ΔMFI medido para o Ca V α2δ1 construir mutantes para o valor obtido para ΔMFI mCherry FITC e com o construto WT.
NOTA: O valor absoluto da intensidade de fluorescência pode variar acentuadamente dependendo do lote de anticorpos e as capacidades técnicas de cada técnico de laboratório, pelo que é necessário para normalizar a intensidade de fluorescência da construção mutante utilizando a construção WT.
Resultados
Este artigo descreve um protocolo fiável de quantificar a superfície total e células de canais de iões recombinantes expressas em TSA-201cells por um fluxo de duas cores ensaio de citometria. Como um exemplo, a superfície de célula relativa e expressão de proteína total foi quantificada para o V α2δ1subunit Ca. A fim de executar o fluxo de duas cores citometria de ensaio, Ca V α2δ1 foi duplamente marcado para expressar um epitopo de HA não fluorescente ...
Discussão
This flow cytometry-based assay was successfully applied to the measurement of relative total and cell surface levels of fluorescently-labelled pore-forming and associated subunits of voltage-gated calcium channels14,22,26. It is best used when investigating the impact of genetic mutations and thus requires that the intrinsic fluorescence intensity of the fluorescently-labelled tagged wild-type construct be at least 10 to 100-fold larger than for the fluorescence intensity of the fluorescently-labelled untagge...
Divulgações
The authors declare that they have no competing financial interests.
Agradecimentos
We thank Mr. Serge Sénéchal and Dr. Jacques Thibodeau for sharing their expertise and granting us access to their flow cytometry and cell sorting platform. This work was completed with the operating grant 130256 from the Canadian Institutes of Health Research, a grant-in-aid from the Canadian Heart and Stroke Foundation, and support from the "Fondation de l'Institut de Cardiologie de Montréal" to L.P.
Materiais
| Name | Company | Catalog Number | Comments |
| Q5 Site-Directed Mutagenesis Kit | New England Biolabs | E0554S | Can be substitute with QuickChange site-directed mutagenesis Kit (Agilent, #200523). |
| Tubes 1.5 ml | Sarstedt | 72-690-001 | |
| Tubes 15 ml | Sarstedt | 62-554-002 | |
| Disposable graduated Tranfer Pipets | VWR | 160001-192 | |
| 100 mm culture dish | Corning | 430167 | For standard culture of HEKT cells. |
| 35 mm culture dish | Falcon | 353001 | For standard culture of HEKT cells. |
| Serological pipette 1 ml | Sarstedt | 86.1251.001 | |
| Serological pipette 5 ml | Sarstedt | 86.1253.001 | |
| Serological pipette 10 ml | Sarstedt | 86.1254.001 | |
| Serological pipette 25 ml | Sarstedt | 86.1285.001 | |
| Dulbecco's high-glucose medium | Life Technologies | 12100-046 | Warm in 37 °C water bath before use. |
| Fetal Bovine Serum, qualified, heat inactivated, US origin | Life Technologies | 16140-071 | |
| Penicillin-Streptomycin (10,000 U/ml) | Life Technologies | 15140-122 | |
| Lipofectamine 2000 | Life Technologies | 11668-019 | For liposomal transfection. Can be substituted with calcium phosphate transfection. |
| Opti-MEM I Reduced Serum Medium | Life Technologies | 31985-070 | Warm in 37 °C water bath before use. |
| Trypsin-EDTA (1x) 0.05%, phenol red | Life Technologies | 25300-062 | |
| 1.5 ml microtubes | Sarstedt | 72.690.001 | |
| Phosphate Buffered Saline 1x | Fisher | BP661-10 | Can be "home-made". |
| Anti-HA FITC conjugated antibody | Sigma | H7411 | |
| IgG1−FITC Isotype Control antibody | Sigma | F6397 | |
| BD Cytofix/Cytoperm Fixation/Permeabilization Solution Kit | BD Biosciences | 554714 | Fixation/Permeabilization. Permeabilization/Wash solution, store at 4 °C. |
| Hemacytometer | Fisher | 49105161 | |
| Trypan Blue | Fisher | 15250061 | To access cell viability. |
| Refrigerated Microcentrifuge, 5430R | Eppendorf | A14H172200 | |
| Forma Steri-Cycle CO2 Incubator | Fisher | 370 | |
| Laboratory Platform Rocker | Fisher | 545034 | |
| Water Bath | VWR | 89032-216 | |
| BD FACSARIA III | BD Biosciences | 648282 | Flow cytometer. |
| FlowJo Software v10 | FlowJo | FlowJo v10 Dongle | For data analysis. |
Referências
- Delisle, B. P., Anson, B. D., Rajamani, S., January, C. T. Biology of Cardiac Arrhythmias: Ion Channel Protein Trafficking. Circ. Res. 94, 1418-1428 (2004).
- Birault, V., Solari, R., Hanrahan, J., Thomas, D. Y. Correctors of the basic trafficking defect of the mutant F508del-CFTR that causes cystic fibrosis. Curr Opin Chem Biol. 17, 353-360 (2013).
- Balijepalli, S. Y., Anderson, C. L., Lin, E. C., January, C. T. Rescue of Mutated Cardiac Ion Channels in Inherited Arrhythmia Syndromes. J. Cardiovas Pharm. 56, 113-122 (2010).
- Gargus, J. J. Unraveling Monogenic Channelopathies and Their Implications for Complex Polygenic Disease. Am. J. Hum. Genet. 72, 785-803 (2003).
- Abriel, H., Zaklyazminskaya, E. V. Cardiac channelopathies: Genetic and molecular mechanisms. Gene. 517, 1-11 (2013).
- Behr, E. R., et al. Sudden arrhythmic death syndrome: familial evaluation identifies inheritable heart disease in the majority of families. Eur Heart J. 29, 1670-1680 (2008).
- Catterall, W. A. Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev.Biol. 16, 521-555 (2000).
- Peterson, B. Z., DeMaria, C. D., Adelman, J. P., Yue, D. T. Calmodulin is the Ca2+ sensor for Ca2+ -dependent inactivation of L- type calcium channels. Neuron. 22, 549-558 (1999).
- Dolphin, A. C. Calcium channel diversity: multiple roles of calcium channel subunits. Curr.Opin.Neurobiol. 19, 237-244 (2009).
- Dai, S., Hall, D. D., Hell, J. W. Supramolecular assemblies and localized regulation of voltage-gated ion channels. Physiol Rev. 89, 411-452 (2009).
- Gao, T., et al. Identification and subcellular localization of the subunits of L-type calcium channels and adenylyl cyclase in cardiac myocytes. J. Biol. Chem. 272, 19401-19407 (1997).
- Carl, S. L., et al. Immunolocalization of sarcolemmal dihydropyridine receptor and sarcoplasmic reticular triadin and ryanodine receptor in rabbit ventricle and atrium. J. Cell Biol. 129, 673-682 (1995).
- Abriel, H., Rougier, J. S., Jalife, J. Ion Channel Macromolecular Complexes in Cardiomyocytes: Roles in Sudden Cardiac Death. Circ. Res. 116, 1971-1988 (2015).
- Bourdin, B., et al. Molecular Determinants of the Cavb-induced Plasma Membrane Targeting of the Cav1.2 Channel. J. Biol. Chem. 285, 22853-22863 (2010).
- Raybaud, A., et al. The Role of the GX9GX3G Motif in the Gating of High Voltage-activated Calcium Channels. J. Biol. Chem. 281, 39424-39436 (2006).
- Burashnikov, E., et al. Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm. 7, 1872-1882 (2010).
- Hennessey, J. A., et al. A CACNA1C Variant Associated with Reduced Voltage-Dependent Inactivation, Increased Cav1.2 Channel Window Current, and Arrhythmogenesis. PLoS ONE. 9, e106982 (2014).
- Adan, A., Alizada, G., Kiraz, Y., Baran, Y., Nalbant, A. Flow cytometry: basic principles and applications. Crit Rev Biotechnol. , 1-14 (2016).
- Graham, M. D. The Coulter Principle: Foundation of an Industry. J. Lab. Autom. 8, 72-81 (2003).
- Baumgarth, N., Roederer, M. A practical approach to multicolor flow cytometry for immunophenotyping. J. Immunol. Methods. 243, 77-97 (2000).
- Rothe, G., Sack, U., Tarnok, A., Rothe, G. . Cellular Diagnostics. Basics, Methods and Clinical Applications of Flow Cytometry. , 53-88 (2009).
- Bourdin, B., et al. Functional Characterization of Cavalpha2delta Mutations Associated with Sudden Cardiac Death. J. Biol. Chem. 290, 2854-2869 (2015).
- Tetreault, M. P., et al. Identification of glycosylation sites essential for surface expression of the Cavalpha2delta1 subunit and modulation of the cardiac Cav1.2 channel activity. J. Biol. Chem. 291, 4826-4843 (2016).
- Senatore, A., Boone, A. N., Spafford, J. D. Optimized Transfection Strategy for Expression and Electrophysiological Recording of Recombinant Voltage-Gated Ion Channels in HEK-293T Cells. J Vis Exp. (47), (2011).
- Herzenberg, L. A., Tung, J., Moore, W. A., Herzenberg, L. A., Parks, D. R. Interpreting flow cytometry data: a guide for the perplexed. Nat.Immunol. 7, 681-685 (2006).
- Shakeri, B., Bourdin, B., Demers-Giroux, P. O., Sauve, R., Parent, L. A quartet of Leucine residues in the Guanylate Kinase domain of Cavbeta determines the plasma membrane density of the Cav2.3 channel. J Biol Chem. 287, 32835-32847 (2012).
- Morton, R. A., Baptista-Hon, D. T., Hales, T. G., Lovinger, D. M. Agonist- and antagonist-induced up-regulation of surface 5-HT3A receptors. Br. J. Pharmacol. 172, 4066-4077 (2015).
- Hoffmann, C., et al. Fluorescent labeling of tetracysteine-tagged proteins in intact cells. Nat. Protocols. 5, 1666-1677 (2010).
- Cockcroft, C. J., Gamper, N. . Ion Channels: Methods and Protocols. , 233-241 (2013).
- Gonzalez-Gutierrez, G., Miranda-Laferte, E., Neely, A., Hidalgo, P. The Src Homology 3 Domain of the beta-Subunit of Voltage-gated Calcium Channels Promotes Endocytosis via Dynamin Interaction. J. Biol.Chem. 282, 2156-2162 (2007).
- Galizzi, J. P., Borsotto, M., Barhanin, J., Fosset, M., Lazdunski, M. Characterization and photoaffinity labeling of receptor sites for the Calcium channel inhibitors d-cis-diltiazem, (+/-)-bepridil, desmethoxyverapamil, and (+)-PN 200-110 in skeletal muscle transverse tubule membranes. J. Biol.Chem. 261, 1393-1397 (1986).
- Bezanilla, F. The voltage sensor in voltage-dependent ion channels. Physiol.Rev. 80, 555-592 (2000).
- Sigworth, F. J. The variance of sodium current fluctuations at the node of Ranvier. J Physiol. 307, 97-129 (1980).
- Bailey, M. A., Grabe, M., Devor, D. C. Characterization of the PCMBS-dependent modification of KCa3.1 channel gating. J. Gen. Physiol. 136, 367-387 (2010).
- Fletcher, P. A., Scriven, D. R., Schulson, M. N., Moore, E. D. Multi-Image Colocalization and Its Statistical Significance. Biophys. J. 99, 1996-2005 (2010).
- Lizotte, E., Tremblay, A., Allen, B. G., Fiset, C. Isolation and characterization of subcellular protein fractions from mouse heart. Anal. Biochem. 345, 47-54 (2005).
- Mattheyses, A. L., Simon, S. M., Rappoport, J. Z. Imaging with total internal reflection fluorescence microscopy for the cell biologist. J. Cell Sci. 123, 3621-3628 (2010).
- Yamamura, H., Suzuki, Y., Imaizumi, Y. New light on ion channel imaging by total internal reflection fluorescence (TIRF) microscopy. J. Pharmacol. Sci. 128, 1-7 (2015).
- Wible, B. A., et al. HERG-Lite-R: A novel comprehensive high-throughput screen for drug-induced hERG risk. J. Pharmacol. Toxicol. Methods. 52, 136-145 (2005).
- Wilde, A. A. M., Brugada, R. Phenotypical Manifestations of Mutations in the Genes Encoding Subunits of the Cardiac Sodium Channel. Circ. Res. 108, 884-887 (2011).
- Milano, A., et al. Sudden Cardiac Arrest and Rare Genetic Variants in the Community. Circ Cardiovasc Genet. , (2016).
- Schnell, U., Dijk, F., Sjollema, K. A., Giepmans, B. N. G. Immunolabeling artifacts and the need for live-cell imaging. Nat. Meth. 9, 152-158 (2012).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados