
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Concanavalin A-basierter Sedimentationsassay zur Messung der Substratbindung von Glucanphosphatasen
* Diese Autoren haben gleichermaßen beigetragen
In diesem Artikel
Zusammenfassung
Diese Methode beschreibt einen lektinbasierten in vitro Sedimentationsassay zur Quantifizierung der Bindungsaffinität von Glucanphosphatase und Amylopektin. Dieser Co-Sedimentations-Assay ist zuverlässig für die Messung der Glucan-Phosphatase-Substratbindung und kann auf verschiedene solubilisierte Glucan-Substrate angewendet werden.
Zusammenfassung
Glucan-Phosphatasen gehören zur größeren Familie der Dual-Spezifity-Phosphatasen (DSP), die Glucansubstrate wie Glykogen bei Tieren und Stärke bei Pflanzen dephosphorylieren. Die Kristallstrukturen der Glucan-Phosphatase mit Modell-Glucan-Substraten zeigen unterschiedliche Glucan-bindende Grenzflächen aus DSP- und Kohlenhydrat-bindenden Domänen. Quantitative Messungen von Glucan-Glucan-Phosphatase-Wechselwirkungen mit physiologisch relevanten Substraten sind jedoch grundlegend für das biologische Verständnis der Glucan-Phosphatase-Enzymfamilie und die Regulation des Energiestoffwechsels. Dieses Manuskript berichtet über einen Concanavalin A (ConA)-basierten in vitro Sedimentationsassay, der entwickelt wurde, um die Substratbindungsaffinität von Glucanphosphatasen gegen verschiedene Glucansubstrate nachzuweisen. Als Proof of Concept wurde die Dissoziationskonstante (KD) der Glucanphosphatase Arabidopsis thaliana Starch Excess4 (SEX4) und Amylopektin bestimmt. Die Charakterisierung von SEX4-Mutanten und anderen Mitgliedern der Glucan-Phosphatase-Familie von Enzymen demonstriert die Nützlichkeit dieses Assays zur Beurteilung der differentiellen Bindung von Protein-Kohlenhydrat-Interaktionen. Diese Daten zeigen die Eignung dieses Assays zur Charakterisierung eines breiten Spektrums von Stärke- und Glykogen-interagierenden Proteinen.
Einleitung
Glucan-Phosphatasen sind Mitglieder einer funktionell vielfältigen Unterfamilie von Dual-Spezifitäts-Phosphatasen (DSPs) innerhalb der Protein-Tyrosin-Phosphatase (PTP)-Superfamilie1. Sie wurden in den meisten Lebensformen gefunden, einschließlich sehr unterschiedlicher photosynthetischer Organismen, Menschen, Wirbeltieren und einigen wirbellosen Tieren und Protisten 2,3,4. Pflanzen enthalten drei bekannte Glucan-Phosphatasen: Stärkeüberschuss4 (SEX4), Like Sex Four1 (LSF1) und Like Sex Four2 (LSF2)5,6,7. Pflanzen, denen Glucanphosphatasen fehlen, zeigen eine verringerte Rate des vorübergehenden Stärkeabbaus und der Anreicherung von Stärke in den Blättern 8,9. Laforin ist das Gründungsmitglied der Glucan-Phosphatase-Familie, die Glykogen bei Wirbeltieren und Menschen dephosphoryliert 3,10. Die Mutationen von Laforin führen zur neurodegenerativen Lafora-Krankheit, einer tödlich verlaufenden autosomal-rezessiven Form der Epilepsie11. Glucanphosphatasen sind für den Glykogen- und Stärkestoffwechsel notwendig und haben sich als wichtige Enzyme für die Modulation des Stärkegehalts in Pflanzen und die Behandlung der neurodegenerativen Lafora-Krankheit herauskristallisiert12,13. Neuere Röntgenkristallographie-Untersuchungen an Glucanphosphatasen mit Modell-Glucan-Substraten haben Aufschluss über die Substratbindung und den katalytischen Mechanismus der Glucan-Dephosphorylierunggegeben 14,15,16,17. Das derzeitige Verständnis darüber, wie Glucanphosphatasen an ihre physiologischen Substrate binden, ist jedoch unvollständig.
Stärke ist ein unlösliches Glukosepolymer, das zu 80%-90% aus Amylopektin und zu 10%-20% aus Amylosebesteht 18. Die Substrate für pflanzliche Glucanphosphatasen sind phosphorylierte Kohlenhydratmoleküle wie Glykogen- und Stärkekörner. Die phosphorylierten Glucosylreste liegen in einem Phosphat:Glucosyl-Restverhältnis von 1:600 vor. Interessanterweise sind die Phosphate nur auf den Amylopektinmolekülen19 vorhanden. Die pflanzliche Haupt-Glucan-Phosphatase SEX4 wirkt auf die Stärkekörner, um Amylopektinmoleküle zu dephosphorylieren. Die Röntgenkristallstruktur von SEX4 in Kombination mit strukturgesteuerten Mutagenesestudien hat die einzigartigen Substratspezifitäten von SEX4 für verschiedene Positionen innerhalb einer Glucanstruktur gezeigt15. Kürzlich konnten wir zeigen, dass die biologisch relevante Aktivität von SEX4 nur beobachtet werden kann, wenn es auf seine solubilisierten Amylopektinsubstrateeinwirkt 20. Das Verständnis der Glucan-SEX4-Wechselwirkungen hat sich jedoch aufgrund der strukturellen Komplexität des Substrats, der breiteren Bindungsspezifität und der geringen Bindungsaffinitäten zwischen dem Protein und seinen Substraten als schwierig erwiesen. Diese Probleme haben die Fähigkeit behindert, Methoden zu nutzen, die üblicherweise in Protein-Ligand-Interaktionen verwendet werden, wie z. B. die isotherme Titrationskalorimetrie (ITC), die Kernspinresonanzspektroskopie (NMR) und ELISA-basierte Assays (Enzyme-linked Immunosorbent Assay).
Interessanterweise stammt ein Großteil unseres Verständnisses der Kohlenhydrat-Protein-Wechselwirkungen aus der Untersuchung von Lektinen. Concanavalin A (ConA) ist eine Familie von Leguminosen-Lektinen, die ursprünglich aus der Jackbohne gewonnen wurden. ConA bindet Kohlenhydrate mit hoher Spezifität, was für den Einsatz in Drug-Targeting- und Drug-Delivery-Anwendungen von Vorteil ist. Die Bindung von ConA an eine Vielzahl von Substraten, die nicht-reduzierendes α-D-Mannosyl und α-D-Glucosyl enthalten, wurde umfassend untersucht19,20. Kommerziell erhältliche ConA-gebundene Sepharose-Kügelchen werden üblicherweise zur Reinigung von Glykoproteinen und Glykolipiden verwendet21. ConA bindet an diese Glucane über C3-, C4- und C6-Hydroxylgruppen der Glukosereste. ConA-Sepharose-Beads wurden auch erfolgreich verwendet, um die Bindung von Glykogen-Protein- und Stärke-Protein-Wechselwirkungen zu messen22,23. In dieser Studie haben wir ConA-Sepharose-Beads verwendet, um einen Bindungsassay zu entwickeln, um die Bindungsspezifität von Glucan-Phosphatase-Amylopektin-Interaktionen zu messen.
Zuvor wurde ein ConA-basierter Sedimentationsassay verwendet, um die Bindungsfähigkeit des Glucanphosphatase-Substrats zu beurteilen14,20,24. In dieser Arbeit wurde die gleiche Strategie verwendet, um eine neuartige Methode zur Bestimmung der Bindungsaffinität von Glucan-Glucan-Phosphatase- und Kohlenhydrat-Interaktionen zu entwickeln. Diese Methode hat auch einen Vorteil für die Untersuchung verschiedener solubilisierter Kohlenhydrat-Protein-Wechselwirkungen.
Protokoll
1. Herstellung von ConA-Sepharose-Kügelchen
- Stellen Sie 250 ml eines Bindepuffers her, der 67 mM HEPES (pH 7,5), 10 mMMgCl2 und 0,2 mM CaCl2enthält. Stellen Sie den pH-Wert mit 1 M NaOH-Lösung ein.
- Pipettieren Sie 250 μl ConA-Sepharose-Bead-Suspension in ein 1,5-ml-Mikrozentrifugenröhrchen. Zentrifugieren Sie den Inhalt bei 10.000 x g für 30 s bei 4 °C. Entsorgen Sie den Überstand.
HINWEIS: Für jede für den Assay verwendete Amylopektinkonzentration werden 250 μl ConA-Sepharose-Beads in einem 1,5-ml-Mikrozentrifugenröhrchen benötigt. - Geben Sie 750 μl des Bindungspuffers in jedes Röhrchen, das 250 μl ConA-Sepharose-Kügelchen enthält. Zentrifugieren Sie die Röhrchen bei 10.000 x g für 1 min bei 4 °C. Entfernen Sie den Überstand. Wiederholen Sie diesen Schritt 2x, um sicherzustellen, dass die Perlen ordnungsgemäß gewaschen und mit dem Bindepuffer im Gleichgewicht sind.
2. Herstellung von Amylopektinlösungen
- Stellen Sie eine Stammlösung von 10 mg/ml Kartoffelamylopektin her. Amylopektin ist wasserunlöslich und kann durch Hitze aufgelöst werden. Zur Solubilisierung 0,1 g Kartoffel-Amylopektin in 10 ml destilliertes Wasser geben. Erhitzen Sie die Suspension im Wasserbad bei 80 °C für 1 h oder bis die Lösung nicht mehr trüb ist.
- Lassen Sie die Lösung wieder auf Raumtemperatur (RT) kommen, wobei Sie wiederholt vortexen, um ein Verklumpen zu vermeiden.
- Die alkoholalkalische Behandlung ist eine alternative Methode zur Solubilisierung von Amylopektinsubstraten. Um mit dieser Methode zu solubilisieren, führen Sie die folgenden Schritte aus.
- 0,5 g Amylopektinsubstrat werden in 5 ml 20%igem Ethanol und 5 ml 2 M NaOH suspendiert. Rühren Sie den Inhalt 15-20 min bei RT kräftig um.
- Als nächstes fügen Sie 10 ml Wasser hinzu und stellen den pH-Wert der Lösung auf 6,5 ein, indem Sie 2 M HCl hinzufügen. Bringen Sie das Volumen der resultierenden Lösung mit destilliertem Wasser auf 50 ml, um eine 10 mg/ml Amylopektinlösung herzustellen.
- Verdünnen Sie die 10 mg/ml gelöste Amylopektinlösung, um eine Reihe von 2 ml verdünnten Amylopektinlösungen herzustellen. Führen Sie beispielsweise halbe Verdünnungen von 10 mg/ml durch, um eine Reihe von Amylopektinkonzentrationen herzustellen (5 mg/ml, 2,5 mg/ml, 1,25 mg/ml, 0,625 mg/ml, 0,3125 mg/ml, 0,156 mg/ml, 0,078 mg/ml, 0,039 mg/ml, 0,019 mg/ml und 0 mg/ml).
3. Herstellung von ConA-Sepharose: Amylopektin-Kügelchen
- Geben Sie 250 μl jeder verdünnten Amylopektinlösung in 1,5-ml-Mikrozentrifugenröhrchen mit 250 μl ConA-Sepharose-Kügelchen, die in Bindepuffer voräquilibriert sind. Mischen Sie den Inhalt gut. Beschriften Sie die Röhrchen mit der entsprechenden Amylopektinkonzentration.
- Den Inhalt auf einem rotierenden Rad bei 4 °C für 30 min inkubieren.
HINWEIS: Der an ConA-Sepharose:Amylopektin-gebundene Komplex ändert sich nach 20 Minuten nicht im Laufe der Zeit. Die Inkubationszeit von 30 Minuten wurde gewählt, indem die Inkubationszeiten von 10 Minuten bis 1 Stunde variiert wurden, um sicherzustellen, dass ein Gleichgewicht erreicht wurde. - Zentrifugieren Sie die Röhrchen bei 10.000 x g für 1 Min. Sammeln Sie den Überstand in einem neu gekennzeichneten 1,5-ml-Mikrozentrifugenröhrchen. Bewahren Sie diese überstehenden Fraktionen auf, um den D-Glukose-Assay12 durchzuführen (saure Hydrolyse von Amylopektin, gefolgt von UV-Bestimmung der Glukose mittels enzymatischem Assay). Dieser Schritt ist notwendig, um sicherzustellen, dass das gesamte Amylopektin an die Kügelchen gebunden ist.
- Fügen Sie 750 μl Bindungspuffer zu den ConA-Sepharose:Amylopektin-Beads hinzu. Zentrifugieren Sie die Röhrchen bei 10.000 x g für 1 Min. Verwerfen Sie den Überstand, um alle ungebundenen Amylopektinmoleküle zu entfernen.
- Wiederholen Sie Schritt 3.4, um eine ausreichende Wäsche zu gewährleisten. Jedes Röhrchen enthält nun ConA-Sepharose-Kügelchen, die an unterschiedliche Mengen an Amylopektin-Substraten gebunden sind.
4. Inkubation von SEX4 mit ConA-Sepharose:Amylopektin-Beads
- Mischen Sie 250 μl ConA-Sepharose:Amylopektin-Beads mit 100 μl des Bindungspuffers, der 10 μg SEX4-Protein, 10 mM Dithiothreitol (DTT) und 10 μM Proteaseinhibitor-Cocktail (PIC) enthält. Beachten Sie, dass das Gesamtvolumen in jedem Röhrchen 350 μl beträgt.
HINWEIS: Ein Proteasehemmer-Cocktail wird vorsorglich hinzugefügt, um einen unnötigen SEX4-Abbau zu vermeiden. Dies ist ein optionaler Schritt. In diesem Assay wird das rekombinante Protein Arabidopsis thaliana SEX4 (AtSEX4) verwendet. Das gereinigte Protein enthält einen N-terminalen Histidin-Tag, der für den Nachweis des Proteins mittels Chemilumineszenz erforderlich ist. Detaillierte Informationen zur Aufreinigung von Glucanphosphatasen sind in früheren Veröffentlichungen 14,20,24 beschrieben. - Inkubieren Sie das Protein und die ConA-Sepharose:Amylopektin-Bead-Suspension bei 4 °C für 45 min unter sanfter Rotation.
Anmerkungen: Die Inkubationszeit von 45 Minuten wird gewählt, um sicherzustellen, dass das Gleichgewicht für den Komplex erreicht wird. - Zentrifugieren Sie die Röhrchen bei 10.000 x g für 1 Min. Pipettieren Sie 50 μl des Überstandes vorsichtig mit einer Gelladespitze in ein neues 1,5-ml-Mikrozentrifugenröhrchen. Geben Sie 20 μl 4x SDS-PAGE-Farbstoff und 10 μl Wasser in jedes Röhrchen, das 50 μl der gesammelten Überstandsfraktionen enthält. Erhitzen Sie die Proben bei 95 °C für 10 min. Speichern Sie diese Beispiele für die Ausführung der SDS-PAGE-Gele. Stellen Sie sicher, dass 10 neue Röhrchen mit der Aufschrift "Überstand (S)" die entsprechenden Substratkonzentrationen aufweisen.
- Geben Sie 750 μl des Bindungspuffers in die ConA-Sepharose:amylopectin: SEX4-Kügelchen, um ungebundenes Protein aus den Kügelchen zu entfernen. Zentrifugieren Sie die Röhrchen bei 10.000 x g für 1 Min. Wiederholen Sie diesen Schritt noch einmal, um eine ordnungsgemäße Wäsche zu gewährleisten. Entsorgen Sie den Überstand.
- Geben Sie 20 μl 4x SDS-PAGE-Farbstoff und 80 μl destilliertes Wasser in die Röhrchen mit gewaschenen ConA-Sepharose:Amylopektin:SEX4-Kügelchen. Erhitzen Sie die Proben 10 min lang bei 95 °C und zentrifugieren Sie sie 1 min lang bei 10.000 x g .
- Entsorgen Sie das Pellet und bewahren Sie den Überstand für den Betrieb der SDS-PAGE-Gele auf. 80 μl des Überstandes in neue Röhrchen pipettieren und als "Pellet (P)" kennzeichnen.
5. Ausführen von SDS-PAGE-Gelen
- Laden Sie 40 μl der ungebundenen Proteinproben (hergestellt in Schritt 2.3, mit S gekennzeichnet) in 4%-12%ige vorgefertigte Polyacrylamid-Gelvertiefungen von der niedrigsten bis zur höchsten, aber lassen Sie die erste Spur frei, um den Proteinmolekulargewichtsmarker zu laden. Verwenden Sie ein zweites Gel, um 10 gebundene Proteinproben zu laden, die in Schritt 2.5 hergestellt wurden (gekennzeichnet als P).
- Frisch zubereiteten SDS-PAGE Laufpuffer in beide Kammern des Gerätes geben. Lassen Sie das Gel 35 Minuten lang bei 150 V laufen oder bis die Farbstofffront den Boden des Gels erreicht.
- Entfernen Sie das Laufgel aus dem Gerät und entfernen Sie die Abstandshalter und Glasplatten. Verwenden Sie das separierte Gel, um eine Western-Blot-Analyse durchzuführen.
6. Western Blot zur Chemilumineszenz-Detektion14,15
HINWEIS: Diese Methode kann leicht modifiziert/angepasst werden, je nachdem, welche Western-Blotting-Geräte die Benutzer in ihren Laboren haben.
- 1 l Transferpuffer mit 5,8 g Tris-Base, 2,9 g Glycin, 0,37 g SDS und 200 ml Methanol herstellen.
- Übertragen Sie die größengetrennten Proteine aus dem Polyacrylamidgel auf eine Nitrozellulosemembran. Schwämme, Filterpapiere, Gel und Nitrozellulosemembran nach westlichem Transferprotokoll14,15 kurz zusammenbauen. 1 h bei 70 V laufen lassen.
- Um eine unspezifische Proteinbindung zu verhindern, inkubiert man die Nitrocellulosemembran, die die Proteinlösung aus 1%-5% Rinderserumalbumin (BSA) oder Milchprotein enthält, in 50 ml TBST-Puffer (20 mM Tris [pH 7,5], 150 mM NaCl, 0,1% Tween 20) für 1 h. Waschen Sie die Membran 3x mit TBST-Puffer, um ungebundene Blockierlösung zu entfernen.
- Inkubieren Sie die Membran 1 h lang mit einem Meerrettichperoxidase (HRP)-gekoppelten Antikörper, der spezifisch für das His-markierte Protein ist. Waschen Sie die Membran 3x in TBST-Puffer, um ungebundene Antikörper zu entfernen. Verwenden Sie eine 1:2.000-Verdünnung des Antikörpers gegen TBST für optimale Reproduzierbarkeit und Sensitivität.
- Der HRP-Enzym-gekoppelte Antikörper bindet spezifisch an den Histidin-Tag des SEX4-Proteins, was in Gegenwart von Chemilumineszenz-Reagenzien eine Bande ergibt. Stellen Sie für die digitale Bildgebung eine Lösung aus gleichen Teilen chemilumineszierender Substratlösungen (jeweils 750 μl) in einem 1,5-ml-Röhrchen her. Inkubieren Sie die Membran mindestens 5 Minuten lang in der Lösung.
- Legen Sie das Membranprotein mit der Seite nach unten auf den Blot-Scanner und führen Sie die Erfassungssoftware aus, um das Protein sowohl in der Pellet- als auch in der Überstandsfraktion zu quantifizieren.
7. Datenanalyse
- Führen Sie die quantitativen Signalmessungen mit der Erfassungssoftware mit dem Blot-Scanner durch. Normalisieren Sie alle quantitativen Messungen in den Überstands- und Pelletfraktionen auf das insgesamt beladene Protein.
HINWEIS: Die Software ermöglicht die Quantifizierung der Intensität jeder Proteinbande in den Überstands- und Pelletfraktionen. - Im Experiment zur Sättigungsbindung wird der Prozentsatz der proteingebundenen im Vergleich zur Amylopektinkonzentration aufgetragen. Passen Sie die Daten an Y = Bmax x X/(K D + X) an, indem Sie die Datenanalysesoftware verwenden, um KD zu berechnen.
HINWEIS: Bmax ist die maximale spezifische Bindung, die Y-Achse ist der Prozentsatz des gebundenen Proteins, die und die X-Achse ist die Amylopektinkonzentration.

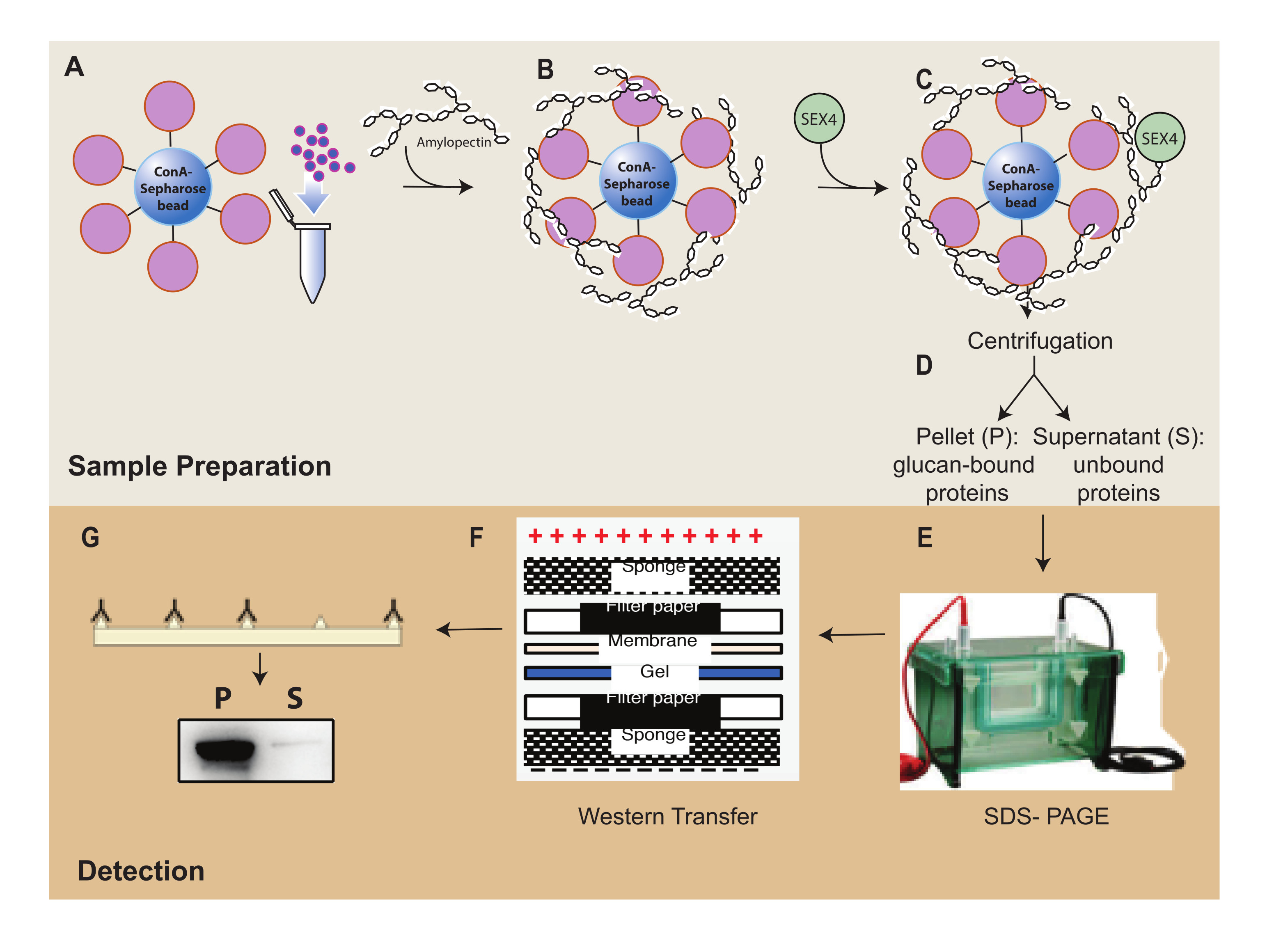
Abbildung 1: Überblick über den Arbeitsablauf des ConA-Sepharose-Sedimentationsassays. (A) Herstellung von ConA-Sepharose-Beads. (B) Inkubation mit Amylopektinsubstrat. (C) Inkubation mit SEX4-Protein. (D) Trennung von gebundenen und ungebundenen Proteinfraktionen durch Zentrifugation. (E) Trennung von Proteinen durch SDS-PAGE. (F) Western-Blot-Analyse. (G) Chemilumineszenz-Nachweis von His-markiertem SEX4-Protein. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
Ergebnisse
Eines der Hauptmerkmale der Glucan-Phosphatase-Proteinfamilie ist ihre Fähigkeit, an Glucansubstrate zu binden. Zunächst wurde die Bindungskapazität von SEX4 an ConA-Sepharose:Amylopektin-Beads mittels SDS-PAGE analysiert (Abbildung 2A). Rinderserumalbumin (BSA) diente als Negativkontrolle, um eine unspezifische Bindung von Proteinen an die ConA-Sepharose:Amylopektin-Kügelchen nachzuweisen. Die SDS-PAGE-Analyse von Proteinen zeigte das Vorhandensein von SEX4-Protein in der Pelletfraktion...
Diskussion
Diese Arbeit demonstriert die erfolgreiche Entwicklung eines neuartigen in vitro Sedimentationsassays, der die Bestimmung der Bindungsaffinität von Glucan-Glucan-Phosphatase-Interaktionen ermöglicht. Das Assay-Design nutzt die spezifische Bindung von Lektin ConA an Glucane über die Hydroxylreste der Glukose, um gelöste Kohlenhydratsubstrate indirekt auf Sepharose-Beads einzufangen. Dies ermöglicht die Trennung von gebundenen und ungebundenen Proteinfraktionen durch Zentrifugation und Besti...
Offenlegungen
Die Autoren erklären keine Interessenkonflikte.
Danksagungen
Diese Studie wurde durch den Preis der National Science Foundation MCB-2012074 unterstützt. Die Autoren danken Dr. Craig W. Vander Kooi vom Department of Biochemistry and Molecular Biology der University of Florida für wertvolle Gespräche und Unterstützung. Die Autoren danken auch Dr. Matthew S. Gentry vom Department of Biochemistry and Molecular Biology der University of Florida für seine Unterstützung. Wir bedanken uns bei Dr. Sara Lagalwar, Vorsitzende des Neurowissenschaftsprogramms des Skidmore College, für die Erlaubnis, den LICOR C-Digit-Blot-Scanner für die Western-Blot-Bildgebung zu verwenden.
Materialien
| Name | Company | Catalog Number | Comments |
| 6x-His Tag monoclonal antibody (HIS.H8), HRP | Therm Fisher Scientific | MA1-21315-HRP | |
| Biorad gel electrophoresis and Western blot kit | Biorad | 1703930 | |
| Calcium chloride | Sigma-Aldrich | 208291 | |
| C-Digit blot scanner | LICOR | 3600-00 | Blot scanner |
| Complete protease inhibitor cocktail | Sigma-Aldrich | 11836170001 | |
| Concanavalin A-sepharose beads | Sigma-Aldrich | C9017 | This product contains in 0.1 M acetate buffer, pH 6, containing 1 M NaCl, 1 mM CaCl2, 1 mM MnCl2, and 1 mM MgCl2 in 20% ethanol |
| Centrifuge | Eppendorf | 5425R | |
| Glycine | Fisher Scientific | BP381-5 | |
| GraphPad Prism 8.0 software | GraphPad | Version 8.0 | Data analysis software |
| HEPES | Sigma-Aldrich | H8651 | |
| Image Studio | LICOR | 3600-501 | Acquisition Software |
| Magnesium chloride | Sigma-Aldrich | M2670 | |
| Methanol | Fisher Scientific | A452SK-4 | |
| Sodium dodecyl sulfate | Fisher Scientific | PI28312 | |
| Potato amylopectin | Sigma-Aldrich | A8515 | |
| Precast SDSPAGE Gels | Genscript | M00653S | |
| Tris base | Fisher Scientific | BP154-1 | |
| Tween 20 | Fisher Scientific | MP1TWEEN201 | |
| Westernsure premium chemiluminescence substrate | LI-COR | 926-95000 |
Referenzen
- Meekins, D. A., Vander Kooi, C. W., Gentry, M. S. Structural mechanisms of plant glucan phosphatases in starch metabolism. The FEBS Journal. 283 (13), 2427-2447 (2016).
- Gentry, M. S., et al. The phosphatase laforin crosses evolutionary boundaries and links carbohydrate metabolism to neuronal disease. The Journal of Cell Biology. 178 (3), 477-488 (2007).
- Worby, C. A., Gentry, M. S., Dixon, J. E. Laforin, a dual specificity phosphatase that dephosphorylates complex carbohydrates. The Journal of Biological Chemistry. 281 (41), 30412-30418 (2006).
- Gentry, M. S., Pace, R. M. Conservation of the glucan phosphatase laforin is linked to rates of molecular evolution and the glucan metabolism of the organism. BMC Evolutionary Biology. 9, 138 (2009).
- Niittyla, T., et al. Similar protein phosphatases control starch metabolism in plants and glycogen metabolism in mammals. The Journal of Biological Chemistry. 281 (17), 11815-11818 (2006).
- Kotting, O., et al. STARCH-EXCESS4 is a laforin-like Phosphoglucan phosphatase required for starch degradation in Arabidopsis thaliana. The Plant Cell. 21 (1), 334-346 (2009).
- Comparot-Moss, S., et al. A putative phosphatase, LSF1, is required for normal starch turnover in Arabidopsis leaves. Plant Physiology. 152 (2), 685-697 (2010).
- Zeeman, S. C., Northrop, F., Smith, A. M., Rees, T. A starch-accumulating mutant of Arabidopsis thaliana deficient in a chloroplastic starch-hydrolysing enzyme. The Plant Journal: For Cell and Molecular Biology. 15 (3), 357-365 (1998).
- Kotting, O., et al. Identification of a novel enzyme required for starch metabolism in Arabidopsis leaves. The phosphoglucan, water dikinase. Plant Physiology. 137 (1), 242-252 (2005).
- Tagliabracci, V. S., et al. Laforin is a glycogen phosphatase, deficiency of which leads to elevated phosphorylation of glycogen in vivo. Proceedings of the National Academy of Sciences. 104 (49), 19262-19266 (2007).
- Gentry, M. S., Guinovart, J. J., Minassian, B. A., Roach, P. J., Serratosa, J. M. Lafora disease offers a unique window into neuronal glycogen metabolism. The Journal of Biological Chemistry. 293 (19), 7117-7125 (2018).
- Brewer, M. K., et al. Targeting pathogenic lafora bodies in lafora disease using an antibody-enzyme fusion. Cell Metabolism. 30 (4), 689-705 (2019).
- Santelia, D., Zeeman, S. C. Progress in Arabidopsis starch research and potential biotechnological applications. Current Opinion in Biotechnology. 22 (2), 271-280 (2011).
- Raththagala, M., et al. Structural mechanism of laforin function in glycogen dephosphorylation and lafora disease. Molecular Cell. 57 (2), 261-272 (2015).
- Meekins, D. A., et al. Phosphoglucan-bound structure of starch phosphatase Starch Excess4 reveals the mechanism for C6 specificity. Proceedings of the National Academy of Sciences. 111 (20), 7272-7277 (2014).
- Vander Kooi, C. W., et al. Structural basis for the glucan phosphatase activity of Starch Excess4. Proceedings of the National Academy of Sciences. 107 (35), 15379-15384 (2010).
- Meekins, D. A., et al. Structure of the Arabidopsis glucan phosphatase like sex four2 reveals a unique mechanism for starch dephosphorylation. The Plant Cell. 25 (6), 2302-2314 (2013).
- Smith, A. M., Zeeman, S. C. Starch: A flexible, adaptable carbon store coupled to plant growth. Annual Review of Plant Biology. 71, 217-245 (2020).
- Jane, J., Kasemuwan, T., Chen, J. F., Juliano, B. O. Phosphorus in rice and other starches. Cereal Foods World. 41 (11), 827-832 (1996).
- Mak, C. A., et al. Cooperative kinetics of the glucan phosphatase starch excess4. Biochemistry. 60 (31), 2425-2435 (2021).
- Campbell, K. P., MacLennan, D. H. Purification and characterization of the 53,000-dalton glycoprotein from the sarcoplasmic reticulum. The Journal of Biological Chemistry. 256 (9), 4626-4632 (1981).
- Campbell, K. P., MacLennan, D. H., Jorgensen, A. O., Mintzer, M. C. Purification and characterization of calsequestrin from canine cardiac sarcoplasmic reticulum and identification of the 53,000 dalton glycoprotein. The Journal of Biological Chemistry. 258 (2), 1197-1204 (1983).
- Davey, M. W., Sulkowski, E., Carter, W. A. Binding of human fibroblast interferon to concanavalin A-agarose. Involvement of carbohydrate recognition and hydrophobic interaction. Biochemistry. 15 (3), 704-713 (1976).
- Meekins, D. A., et al. Mechanistic insights into glucan phosphatase activity against polyglucan substrates. The Journal of Biological Chemistry. 290 (38), 23361-23370 (2015).
- Wilkens, C., et al. Plant α-glucan phosphatases SEX4 and LSF2 display different affinity for amylopectin and amylose. FEBS Letters. 590 (1), 118-128 (2016).
- Atanasova, M., Bagdonas, H., Agirre, J. Structural glycobiology in the age of electron cryo-microscopy. Current Opinion in Structural Biology. 62, 70-78 (2020).
- Doyle, M. L. Characterization of binding interactions by isothermal titration calorimetry. Current Opinion in Biotechnology. 8 (1), 31-35 (1997).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten