
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Montaje y Seguimiento de Desarrollo Comunitario Microbiano dentro de una Plataforma de Matriz de Micropedulos
En este artículo
Resumen
El desarrollo de las comunidades microbianas depende de una combinación de factores, incluyendo la arquitectura ambiental, la abundancia de los miembros, los rasgos y las interacciones. Este protocolo describe un ambiente sintético y microfabricado para el seguimiento simultáneo de miles de comunidades contenidas en pozos de femtoliter, donde factores clave como el tamaño de nicho y el confinamiento pueden ser aproximados.
Resumen
El desarrollo de las comunidades microbianas depende de una combinación de factores deterministas y estocásticos complejos que pueden alterar drásticamente la distribución espacial y las actividades de los miembros de la comunidad. Hemos desarrollado una plataforma de matriz de micropocillos que se puede utilizar para montar y rastrear rápidamente miles de comunidades bacterianas en paralelo. Este protocolo destaca la utilidad de la plataforma y describe su uso para supervisar ópticamente el desarrollo de comunidades simples de dos miembros dentro de un conjunto de matrices dentro de la plataforma. Esta demostración utiliza dos mutantes de Pseudomonas aeruginosa , parte de una serie de mutantes desarrollados para estudiar la patogenicidad de la secreción de Tipo VI. Las inserciones cromosómicas de los genes mCherry o GFP facilitan la expresión constitutiva de proteínas fluorescentes con distintas longitudes de onda de emisión que pueden usarse para monitorear la abundancia y ubicación de los miembros de la comunidad dentro de cada micropocillo. Este protocolo describe una metodologíaD para ensamblar mezclas de bacterias en los pocillos de la matriz y usar imágenes de fluorescencia en intervalos de tiempo y análisis cuantitativo de imágenes para medir el crecimiento relativo de cada población miembro a lo largo del tiempo. Se discute la siembra y el ensamblaje de la plataforma de micropocillos, los procedimientos de imagen necesarios para el análisis cuantitativo de las comunidades microbianas dentro de la matriz y los métodos que pueden usarse para revelar interacciones entre el área de las especies microbianas.
Introducción
Las comunidades microbianas están formadas por factores deterministas, como la estructura del medio ambiente y los procesos estocásticos, que están asociados con la muerte celular, la división, la concentración de proteínas, el número de organelos y la mutación 1 . Dentro del entorno natural, puede ser casi imposible analizar el impacto individual de estas influencias en la composición y actividad de la comunidad. Obscurecido por las estructuras naturales y enterrado dentro de un medio químico y biológico, la identificación de los miembros de la comunidad y la resolución de su distribución espaciotemporal en el medio ambiente natural es extremadamente difícil. Sin embargo, los esfuerzos recientes han subrayado la importancia de la organización espacial en la función de la comunidad y señalan la necesidad de tener en cuenta tanto la abundancia como la organización de los miembros en los estudios en curso 2 , 3 , 4 .
Eso( Es decir, la disponibilidad de nutrientes y metabolitos secundarios), la estructura física ( por ejemplo, la arquitectura del suelo, las raíces de las plantas, las partículas oceánicas o los microvellos intestinales), la presencia o ausencia de oxígeno y la introducción de Las especies patógenas afectan a la composición, la arquitectura y la función de las comunidades microbianas 5 , 6 , 7 , 8 , 9 , 10 , 11 . Sin embargo, las técnicas tradicionales para las culturas que descuidan capturar estos factores continúan prevaleciendo. La composición de la comunidad ( por ejemplo, la presencia de especies co-dependientes), la unión física, la concentración de la molécula de señalización y el contacto directo célula-célula son factores importantes para formar una comunidad microbiana y pueden perderse en cCondiciones de cultivo convencionales. Estas propiedades son difíciles de replicar en un cultivo líquido a granel o en una placa de agar. Sin embargo, la disponibilidad de técnicas microfluídicas, micropatternas y de nanofabricación que permiten la replicación de características físicas y químicas claves de entornos naturales ha permitido a muchos investigadores construir comunidades bacterianas para estudiar sus interacciones 12 , 13 , 14 y desarrollar ambientes sintéticos que Imitan las condiciones naturales 4 , 15 , 16 , 17 , 18 , 19 , 20 .
Este protocolo describe un método para fabricar un dispositivo de matriz de micropocillos y proporciona procedimientos experimentales detallados que se pueden utilizar paraE en la matriz y para cultivar bacterias, tanto como colonias de una sola especie como en comunidades de múltiples miembros. Este trabajo también demuestra cómo se pueden usar bacterias modificadas para producir proteínas informadoras fluorescentes para monitorear el crecimiento bacteriano dentro de los pozos con el tiempo. Una matriz similar se presentó anteriormente y mostró que es posible rastrear el crecimiento de colonias de una sola especie de Pseudomonas aeruginosa ( P. aeruginosa) en micropocillos. Mediante la modulación del tamaño del pocillo y la densidad de siembra, las condiciones de partida de miles de experimentos de crecimiento se pueden variar en paralelo para determinar cómo las condiciones de inoculación inicial afectan a la capacidad de crecimiento de las bacterias 21 . El trabajo actual utiliza una versión ligeramente modificada de la matriz de micropocillos que se basa en el trabajo anterior, permitiendo la comparación simultánea de múltiples matrices y utilizando un protocolo experimental más robusto. La matriz utilizada en este trabajo contiene múltiples subarrays, o array ensemQue contienen pozos de diferentes tamaños, que van desde 15 - 100 μm de diámetro, que están dispuestos en tres pasos diferentes ( es decir , 2x, 3x y 4x el diámetro del pozo). Las matrices se graban en silicio y el crecimiento de las bacterias sembradas en las matrices de silicio se habilita sellando las matrices con un cubreobjetos que se ha recubierto con un gel de agarosa con infusión media. Los mutantes de P. aeruginosa diseñados para estudiar el sistema de secreción de Tipo VI se usan en esta demostración.
Los resultados presentados aquí se construyen hacia el objetivo final de analizar las comunidades de múltiples miembros dentro de las matrices de micropocillos, lo que permite a los investigadores para controlar la abundancia y la organización de las bacterias in situ mientras controla y sondeo del entorno químico. Esto, en última instancia, ofrecerá información sobre las "reglas" que rigen el desarrollo y la sucesión de la comunidad.
Protocolo
1. Fabricación del Microwell-arsenal del silicio
- Revestimiento de parileno
- Depositar entre 1-1,5 μm de parileno N en obleas de silicio utilizando un sistema de revestimiento de parileno comercialmente disponible según las especificaciones e instrucciones del fabricante (ajustes: punto de consigna del vaporizador = 160 ° C, punto de ajuste del horno = 650 ° C).
NOTA: Aproximadamente 6 g de parileno N cargado en una cámara producen recubrimientos de 1-1,5 μm de espesor.
- Depositar entre 1-1,5 μm de parileno N en obleas de silicio utilizando un sistema de revestimiento de parileno comercialmente disponible según las especificaciones e instrucciones del fabricante (ajustes: punto de consigna del vaporizador = 160 ° C, punto de ajuste del horno = 650 ° C).
- Fotolitografía
- Revestir por centrifugación las obleas revestidas con N de parileno con un promotor de adhesión, hexametildisilazano al 20% (HMDS) y acetato de éter monometílico de propilenglicol al 80% (PGMEA) (ver la Tabla de Materiales ) a 3.000 rpm durante 45 s. Llene una pipeta de transferencia de 2 ml con un promotor de adhesión y espolvorearla sobre toda la oblea. Deje que la oblea se sienta durante aproximadamente 10 segundos antes de centrifugarla.
- Llene una pipeta de transferencia de 2 ml conNe (ver la Tabla de Materiales ) y dispensar la fotorresistencia en el centro de la oblea. Girar a 3.000 rpm durante 45 s para producir un revestimiento resistente que tiene aproximadamente 1,5 μm de espesor.
- Hornear las muestras en una placa caliente a 115 ° C durante 1 min.
- Utilice un alineador de contactos y una fotomáscara con el patrón de pozo deseado para exponer la muestra a la luz ultravioleta. Exponer la oblea recubierta por centrifugación a través de la fotomáscara con dibujos durante 6 s, dando una dosis aproximada de 60-80 mJ / cm2 medida a 365 nm.
- Desarrollar el patrón sumergiendo la muestra en el revelador (<3% de hidróxido de tetrametilamonio en agua, véase la Tabla de Materiales ) durante 2 min. Enjuagar con agua DI y secar con nitrógeno limpio y seco.
NOTA: Las áreas de fotorresistencia expuestas a la radiación UV deben ser despejadas durante el desarrollo.
- Grabado con iones reactivos
- Utilice un grabado con plasma de oxígeno para eliminar el parileno expuestoTodo el camino hasta el sustrato de silicio.
NOTA: La receta puede ser modulada para alterar la velocidad de grabado del parileno. Para espesores de parileno entre 1 y 5 μm, use una receta con 60 mTorr, 20 ° C, 100 sccm O 2 , 10 W RF, y 2.000 W ICP en una herramienta de Ion Etching Reactivo (RIE). Después de grabar y retirar la capa de parileno expuesta, el área estampada ( es decir , el silicio expuesto) debe lucir brillante y plateada. - Utilice un proceso de grabado profundo de RIE (DRIE, eg, Bosch DRIE) para grabar en el silicio.
NOTA: La velocidad de grabado y la duración determinarán la profundidad del pozo. Un ciclo completo del proceso Bosch (una etapa de deposición de 3 s: 20 mTorr, 15ºC, 140 sccm C 4 F 8 , 10 W RF y 1,750 W ICP seguido por un proceso de decapado de 10 s: 20 mTorr, 15ºC , 120 sccm SF 6 , 8 W RF y 1,750 W ICP) corresponde a aproximadamente 1 μm de profundidad de ataque. Los pozos utilizados en esta demostración oscilan entre 3 y 3,5 μm de profundidad. - VLa profundidad del grabado utilizando la profilometría física.
- Cargue la muestra en un perfilómetro físico (consulte la Tabla de materiales ).
- Encienda el vacío de muestra y presione el botón de carga manual.
- Enfoque el sistema en la muestra presionando el botón "Focus". Coloque una característica apropiada para la medición en la pantalla de visualización.
- Escanear la muestra. Nivele el perfil y mida la profundidad de la característica.
- Registre la velocidad de grabado y modifique los tiempos de grabado posteriores para lograr la profundidad deseada.
NOTA: Las mediciones incluirán la profundidad del pozo de silicio, el espesor del parileno depositado y el grosor de la fotorresistencia. La verificación del espesor de cada capa durante todo el procedimiento es necesaria para lograr una profundidad de pozo precisa.
- Utilice un grabado con plasma de oxígeno para eliminar el parileno expuestoTodo el camino hasta el sustrato de silicio.
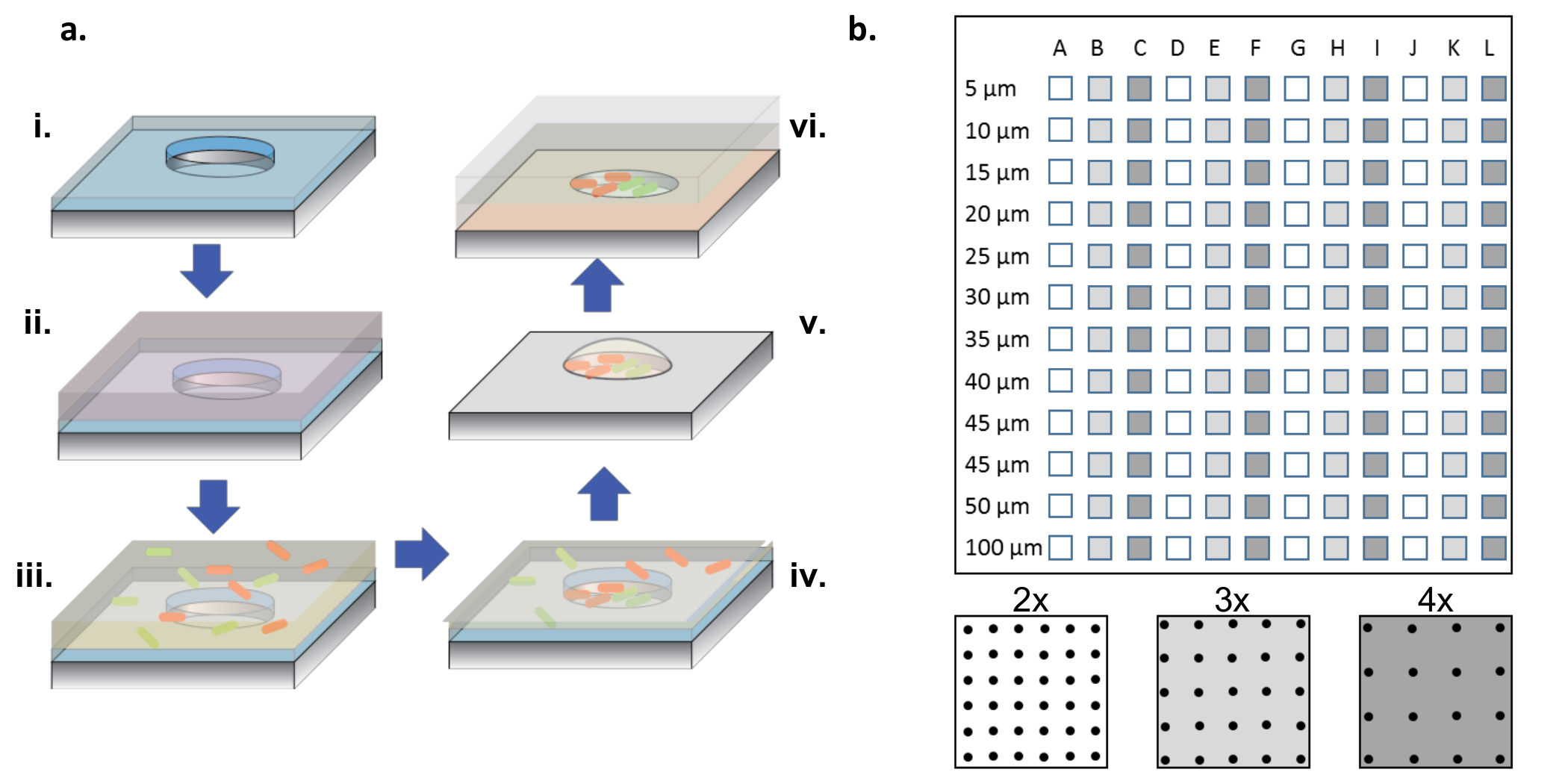
2. Cultivo bacteriano y siembra ( Figura 1a )
- Iniciar colonias en Luria Broth (LB) placas de agar de las existencias de glicerol y el uso dentro de dos semanas. Elije colonias de las cepas deseadas de las placas de agar LB y comience durante la noche los cultivos de P. aeruginosa. Incubar los cultivos durante la noche durante aproximadamente 18 h a 37ºC mientras se agita a 220 rpm en medio R2A.
NOTA: Las colonias deben ser recogidas dentro de las dos semanas de la galjanoplastia para asegurar que las mutaciones y los genes fluorescentes se mantienen. Todos los trabajos de P. aeruginosa deben realizarse en condiciones BSL-2. - Utilice un escriba de diamante para la sección de la oblea de silicio en fichas individuales que contienen los conjuntos de diferentes tamaños y arreglos de tono-pozo. Asegúrese de que cada chip contenga el complemento completo de tamaños de pozos y campos para el estudio.

Figura 1: Procedimiento de fabricación y siembra de células. (A) Arrays de micropocillos aRe grabado en obleas de silicio revestidas con una fina capa de parileno (i). Para humedecer los pocillos y / o para funcionalizar la superficie, se añade una solución de proteína en una gotita encima de las matrices (ii). Se separa la solución de proteína, se secan las obleas y se añade una nueva solución que contiene las bacterias deseadas (iii). La solución bacteriana se elimina después de un período de incubación, y las obleas se dejan secar dejando bacterias en los pocillos y en la superficie (iv). Las bacterias asociadas a la superficie se eliminan con el despegue de parileno, dejando atrás las bacterias que se siembran limpiamente en los micropocillos y siguen siendo viables debido al medio de glicerol al 2%, lo que ayuda a mantener los pocillos hidratados (v). Los chips de silicio se colocan entonces en el lado de la matriz hacia abajo sobre una lámina de vidrio revestida con gel de agarosa, que alimenta el crecimiento bacteriano en los micropocillos (vi). ( B ) Disposición de sub-matrices en un único dispositivo de silicio. Cada sub-matriz contiene un conjunto de pozos idénticos. El diámetro de los micropocillos a través de todos los sub-aLos rayos varían en diámetro desde 5-100 μm y están organizados en 2x, 3x o 4x el paso de diámetro del pozo, que se indica por los colores blanco a gris oscuro en el esquema del panel inferior. Cuando las profundidades de los pozos son poco profundas (<10 μm), los diámetros de los pozos de 5 y 10 μm son raramente útiles, generalmente debido a la falta de células que colonizan estos pocillos muy pequeños. En este trabajo, sólo se analizaron los datos de pozos con diámetros de 15-100 μm. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
NOTA: Como se muestra en la Figura 1b , un chip completo contiene sub-conjuntos de pocillos, con diámetros entre 5 y 100 μm, con tres pasos diferentes ( es decir , 2x, 3x y 4x el diámetro) repitiéndose 4 veces.
- Colocar una gotita de 150 μL de Albumina de Suero Bovino (BSA) de 500 μg / mL en solución de PBSEn la parte superior de la matriz para mojar los micropocillos. Incubar la solución de BSA durante 1 h en el chip a RT en una cámara húmeda.
- Cree la cámara rellenando la parte inferior de una caja de puntas de pipeta vacía con solución salina tamponada con fosfato (PBS).
NOTA: Se pueden usar otras sustancias, como lectinas específicas, en lugar de BSA para funcionalizar la superficie de los micropocillos.
- Cree la cámara rellenando la parte inferior de una caja de puntas de pipeta vacía con solución salina tamponada con fosfato (PBS).
- Mientras se incuban chips de silicio con solución de BSA, se centrifugan los cultivos a 2.500 rpm (correspondientes a un promedio de 950 xg) durante 5 min y después se resuspenden en 500 μl de medio R2A fresco con 2% de glicerol.
- Determinar la DO del cultivo usando un espectrómetro UV-vis a 600 nm. Ajústelo a una DO de 0,02 utilizando 2% de medio glicerol R2A.
NOTA: El glicerol ayuda a evitar que los pocillos se sequen durante el despegue de parileno.
- Determinar la DO del cultivo usando un espectrómetro UV-vis a 600 nm. Ajústelo a una DO de 0,02 utilizando 2% de medio glicerol R2A.
- Después de la incubación, retirar la solución de BSA y enjuagar 3x con PBS eliminando y reemplazando la gota de líquidoT que cubre la matriz de micropocillos de silicio. Secar bajo nitrógeno.
- Añadir 150 μl de 0,02 cultivos OD a cada uno de los conjuntos secos colocados en una cámara húmeda. Incubar durante 1 h a 4 ° C para permitir que las bacterias se adhieran a las paredes del pozo.
NOTA: La refrigeración no es necesaria para la incubación. El tiempo de incubación de 4 ° C se puede utilizar para prevenir el crecimiento de bacterias antes de que comience la imagen, de modo que se pueda visualizar la organización espacial de las comunidades antes del crecimiento. También puede utilizarse la incubación a temperatura ambiente. Ambos protocolos dan como resultado curvas de crecimiento similares.
3. Configuración del Microscopio
- Antes de iniciar la incubación bacteriana en los chips de silicio, encienda la cámara de control medioambiental de la etapa superior (consulte la tabla de materiales) y ajuste los ajustes en la caja de control para que la humedad (~ 100%) y la temperatura (30-32 ° C, véase el paso 3.2) puede equilibrarse antes de añadir las muestras.
- Nivele el soporte de la muestra y alinee elAlrededor de la muestra con toallitas de laboratorio empapadas con PBS (ver Tabla de Materiales ) para aumentar la humedad en la cámara hasta el punto de rocío. Ajuste la temperatura de la cámara a 30 ° C y la de la tapa de la cámara a 32 ° C para reducir la condensación en el plano de imagen.
NOTA: El porta-diapositivas encaja en la cámara de células vivas con una junta de aproximadamente 1 cm de espesor. El soporte de la muestra se nivela con la ayuda de un nivel de burbuja que se coloca en la parte superior del soporte de la muestra. El soporte de la muestra se puede inclinar ligeramente y permanecer sellado en la junta al nivel. - Mientras los cultivos se incuban en los chips de silicio, encienda manualmente el interruptor de alimentación de la lámpara de mercurio por lo menos 30 minutos antes de la formación de imágenes. Activar manualmente la cámara y el escenario automático del microscopio. Abra el software utilizado para controlar el microscopio y el equipo periférico y asegúrese de que el equipo reconoce el equipo.
NOTA: La ampliación es 10X con NA = 0.3.
- Las soluciones de agarosa previamente preparadas en microondas ( es decir, 2% de agarosa en medio R2A) hasta que se alcanza un estado líquido, aproximadamente 60 s.
- Mueva la parte posterior de un portaobjetos de vidrio de 75 mm x 22 mm, # 1.5 con etanol y colóquelo longitudinalmente, centrado, a través de un portaobjetos de vidrio de 2 x 3 "(50 x 75 mm) Coloque dos separadores PDMS (espesor de ~ 1 mm) A lo largo de los bordes largos de la cubreobjetos y desplace el cubreobjetos de vidrio de modo que aproximadamente 1 mm de la cubreobjetos sobresalga del borde de la corredera.
- Vierta 5 ml de solución de agarosa líquida en la parte superior del cubreobjetos de vidrio, lo suficiente para cubrir completamente, y coloque un segundo 2 "de 3" de vidrio en la parte superior del conjunto para "emparedar" el agar líquido entre el cubreobjetos y la diapositiva.
NOTA: Esto controla la profundidad de la agarosa, haciendo que el espesor total de la cubreobjetos y agarosa endurecida sea igual en espesor a los espaciadores PDMS. - Permitir que el cristal de diapositivas-coverslip-agarosa gel-vidrio sándwich de diapositivas para establecer hasta que la solución de agarosa comienza a solidificar; Luego, transferirlo a un refrigerador. Después de 15 minutos, retire el exceso de agarosa sólida y corte alrededor del cubreobjetos de vidrio. Mueva esto a un plato limpio y colóquelo en un refrigerador hasta su uso.
5. Sellado de los pozos con un recubrimiento revestido de agarosa e imágenes
- Una vez finalizado el período de incubación de las bacterias, retirar el cubreobjetos recubierto de agarosa del refrigerador y preparar los chips de silicio, como sigue.
- Sumerja los chips de silicio en agua ultrapura, uno a la vez, durante 10 s cada uno. Póngalos en sus bordes en un trapo o tejido de laboratorio hasta que la mayor parte del exceso de líquido se haya drenado de los bordes de las virutas.
- Corte un pedazo de cinta para que coincida con la longitud del borde de cada chip de silicio. Coloque la cinta sobre el parileno que cubre el silicio y utilícelo para pelar rápidamente el revestimiento de parileno.
- ImmedInvertir inmediatamente cada viruta pelada y colocar cada chip de tal manera que el lado de la matriz de micropocillos se enfrenta (y hace contacto) con el lado revestido con agarosa de un cubreobjetos revestido con agarosa. Tenga cuidado de no mover o cambiar el chip después de que toque la agarosa para evitar el crecimiento de bacterias fuera de los pozos.
- Coloque el cubreobjetos de micropocillos / agarosa ensamblados en el porta-diapositivas de la cámara de control ambiental de nivel superior en el miscroscopio.
- Utilice luz ambiental o luz dirigida ( por ejemplo, una linterna) para localizar matrices de interés. Utilice el software comercial que controla la etapa automatizada para guardar esas posiciones (consulte la tabla de materiales). Apague la luz ambiente o la luz dirigida después de almacenar las posiciones.
- En el software comercial, abra el panel "ND Acquisition".
NOTA: Este panel incluye un menú para guardar automáticamente en un directorio específico, así como la adquisición de imágenes programables. Para estos experimentos, Se utilizan los menús "Hora", "XY" y "λ". - Para guardar las ubicaciones en el software, haga clic en el "menú XY" y luego marque una casilla vacía en el lado izquierdo para cada posición que necesita ser guardada. También, haga clic en el botón "Incluir Z".
- Adquirir imágenes a lo largo del tiempo a las longitudes de onda deseadas y 10 aumentos utilizando los cubos de filtro de fluorescencia adecuados (ver Tabla de Materiales).
- Utilice el software de control y las posiciones de matriz guardadas para moverse a cada ubicación guardada y enfocar los pozos. Haga clic en cada ubicación XY en la lista guardada y ajuste el enfoque utilizando el filtro Green Flourescence Protein (GFP). Guarde la nueva posición z haciendo clic en la flecha que apunta a la ubicación z.
NOTA: Este proceso puede tardar mucho tiempo. Considere tomar la precaución de aumentar la ganancia y usar el filtro de densidad neutra para reducir la intensidad de la luz para evitar el fotoblanqueo. - Determine la distancia del eje z entre planos focales para cada longitud de onda, anotando la diferencia en la posición del eje z cuando se enfoca en la superficie de la matriz. Elija 2-3 ubicaciones de la matriz con la población mixta de bacterias rojas / verdes y enfoque utilizando el filtro de Proteína de Fluorescencia Roja (RFP).
- Reste la distancia entre los planos focales utilizando los filtros de fluorescencia GFP y RFP y añada el ajuste del plano focal en el menú "λ".
NOTA: Por ejemplo, si la matriz aparece enfocada en el canal GFP en una ubicación z de 50 μm, y la misma matriz aparece enfocada en el canal RFP a 55 μm, añada +5 junto a la configuración óptica RFP en la "λ ".
- Reste la distancia entre los planos focales utilizando los filtros de fluorescencia GFP y RFP y añada el ajuste del plano focal en el menú "λ".
- Comience la adquisición de imágenes de lapso de tiempo.
NOTA: Para los experimentos mostrados aquí, las imágenes RFP y GFP se adquirieron para cada posición de la matriz a intervalos de 30 minutos usando adquisición de imágenes multidimensional a través de un software comercial que controlaLa cámara, el obturador, la rueda del filtro y la etapa motorizada.- Ajuste el "intervalo" a 30 min y la "duración del experimento" a 24 h en el menú "Tiempo". Haga clic en "Ejecutar ahora".
NOTA: Con las casillas "Time", "XY" y "λ" marcadas, ejecutar el programa moverá el escenario a la imagen de cada ubicación ( es decir, las ubicaciones XYZ guardadas), tomará una imagen en una longitud de onda, moverá la posición z Para tomar en cuenta las diferencias de plano focal ( es decir, control de la longitud de onda o lambda), tomar la segunda imagen, pasar a la siguiente ubicación de la matriz (multipunto) y realizar un bucle a intervalos de 30 minutos.
- Ajuste el "intervalo" a 30 min y la "duración del experimento" a 24 h en el menú "Tiempo". Haga clic en "Ejecutar ahora".
- Utilice el software de control y las posiciones de matriz guardadas para moverse a cada ubicación guardada y enfocar los pozos. Haga clic en cada ubicación XY en la lista guardada y ajuste el enfoque utilizando el filtro Green Flourescence Protein (GFP). Guarde la nueva posición z haciendo clic en la flecha que apunta a la ubicación z.
- Adquirir imágenes de control de iluminación.
NOTA: Utilice los menús "Adquisición ND", "Tiempo" y "XY" para tomar imágenes de 4 ubicaciones, 25 veces cada una.- Tome una serie de 100 imágenes "darkfield" apagando todas las fuentes de luz y tomandoUna "imagen" de una diapositiva estándar. Estas imágenes capturarán el ruido de la cámara. Utilice el tiempo de exposición más largo utilizado durante el tiempo de espera (paso 5.3.3).
- Tomar una serie de 100 "imágenes de campo de iluminación" mediante la proyección de imagen de una diapositiva estándar ( es decir, uniforme RFP o GFP intensidad) en algunos lugares diferentes para capturar la iluminación desigual en las condiciones experimentales dado. Elija un tiempo de exposición que maximice la señal sin alcanzar la saturación.
6. Análisis
- Procese las pilas de imágenes utilizando un software de análisis de imágenes ( por ejemplo, ImageJ).
- Convierte las imágenes adquiridas en formato de archivo tiff utilizando el software comercial. Suba imágenes al software de análisis de imágenes haciendo clic en "Archivo"> "Importar"> "Secuencia de imagen".
- Cree una "imagen de corrección" haciendo un promedio de todas las imágenes de "campo oscuro" y "campo de iluminación". SubtracT la imagen "darkfield" promedio de la imagen promedio de "campo de iluminación" eligiendo "Process"> "Image Calculator". Seleccione las dos imágenes, "Image1" e "Image2", y luego "Subtract" en el campo "Operation". Haga clic en Aceptar."
- Para calcular el promedio, cargue las imágenes de corrección (o campo oscuro), haga clic en "Imagen"> "Pilas"> "Proyecto Z"> "Proyección promedio".
- Realice el registro de la imagen si es necesario. A continuación, realice la resta de fondo haciendo clic en "Proceso"> "Restar fondo". Introduzca un radio ( por ejemplo, 125) en el campo "radio" y seleccione "deslizante paraboloide".
- Realice la corrección de iluminación usando "Proceso"> "Calculadora Plus". Elija los siguientes parámetros: operación, división; I1, imagen del pozo; I2, imagen de corrección; K1, media de imagen de corrección; Y k2, 0. Haga clic en "CreComió Nueva Ventana. "
NOTA: Este conjunto de datos no requiere registro, pero en otro trabajo, ImageJ Plugin StackReg se utilizó con la transformación "Traducción". Para la sustracción de fondo, use el mismo radio deslizante de paraboloide para cada conjunto de imágenes. Por ejemplo, si los pozos más grandes tienen un radio de pixel de 100, utilice un radio mayor que 100 ( por ejemplo, 125) para cada conjunto de imágenes.
- Determinar el crecimiento de cada cepa en los micropocillos.
- Seleccione las regiones de interés (ROI) alrededor de cada micropocillo en las matrices deseadas usando el complemento ImageJ "MicroArray".
- En el menú "MAP", haga clic en "Restablecer cuadrícula". Especifique filas, columnas y diámetro (basado en el tamaño y el número del pozo en la matriz, consulte la Figura 1b ). Seleccione "círculo" en el menú "ROI forma".
- Mantenga pulsada la tecla "Alt" mientras selecciona el ROI superior izquierdo con el ratón para mover tMatriz del ROI. Mantenga pulsada la tecla "shift" mientras selecciona el ROI inferior izquierdo para cambiar el tamaño de la matriz. Mantenga pulsada la tecla "shift" mientras selecciona un ROI del lado derecho de la matriz, pero no en las esquinas, para cambiar el espaciado de los ROI.
- Utilice los comandos anteriores para ajustar la matriz ROI sobre los pozos de una imagen. Haga clic en "Medir RT".
NOTA: El complemento exportará las mediciones deseadas de cada ROI. Utilice tres tamaños ROI, creando anillos concéntricos alrededor de los pozos para recoger localmente la señal de fondo ( es decir, la señal del anillo central restado del anillo externo) y mediciones de fluorescencia ( es decir, la señal del anillo interno).
- Recopilar los datos en un software de hoja de cálculo y calcular la señal de fondo. Importar a un software de scripting personalizado para un análisis más detallado.
- Seleccione las regiones de interés (ROI) alrededor de cada micropocillo en las matrices deseadas usando el complemento ImageJ "MicroArray".
- Organización y análisis de datos
- Importe los datos y organice los datosRecogidos en ImageJ en una matriz en el siguiente orden para todos los tiempos: columna 1, sub-array número; Columna 2, fila de pozos; Columna 3, columna de pozo; Columna 4, intensidad media; Columna 5, intensidad de fondo; Y columna 6, intensidad media - intensidad de fondo.
- Separe los resultados de la adquisición mCherry y GFP en diferentes matrices. Guarde los resultados de cada sub-matriz y cada color en una celda diferente en una matriz de celdas.
NOTA: Esta organización facilita el desplazamiento entre datos de imagen y resultados de medición, la limpieza de los datos y la garantía de que las mediciones representan con precisión los datos.
- Separe los resultados de la adquisición mCherry y GFP en diferentes matrices. Guarde los resultados de cada sub-matriz y cada color en una celda diferente en una matriz de celdas.
- Ajuste para la autofluorescencia de P. aeruginosa .
NOTA: En experimentos que involucran el co-cultivo de cepas GFP y mCherry, se debe analizar un chip mCherry sólo para determinar la relación entre mCherry y autofluorescencia verde.- Trazar la señal mCherry-versus-GFP de todos los mCherry ΔretS &# 916; tse / i1-6 en todos los puntos temporales para determinar la relación entre la señal mCherry y la autofluorescencia en el canal GFP. Reste la señal de autofluorescencia de los co-cultivos.
- Trace las trayectorias y ajuste una ecuación logística modificada a cada trayectoria para extraer los parámetros usando el ajuste de mínimos cuadrados en un software de hoja de cálculo o un software de scripting personalizado.
- Busque correlaciones entre y entre los parámetros de trayectoria de GFP y mCherry.
- Importe los datos y organice los datosRecogidos en ImageJ en una matriz en el siguiente orden para todos los tiempos: columna 1, sub-array número; Columna 2, fila de pozos; Columna 3, columna de pozo; Columna 4, intensidad media; Columna 5, intensidad de fondo; Y columna 6, intensidad media - intensidad de fondo.
Resultados
La plataforma experimental presentada aquí está diseñada para estudios de alto rendimiento y alto contenido de comunidades bacterianas. El diseño permite que miles de comunidades, que crecen en pozos de diferentes tamaños, sean analizadas simultáneamente. Con este diseño de la matriz de micropocillos, se puede determinar la dependencia de la composición final de la comunidad sobre las densidades iniciales de siembra, el tamaño del pozo y el ambiente químico. Este trabajo demues...
Discusión
En este artículo se presentó un dispositivo de matriz de micropocillos y protocolos experimentales diseñados para permitir el análisis de alta capacidad y alto contenido de células vivas basados en imágenes del desarrollo de la comunidad bacteriana. Si bien el objetivo de la demostración aquí fue estudiar los efectos de la secreción de tipo VI mediada por contacto en el desarrollo de la comunidad, los arrays fueron diseñados para ser flexible y acomodar el estudio de una amplia gama de comunidades microb...
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Las matrices de micropocillos se fabricaron y caracterizaron en la División de Instalaciones de Usuarios del Centro de Ciencia de Materiales de Nanofase, Oficina de Ciencias Básicas de Energía, Departamento de Energía de los Estados Unidos. El apoyo financiero para este trabajo se proporcionó a través del Fondo de Investigación y Desarrollo del Director del Laboratorio Nacional de Oak Ridge. Los autores también desean agradecer al Laboratorio J. Mougous (Universidad de Washington, Seattle, WA) por el suministro de cepas de P. aeruginosa usadas en estos estudios.
Materiales
| Name | Company | Catalog Number | Comments |
| Parylene N | Specialty Coating Systems | CAS NO.:1633-22-3 | |
| Parylene coater | Specialty Coating Systems | Labcoter 2 Parylene Deposition Unit PDS2010 | |
| Silicon Wafer | WRS Materials | 100mm diameter, 500-550μm thickness, Prime, 10-20 resistivity, N/Phos<100>, | |
| adhesion promoter | Shin-Etsu Microsci | MicroPrime P20 adhesion promoter | |
| postive tone photoresist | Rohm and Haas Electronics Materials LLC (Owned by Dow) | Microposit S1818 Positive Photoresist (code 10018357) | |
| Quintel Contact Aligner | Neutronix Quintel Corp | NXQ 7500 Mask Aligner | |
| Reactive Ion Etching Tool | Oxford Instruments | Plasmalab System 100 Reactive Ion Etcher | |
| R2A Broth | TEKnova | R0005 | |
| Bovine Serum Albumin | Sigma | A9647 | |
| Multimode Plate Reader | Perkin Elmer | Enspire, 2300-0000 | |
| Fluorescent Microscope | Nikon | Eclipse Ti-U | |
| Automated Stage | Prior | ProScan III | |
| CCD camera | Nikon | DS-QiMc | |
| Stage-top environmental control chamber | In Vivo Scientific | STEV ECU-HOC | |
| Phosphate Buffered Saline | ThermoFisher Scientific | 14190144 | |
| UltraPure Agarose | ThermoFisher Scientific | 16500500 | |
| 25 x 75 mm No. 1.5 coverslip | Nexterion | High performance #1.5H coverslips | |
| Fluorescence Reference Slides | Ted Pella | 2273 | |
| Physical Stylus Profilometer | KLA Tencor | P-6 | |
| lab wipes | Kimberly Clark | Kimipe KIMTECH SCIENCE Brand, 34155 | |
| commercial software | Nikon | NIS Elements | |
| Zeiss 710 Confocal Microscope | Zeiss | ||
| filter cubes | Nikon | Nikon FITC (96311), Nikon Texas Red(96313) |
Referencias
- Zhou, J., Deng, Y., et al. Stochasticity, succession, and environmental perturbations in a fluidic ecosystem. Proc Natl Acad Sci. 111, E836-E845 (2014).
- Valm, A. M., Welch, J. L. M., et al. Systems-level analysis of microbial community organization through combinatorial labeling and spectral imaging. Proc Natl Acad Sci USA. 108 (10), 4152-4157 (2011).
- Satoh, H., Miura, Y., Tsushima, I., Okabe, S. Layered structure of bacterial and archaeal communities and their in situ activities in anaerobic granules. Appl Environ Microbiol. 73 (22), 7300-7307 (2007).
- Kim, H. J., Boedicker, J. Q., Choi, J. W., Ismagilov, R. F. Defined spatial structure stabilizes a synthetic multispecies bacterial community. Proc Natl Acad Sci USA. 105 (47), 18188-18193 (2008).
- Nunan, N., Wu, K., Young, I. M., Crawford, J. W., Ritz, K. Spatial distribution of bacterial communities and their relationships with the micro-architecture of soil. FEMS Microbiol Ecol. 44, 203-215 (2003).
- Grundmann, G. L. Spatial scales of soil bacterial diversity - The size of a clone. FEMS Microbiol Ecol. 48, 119-127 (2004).
- Langenheder, S., Lindstrom, E. S., Tranvik, L. J. Structure and Function of Bacterial Communities Emerging from Different Sources under Identical Conditions. Appl Environ Microbiol. 72 (1), 212-220 (2006).
- Camp, J. G., Kanther, M., Semova, I., Rawls, J. F. Patterns and Scales in Gastrointestinal Microbial Ecology. Gastroenterology. 136 (6), 1989-2002 (2009).
- Renner, L. D., Weibel, D. B. Physicochemical regulation of biofilm formation. MRS Bull. 36 (5), 347-355 (2011).
- Wessel, A. K., Hmelo, L., Parsek, M. R., Whiteley, M. Going local: technologies for exploring bacterial microenvironments. Nat Rev Microbiol. 11 (5), 337-348 (2013).
- Stacy, A., McNally, L., Darch, S. E., Brown, S. P., Whiteley, M. The biogeography of polymicrobial infection. Nat Rev Microbiol. 14 (2), 93-105 (2015).
- Hansen, R. R., Shubert, K. R., Morrell-Falvey, J. L., Lokitz, B. S., Doktycz, M. J., Retterer, S. T. Microstructured block copolymer surfaces for control of microbe adhesion and aggregation. Biosensors. 4 (1), 63-75 (2014).
- Hansen, R. R., Hinestrosa, J. P., et al. Lectin-functionalized poly(glycidyl methacrylate)- block -poly(vinyldimethyl azlactone) surface scaffolds for high avidity microbial capture. Biomacromolecules. 14 (10), 3742-3748 (2013).
- Timm, C. M., Hansen, R. R., Doktycz, M. J., Retterer, S. T., Pelletier, D. A. Microstencils to generate defined, multi-species patterns of bacteria. Biomicrofluidics. 9 (6), (2015).
- Keymer, J. E., Galajda, P., Muldoon, C., Park, S., Austin, R. H. Bacterial metapopulations in nanofabricated landscapes. Proc Natl Acad Sci USA. 103 (46), 17290-17295 (2006).
- Zhang, Q., Lambert, G., et al. Acceleration of Emergence of Bacterial Antibiotic Resistance in Connected Microenvironments. Science. 333 (6050), 1764-1767 (2011).
- Friedlander, R. S., Vlamakis, H., Kim, P., Khan, M., Kolter, R., Aizenberg, J. Bacterial flagella explore microscale hummocks and hollows to increase adhesion. Proc Natl Acad Sci USA. 110 (14), 5624-5629 (2013).
- Zhou, J., Liu, W., et al. Stochastic Assembly Leads to Alternative Communities with Distinct Functions in a Bioreactor Microbial Community. MBio. 4 (2), 1-8 (2013).
- van Vliet, S., Hol, F. J., Weenink, T., Galajda, P., Keymer, J. E. The effects of chemical interactions and culture history on the colonization of structured habitats by competing bacterial populations. BMC Microbiol. 14 (1), 116 (2014).
- Niepa, T. H. R., Hou, L., et al. Microbial Nanoculture as an Artificial Microniche. Sci Rep. 6, 30578 (2016).
- Hansen, R. H., Timm, A. C., et al. Stochastic Assembly of Bacteria in Microwell Arrays Reveals the Importance of Confinement in Community Development. PLoS ONE. 11 (5), e0155080 (2016).
- Hood, R. D., Singh, P., et al. A Type VI Secretion System of Pseudomonas aeruginosa Targets a Toxin to Bacteria. Cell Host Microbe. 7 (1), 25-37 (2010).
- LeRoux, M., Ja De Leon, ., et al. Quantitative single-cell characterization of bacterial interactions reveals type VI secretion is a double-edged sword. Proc Natl Acad Sci. 109 (48), 19804-19809 (2012).
- Whitney, J. C., Beck, C. M., et al. Genetically distinct pathways guide effector export through the type VI secretion system. Mol Microbiol. 92 (3), 529-542 (2014).
- Warrick, J. W., Timm, A., Swick, A., Yin, J. Tools for Single-Cell Kinetic Analysis of Virus-Host Interactions. PLoS ONE. 11 (1), e0145081 (2016).
- Zwietering, M. H., Jongenburger, I., Rombouts, F. M., Van't Riet, K. Modeling of the Bacterial Growth Curve. Appl Environ Microbiol. 56 (6), 1875-1881 (1990).
- Halsted, M., Wilmoth, J. L., et al. Development of transparent microwell arrays for optical monitoring and dissection of microbial communities. J Vac Sci Technol B Nanotechnol Microelectron. 34 (6), 06KI03 (2016).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados