
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Montagem e Rastreamento do Desenvolvimento de Comunidade Microbiana em uma Plataforma Microwell Array
Neste Artigo
Resumo
O desenvolvimento de comunidades microbianas depende de uma combinação de fatores, incluindo arquitetura ambiental, abundância de membros, traços e interações. Este protocolo descreve um ambiente sintético e microfabricado para o rastreamento simultâneo de milhares de comunidades contidas em poços de femtoliter, onde fatores-chave como tamanho de nicho e confinamento podem ser aproximados.
Resumo
O desenvolvimento de comunidades microbianas depende de uma combinação de fatores deterministas e estocásticos complexos que podem alterar drasticamente a distribuição espacial e as atividades dos membros da comunidade. Nós desenvolvemos uma plataforma de matriz de microwell que pode ser usada para montar e rastrear rapidamente milhares de comunidades bacterianas em paralelo. Este protocolo destaca a utilidade da plataforma e descreve seu uso para monitorar opticamente o desenvolvimento de comunidades simples de dois membros dentro de um conjunto de arrays dentro da plataforma. Esta demonstração usa dois mutantes de Pseudomonas aeruginosa , parte de uma série de mutantes desenvolvidos para estudar a patogenicidade da secreção do Tipo VI. As inserções cromossômicas de genes mCherry ou GFP facilitam a expressão constitutiva de proteínas fluorescentes com comprimentos de onda de emissão distintos que podem ser usados para monitorar a abundância e localização de membros da comunidade dentro de cada micropoeira. Este protocolo descreve um metho detalhadoD para montagem de misturas de bactérias nos poços da matriz e uso de imagens de fluorescência por lapso de tempo e análise de imagem quantitativa para medir o crescimento relativo de cada população membro ao longo do tempo. A semeadura e montagem da plataforma de micropoços, os procedimentos de imagem necessários para a análise quantitativa das comunidades microbianas dentro da matriz e os métodos que podem ser usados para revelar as interações entre a área de espécies microbianas discutidas.
Introdução
As comunidades microbianas são moldadas por fatores deterministas, como a estrutura do meio ambiente e os processos estocásticos, associados à morte celular, divisão, concentração de proteína, número de organelas e mutação 1 . Dentro do ambiente natural, pode ser quase impossível analisar o impacto individual dessas influências na composição e atividade da comunidade. Obscurecido por estruturas naturais e enterrado dentro de um meio químico e biológico, identificar membros da comunidade e resolver ainda mais sua distribuição espaciotemporal dentro do ambiente natural é extremamente desafiador. No entanto, os esforços recentes sublinharam a importância da organização espacial na função comunitária e apontam para a necessidade de explicar a abundância e a organização dos membros nos estudos em andamento 2 , 3 , 4 .
istoÉ claro que o ambiente químico local ( ou seja, a disponibilidade de nutrientes e metabolitos secundários), a estrutura física ( por exemplo, arquitetura do solo, raízes da planta, partículas do oceano ou microvília intestinal), a presença ou ausência de oxigênio e a introdução de As espécies patogênicas afetam a composição, a arquitetura e a função das comunidades microbianas 5 , 6 , 7 , 8 , 9 , 10 , 11 . No entanto, as técnicas tradicionais para culturas que negligenciam a captura desses fatores continuam a prevalecer. A composição da comunidade ( por exemplo, a presença de espécies co-dependentes), a ligação física, a concentração da molécula de sinalização e o contato direto com células-células são fatores importantes para a formação de uma comunidade microbiana e podem ser perdidos em cCondições culturais convencionais. Essas propriedades são difíceis de replicar em uma cultura líquida em massa ou em uma placa de ágar. A disponibilidade de técnicas microfluídicas, micropatternadoras e de nanofabricação que permitem a replicação de características físicas e químicas fundamentais de ambientes naturais, no entanto, permitiu que muitos pesquisadores construíssem comunidades bacterianas para estudar suas interações 12 , 13 , 14 e desenvolver ambientes sintéticos que Imita as condições naturais 4 , 15 , 16 , 17 , 18 , 19 , 20 .
Este protocolo descreve um método para fabricar um dispositivo de matriz de micropoços e fornece procedimentos experimentais detalhados que podem ser usados para funcionar.E poços na matriz e para cultivar bactérias, tanto como colônias de uma única espécie como em comunidades multi-membros. Este trabalho também demonstra como as bactérias modificadas para produzir proteínas repórter fluorescentes podem ser usadas para monitorar o crescimento bacteriano dentro dos poços ao longo do tempo. Uma matriz semelhante foi apresentada anteriormente e mostrou que é possível rastrear o crescimento de colônias de uma única espécie de Pseudomonas aeruginosa ( P. aeruginosa) em micropoços. Ao modular o tamanho do poço e densidade de semeadura, as condições iniciais de milhares de experiências de crescimento podem ser variadas em paralelo para determinar como as condições iniciais de inoculação afetam a capacidade das bactérias de crescer 21 . O trabalho atual usa uma versão ligeiramente modificada da matriz de micropoços que se baseia no trabalho anterior, permitindo a comparação simultânea de matrizes múltiplas e usando um protocolo experimental mais robusto. A matriz usada neste trabalho contém múltiplas subarquias, ou matriz ensemCom poços de diferentes tamanhos, que variam de 15 a 100 μm de diâmetro, que estão dispostos em três campos diferentes ( ou seja , 2x, 3x e 4x o diâmetro do poço). As matrizes são gravadas em silício e o crescimento das bactérias semeadas nas matrizes de silício é habilitado selando as matrizes com uma lamínula que foi revestida com um gel de agarose com infusão média. Os mutantes de P. aeruginosa projetados para estudar o sistema de secreção de Tipo VI são utilizados nesta demonstração.
Os resultados apresentados aqui se baseiam no objetivo final de analisar as comunidades de multimídia dentro dos arrays de micropoços, permitindo aos pesquisadores monitorar a abundância ea organização das bactérias in situ enquanto controlam e examinam o ambiente químico. Isso deve, em última análise, fornecer informações sobre as "regras" que regem o desenvolvimento e a sucessão da comunidade.
Protocolo
1. Silicon Microrowell-array Fabrication
- Revestimento de parileno
- Depósito entre 1-1,5 μm de parileno N em bolachas de silício usando um sistema de revestimento de parileno comercialmente disponível de acordo com as especificações e instruções do fabricante (configurações: ponto de ajuste do vaporizador = 160 ° C, ponto de ajuste do forno = 650 ° C).
NOTA: Aproximadamente 6 g de parileno N carregado em uma câmara produz reveses de 1-1,5 μm de espessura.
- Depósito entre 1-1,5 μm de parileno N em bolachas de silício usando um sistema de revestimento de parileno comercialmente disponível de acordo com as especificações e instruções do fabricante (configurações: ponto de ajuste do vaporizador = 160 ° C, ponto de ajuste do forno = 650 ° C).
- Fotolitografia
- Revestir as bolachas revestidas com parileno N com promotor de adesão, 20% de hexametildisilazano (HMDS) e 80% de acetato de éter monometílico de propileno glicol (PGMEA) (ver Tabela de Materiais ) a 3.000 rpm durante 45 s. Encha uma pipeta de transferência de 2 mL com o promotor de adesão e polvilhe-a sobre toda a bolacha. Permita que a bolacha se sente durante aproximadamente 10 s antes de girar para secar.
- Preencha uma pipeta de transferência de 2 mL comNe fotorresistência (veja a Tabela de Materiais ) e dispensar a fotorresistência no centro da bolacha. Girar a 3.000 rpm por 45 s para produzir um revestimento de resistência com aproximadamente 1,5 um de espessura.
- Cozinhe as amostras em uma placa quente a 115 ° C durante 1 min.
- Use um alinhador de contato e uma fotomáquica com o padrão de poço desejado para expor a amostra à luz ultravioleta. Exponha a bolacha revestida por centrifugação através da fotomáscara padronizada por 6 s, dando uma dose aproximada de 60-80 mJ / cm2 medida a 365 nm.
- Desenvolva o padrão submergindo a amostra no desenvolvedor (<3% de hidróxido de tetrametil amônio na água, veja a Tabela de Materiais ) durante 2 min. Enxaguar com água DI e secar com nitrogênio limpo e seco.
NOTA: As áreas de fotorresistentes expostas aos UV devem ser limpas durante o desenvolvimento.
- Gravação de iões reativa
- Use uma gravação de plasma de oxigênio para remover o parileno expostoTodo o caminho para o substrato de silício.
NOTA: A receita pode ser modulada para alterar a taxa de etch do parileno. Para espessuras de parileno entre 1 e 5 μm, use uma receita com 60 mTorr, 20 ° C, 100 sccm O 2 , 10 W RF e 2,000 W ICP em uma ferramenta Reactive Ion Etching (RIE). Após a gravação e remoção da camada de parileno exposta, a área padronizada ( ou seja, o silício exposto) deve parecer brilhante e prata. - Use um processo de gravação profunda RIE (DRIE, por exemplo, Bosch DRIE) para gravar no silício.
NOTA: A taxa e a duração do etch determinam a profundidade do poço. Um ciclo completo do processo Bosch (um passo de deposição de 3 s: 20 mTorr, 15 ° C, 140 sccm C 4 F 8 , 10 W RF e 1.750 W ICP seguido de um processo de gravação de 10 s: 20 mTorr, 15 ° C , 120 sccm SF 6 , 8 W RF e 1.750 W ICP) corresponde a aproximadamente 1 μm de profundidade de gravação. Os poços utilizados nesta demonstração variam de 3 a 3,5 μm de profundidade. - VErite a profundidade do ataque usando a profilometria física.
- Coloque a amostra em um perfilômetro físico (veja a Tabela de Materiais ).
- Ligue o vácuo de amostra e pressione o botão de carga manual.
- Focalize o sistema na amostra pressionando o botão "Foco". Posicione uma característica apropriada para a medição na tela de exibição.
- Digitalize a amostra. Nivele o perfil e mire a profundidade do recurso.
- Registre a taxa de gravura e module os tempos de gravação subsequentes para atingir a profundidade desejada.
NOTA: As medidas incluirão a profundidade do poço de silício, a espessura do parileno depositado e a espessura da fotorresistência. Verificando a espessura de cada camada ao longo do procedimento é necessário para obter uma profundidade de poço precisa.
- Use uma gravação de plasma de oxigênio para remover o parileno expostoTodo o caminho para o substrato de silício.
2. Cultura bacteriana e sementeira ( Figura 1a )
- Comece colônias em Luria Broth (LB) placas de ágar a partir de estoques de glicerol e use dentro de duas semanas. Escolha as colônias das estirpes desejadas das placas de agar LB e comece as culturas durante a noite de P. aeruginosa. Incubar as culturas durante a noite durante aproximadamente 18 h a 37 ° C enquanto se agita a 220 rpm em meio R2A.
NOTA: As colônias devem ser colhidas dentro de duas semanas de revestimento para garantir que as mutações e os genes repórteres fluorescentes sejam mantidos. Todo o trabalho de P. aeruginosa deve ser feito nas condições de BSL-2. - Use um escriba de diamante para separar a bolacha de silício em pastilhas individuais que contenham conjuntos de diferentes tamanhos e arrays de pitch-well. Certifique-se de que cada chip contém o conjunto completo de tamanhos de poços e lançamentos para o estudo.

Figura 1: Procedimento de fabricação e semente celular. (A) Microwell arrays aSão gravados em bolachas de silício revestidas com uma fina camada de parileno (i). Para molhar os poços e / ou funcionalizar a superfície, uma solução de proteína é adicionada em uma gota sobre as matrizes (ii). A solução de proteína é removida, as bolachas são secas, e uma nova solução contendo as bactérias desejadas é adicionada (iii). A solução bacteriana é removida após um período de incubação, e as bolachas são deixadas a secar, deixando para trás bactérias nos poços e na superfície (iv). As bactérias associadas à superfície são removidas com destilação de parileno, deixando para trás bactérias semeadas limpas nos micropoços e ainda viáveis devido ao meio de glicerol a 2%, o que ajuda a manter os poços hidratados (v). As microplaquetas de silício são então colocadas em um lado de matriz em uma camada de vidro revestida com gel de agarose, que alimenta o crescimento bacteriano nos micropoços (vi). ( B ) Layout de sub-arrays em um único dispositivo de silício. Cada sub-matriz contém um conjunto de poços idênticos. O diâmetro dos micropoços em todos os sub-aAs bandejas variam em diâmetro de 5-100 μm e são organizadas em 2x, 3x ou 4x, o passo do diâmetro do poço, que é denotado pelas cores de branco a cinza escuro no esquema do painel inferior. Quando as profundidades dos poços são superficiais (<10 μm), os diâmetros de 5 e 10 μm são raramente úteis, geralmente devido à falta de células que colonizam esses poços muito pequenos. Neste trabalho, apenas os dados de poços com diâmetros de 15 a 100 μm foram analisados. Clique aqui para ver uma versão maior desta figura.
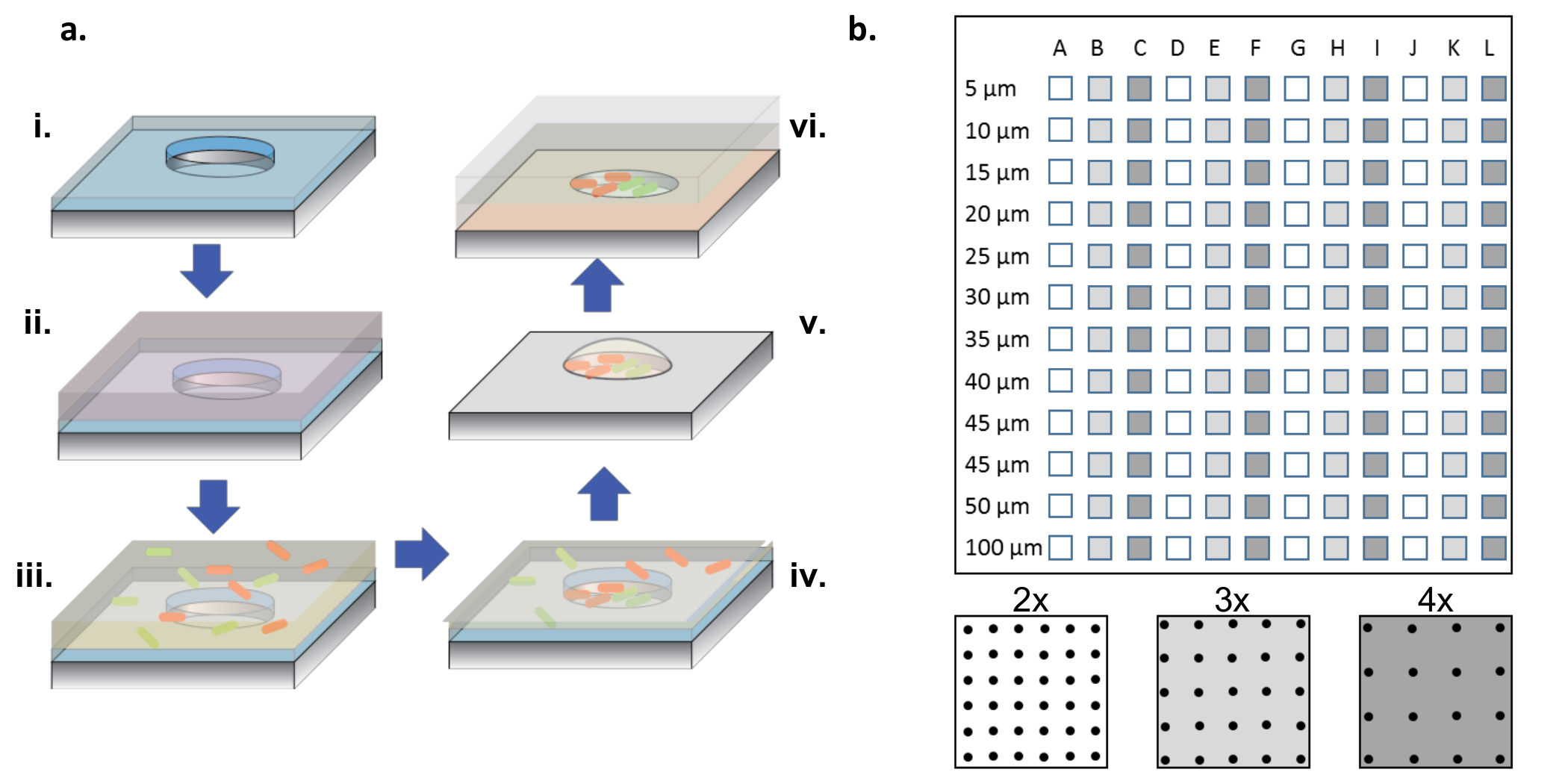{kind=link}
NOTA: Como mostrado na Figura 1b , um chip completo contém sub-arrays de poços, com diâmetros que variam de 5 a 100 μm, com três passos diferentes ( ou seja , 2x, 3x e 4x o diâmetro) repetindo 4 vezes.
- Coloque uma gota de 150 μL de albumina de soro bovino (BSA) de 500 μg / mL em solução de PBSEm cima da matriz para molhar os micropoços. Incubar a solução de BSA por 1 h no chip na RT em uma câmara úmida.
- Crie a câmara preenchendo o fundo de uma caixa de ponta de pipeta vazia com salina tamponada com fosfato (PBS).
NOTA: Outras substâncias, tais como lectinas específicas, podem ser utilizadas em lugar de BSA para funcionarizar a superfície dos micropoços.
- Crie a câmara preenchendo o fundo de uma caixa de ponta de pipeta vazia com salina tamponada com fosfato (PBS).
- Ao incubar chips de silício com solução de BSA, centrifugar as culturas a 2.500 rpm (correspondente a uma média de 950 xg) durante 5 min e depois ressuspendê-las em 500 μL de meio R2A fresco com 2% de glicerol.
- Determine a OD da cultura usando um espectrômetro UV-vis a 600 nm. Ajuste-o para uma DO de 0,02 usando 2% de meio de glicerol R2A.
NOTA: O glicerol ajuda a evitar que os poços se sectem durante a retirada de parileno.
- Determine a OD da cultura usando um espectrômetro UV-vis a 600 nm. Ajuste-o para uma DO de 0,02 usando 2% de meio de glicerol R2A.
- Após a incubação, remover a solução de BSA e enxaguar 3x com PBS removendo e substituindo a gota de líquidoT cobrindo a matriz de micropoços de silício. Secar sob nitrogênio.
- Adicione 150 μL de culturas de 0,02 OD a cada um dos arrays secos colocados em uma câmara úmida. Incube durante 1 h a 4 ° C para permitir que as bactérias adiram às paredes do poço.
NOTA: A refrigeração não é necessária para a incubação. O tempo de incubação de 4 ° C pode ser usado para prevenir o crescimento de bactérias antes do início da imagem, para que se possa visualizar a organização espacial das comunidades antes do crescimento. A incubação à temperatura ambiente também pode ser utilizada. Ambos os protocolos resultam em curvas de crescimento semelhantes.
3. Configuração do microscópio
- Antes de iniciar a incubação bacteriana nas pastilhas de silício, ligue a câmara de controle ambiental do estágio (veja a tabela de materiais) e ajuste as configurações na caixa de controle para que a umidade (~ 100%) e a temperatura (30-32 ° C, ver passo 3.2) podem equilibrar-se antes da adição de amostras.
- Nivele o suporte da amostra e alinhe oTerior em torno da amostra com toalhetes de laboratório embebidos em PBS (veja a Tabela de Materiais ) para aumentar a umidade na câmara para o ponto de condensação . Ajuste a temperatura da câmara para 30 ° C e a da tampa da câmara para 32 ° C para reduzir a condensação no plano de imagem.
NOTA: O suporte de deslizamento se encaixa na câmara de células vivas com uma junta de aproximadamente 1 cm de espessura. O suporte da amostra é nivelado com a ajuda de um nível de bolha que é colocado no topo do suporte da amostra. O suporte da amostra pode ser inclinado ligeiramente e permanecer selado na junta até o nível. - Enquanto as culturas estão incubando nos chips de silício, ligue manualmente o interruptor de energia para a lâmpada de mercúrio pelo menos 30 minutos antes da imagem. Ligue manualmente a câmera e o estágio do microscópio automatizado. Abra o software usado para controlar o microscópio e equipamentos periféricos e garantir que o equipamento seja reconhecido pelo software.
NOTA: A ampliação é 10X com NA = 0.3.
- Soluções de agarose previamente preparadas com microondas ( isto é, 2% de agarose no meio R2A) até atingir um estado líquido, aproximadamente 60 s.
- Molhe a parte de trás de uma lamínula de vidro de 75 mm x 22 mm, nº 1.5 com etanol e colocá-la longitudinalmente, centrada, através de uma corrediça de vidro de 2 x 3 "(50 x 75 mm). Coloque dois espaçadores PDMS (espessura de ~ 1 mm) Ao longo das bordas longas da lamínula e deslize a lamela de vidro de modo que aproximadamente 1 mm da lamínula sobreponha a borda do slide.
- Despeje 5 mL de solução líquida de agarose no topo da lamínula de vidro, apenas o suficiente para cobri-la completamente, e coloque uma segunda corrediça de vidro de 2 x 3 "em cima da montagem para" encaixar "o agar líquido entre a lamínula e a corrediça.
NOTA: Isso controla a profundidade da agarose, fazendo com que a espessura total da lamínula e agarose endurecida seja igual em espessura aos espaçadores PDMS. - Permitir que o sanduíche de deslizamento de vidro e gel de vidro de agarose de vidro para definir até a solução de agarose começar a solidificar; Então, transfira para uma geladeira. Após 15 min, retire o excesso de agarose sólida e corte ao redor da lamínula de vidro. Mova isto para um prato limpo e coloque-o na geladeira até o uso.
5. Selar os poços com uma capa de cobertura revestida de agarose e imagem
- Após o período de incubação das bactérias estar completo, remova a lamínula revestida com agarose da geladeira e prepare as pastilhas de silício, como se segue.
- Mergulhe as pastilhas de silício em água ultrapura, uma por vez, por 10 s cada. Coloque-os em suas bordas em um limpa ou tecido de laboratório até que a maior parte do excesso de líquido tenha escorrido das bordas dos chips.
- Corte um pedaço de fita para combinar o comprimento da borda de cada chip de silício. Coloque a fita no parileno que cobre o silício e use-o para remover rapidamente o revestimento de parileno.
- ImmedInvente imediatamente cada pastilha descascada e coloque cada chip de tal modo que o lado da matriz de micropoços se enfrente (e faça contato) com o lado revestido com agarose de um lamíngulo revestido com agarose. Tenha cuidado para não mover ou deslocar o chip depois que ele toca a agarose para evitar o crescimento de bactérias fora dos poços.
- Coloque o conjunto de micropoços montado / lâmina de agarose no suporte deslizante da câmara de controle ambiental do palco no miscroscópio.
- Use luz ambiente ou luz dirigida ( por exemplo, uma lanterna) para localizar matrizes de interesse. Use o software comercial que controla o estágio automatizado para salvar essas posições (veja a tabela de materiais). Desligue a luz ambiente ou dirigida depois de as posições serem armazenadas.
- No software comercial, abra o painel "ND Acquisition".
NOTA: Este painel inclui um menu para gravação automática em um diretório específico, bem como aquisição de imagem programável. Para esses experimentosOs menus "Time", "XY," e "λ" são usados. - Para salvar as localizações no software, clique no "menu XY" e, em seguida, marque uma caixa vazia no lado esquerdo para cada posição que precisa ser salva. Além disso, clique no botão "Incluir Z".
- Adquira imagens ao longo do tempo aos comprimentos de onda desejados e 10 ampliação usando os cubos de filtro de fluorescência apropriados (veja a Tabela de Materiais).
- Use o software de controle e as posições de matriz salvas para mover para cada local salvo e concentrar-se nos poços. Clique em cada local XY na lista salva e ajuste o foco usando o filtro Green Flourescence Protein (GFP). Salve a nova posição z clicando na seta apontando para o local z.
NOTA: Este processo pode demorar muito tempo. Considere tomar a precaução de aumentar o ganho e usar o filtro de densidade neutra para reduzir a intensidade da luz para evitar a fotovelagem. - Determine a distância do eixo z entre os planos focais para cada comprimento de onda, observando a diferença na posição do eixo z quando focada na superfície da matriz. Escolha 2-3 locais da matriz com a população de bactérias vermelhas / verdes misturadas e foco usando o filtro Red Fluorescence Protein (RFP).
- Subtrair a distância entre os planos focais usando os filtros de fluorescência GFP e RFP e adicionar o ajuste do plano focal no menu "λ".
NOTA: Por exemplo, se a matriz aparecer focada no canal GFP em uma localização z de 50 μm, e a mesma matriz aparece focada no canal RFP a 55 μm, adicione +5 ao lado da configuração ótica RFP na seção "λ " cardápio.
- Subtrair a distância entre os planos focais usando os filtros de fluorescência GFP e RFP e adicionar o ajuste do plano focal no menu "λ".
- Comece a aquisição de imagens em lapso de tempo.
NOTA: Para os experimentos mostrados aqui, as imagens de RFP e GFP foram adquiridas para cada posição de matriz em intervalos de 30 min usando aquisição de imagem multidimensional através de um software comercial que controlaA câmera, o obturador, a roda do filtro e o estágio motorizado.- Defina o "intervalo" para 30 min e a "duração da experiência" para 24 h no menu "Hora". Clique em "Executar agora".
NOTA: Com as caixas "Tempo", "XY" e "λ" marcadas, o programa executará o palco para a imagem de cada local ( ou seja, as localizações XYZ salvas), tirar uma imagem em um comprimento de onda, mover a posição z Para explicar as diferenças do plano focal ( ou seja, o controle de lambda ou o comprimento de onda), pegue a segunda imagem, avance para a próxima localização da matriz (multiponto) e faça um loop a intervalos de 30 minutos (lapso de tempo).
- Defina o "intervalo" para 30 min e a "duração da experiência" para 24 h no menu "Hora". Clique em "Executar agora".
- Use o software de controle e as posições de matriz salvas para mover para cada local salvo e concentrar-se nos poços. Clique em cada local XY na lista salva e ajuste o foco usando o filtro Green Flourescence Protein (GFP). Salve a nova posição z clicando na seta apontando para o local z.
- Adquira imagens de controle de iluminação.
NOTA: use os menus "ND Acquisition", "Time" e "XY" para tirar imagens de 4 locais, 25x cada.- Pegue uma série de 100 imagens de "darkfield" desligando todas as fontes de luz eUma "imagem" de um slide padrão. Essas imagens captarão o ruído da câmera. Use o tempo de exposição mais longo usado durante o timelapse (etapa 5.3.3).
- Pegue uma série de 100 imagens de "campo de iluminação" ao imaginar um slide padrão ( ou seja, uma intensidade RFP ou GFP uniforme) em alguns locais diferentes para capturar a iluminação irregular nas condições experimentais fornecidas. Escolha um tempo de exposição que maximize o sinal sem atingir a saturação.
6. Análise
- Processe as pilhas de imagens usando um software de análise de imagem ( por exemplo, ImageJ).
- Converta as imagens adquiridas em formato de arquivo tiff usando o software comercial. Carregue imagens no software de análise de imagem clicando em "Arquivo"> "Importar"> "Sequência de imagens".
- Crie uma "imagem de correção" com a média de todas as imagens de "campo escuro" e "campo de iluminação". SubtracT a imagem média "darkfield" da imagem média de "campo de iluminação" escolhendo "Processo"> "Calculadora de imagem". Selecione as duas imagens, "Image1" e "Image2", e depois "Subtrair" no campo "Operação". Clique em "OK".
- Para calcular a média, carregue as imagens de correção (ou campo escuro), clique em "Imagem"> "Pilhas"> "Projeto Z"> "Projeção média".
- Execute o registro de imagem, se necessário. Em seguida, execute a subtração de fundo clicando em "Processar"> "Subtrair fundo". Digite um raio ( por exemplo, 125) no campo "raio" e selecione "paraboloide deslizante".
- Execute a correção de iluminação usando "Processo"> "Calculadora Plus". Escolha os seguintes parâmetros: operação, divisão; I1, bem imagem; I2, imagem de correção; K1, imagem de correção significa; E k2, 0. Clique em "CreComeu Nova Janela ".
NOTA: Este conjunto de dados não exigiu registro, mas em outros trabalhos, o ImageJ Plugin StackReg foi usado com a transformação "Tradução". Para a subtração de fundo, use o mesmo raio de paraboloide deslizante para cada conjunto de imagens. Por exemplo, se os maiores poços criados tiverem um raio de pixel de 100, use um raio maior que 100 ( por exemplo, 125) para cada conjunto de imagens.
- Determine o crescimento de cada estirpe nos micropoços.
- Selecione regiões de interesse (ROIs) em torno de cada micropoente nas matrizes desejadas usando o plugin ImageJ "MicroArray".
- No menu "MAP", clique em "Redefinir grade". Especifique linhas, colunas e diâmetro (com base no tamanho e número do poço na matriz, veja a Figura 1b ). Selecione "círculo" no menu "ROI shape".
- Segure a tecla "Alt" enquanto seleciona o ROI superior esquerdo com o mouse para mover tEle ROI array. Segure a tecla "shift" enquanto seleciona o ROI inferior esquerdo para alterar o tamanho da matriz. Mantenha a tecla "shift" enquanto seleciona um ROI do lado direito da matriz, mas não nos cantos, para alterar o espaçamento dos ROIs.
- Use os comandos acima para caber a matriz ROI sobre os poços em uma imagem. Clique em "Medir RT".
NOTA: O plugin irá exportar as medidas desejadas de cada ROI. Use três tamanhos ROI, criando anéis concêntricos ao redor dos poços para coletar local o sinal de fundo ( ou seja, o sinal do anel médio subtraído do anel externo) e medidas de fluorescência ( ou seja, o sinal do anel interno).
- Colecione os dados em um software de planilha eletrônica e calcule o sinal de fundo. Importá-lo para um software de script personalizado para análise posterior.
- Selecione regiões de interesse (ROIs) em torno de cada micropoente nas matrizes desejadas usando o plugin ImageJ "MicroArray".
- Organização e análise de dados
- Importe os dados e organize os dadosColetados em ImageJ em uma matriz na seguinte ordem para todos os tempos: coluna 1, número de sub-matriz; Coluna 2, fileira de poços; Coluna 3, coluna do poço; Coluna 4, intensidade média; Coluna 5, intensidade de fundo; E coluna 6, intensidade média - intensidade de fundo.
- Separe os resultados da aquisição mCherry e GFP em diferentes matrizes. Armazene os resultados de cada sub-matriz e cada cor em uma célula diferente em uma matriz de células.
NOTA: Esta organização facilita a movimentação entre dados de imagem e resultados de medição, limpar os dados e garantir que as medições representem com precisão os dados.
- Separe os resultados da aquisição mCherry e GFP em diferentes matrizes. Armazene os resultados de cada sub-matriz e cada cor em uma célula diferente em uma matriz de células.
- Ajuste para a autofluorescência de P. aeruginosa .
NOTA: Em experimentos envolvendo a co-cultura das cepas GFP e mCherry, um chip apenas de mCherry deve ser analisado para determinar a relação entre mCherry e autofluorescência verde.- Trace o sinal mCherry versus GFP de todos os mCherry ΔretS &# 916; tse / i1-6 wells em todos os pontos de tempo para determinar a relação entre o sinal mCherry e a autofluorescência no canal GFP. Subtrair o sinal de autofluorescência das co-culturas.
- Trace as trajetórias e ajuste uma equação logística modificada para cada trajetória para extrair parâmetros usando o ajuste de mínimos quadrados em um software de planilha ou um software de script personalizado.
- Procure as correlações entre e entre os parâmetros da trajetória GFP e mCherry.
- Importe os dados e organize os dadosColetados em ImageJ em uma matriz na seguinte ordem para todos os tempos: coluna 1, número de sub-matriz; Coluna 2, fileira de poços; Coluna 3, coluna do poço; Coluna 4, intensidade média; Coluna 5, intensidade de fundo; E coluna 6, intensidade média - intensidade de fundo.
Resultados
A plataforma experimental apresentada aqui é projetada para estudos de alto teor de alto teor de conteúdo de comunidades bacterianas. O design permite que milhares de comunidades, crescendo em poços de vários tamanhos, sejam analisados simultaneamente. Com este projeto de matriz de micropoços, pode-se determinar a dependência da composição final da comunidade nas densidades iniciais de semeadura, tamanho do poço e ambiente químico. Este trabalho demonstra o crescimento de...
Discussão
Este artigo apresentou um dispositivo de matriz de micropoços e protocolos experimentais projetados para permitir a análise baseada em imagens de células vivas de alto conteúdo e alto conteúdo de desenvolvimento de comunidades bacterianas. Enquanto o foco da demonstração aqui era estudar os efeitos da secreção de tipo VI mediada pelo contato no desenvolvimento da comunidade, os arrays foram projetados para ser flexíveis e acomodar o estudo de uma ampla gama de comunidades microbianas e interações de micróbi...
Divulgações
Os autores não têm nada a revelar.
Agradecimentos
As matrizes de microwell foram fabricadas e caracterizadas no Centro de Divisão de Instalações do Usuário das Ciências dos Materiais de Nanophase, Escritório de Ciências Básicas da Energia, Departamento de Energia dos EUA. O apoio financeiro para este trabalho foi fornecido através do Fundo de Pesquisa e Desenvolvimento do Diretor de Laboratório Nacional Oak Ridge. Os autores também agradecem ao Laboratório J. Mougous (Universidade de Washington, Seattle, WA) para o fornecimento de cepas de P. aeruginosa usadas nesses estudos.
Materiais
| Name | Company | Catalog Number | Comments |
| Parylene N | Specialty Coating Systems | CAS NO.:1633-22-3 | |
| Parylene coater | Specialty Coating Systems | Labcoter 2 Parylene Deposition Unit PDS2010 | |
| Silicon Wafer | WRS Materials | 100mm diameter, 500-550μm thickness, Prime, 10-20 resistivity, N/Phos<100>, | |
| adhesion promoter | Shin-Etsu Microsci | MicroPrime P20 adhesion promoter | |
| postive tone photoresist | Rohm and Haas Electronics Materials LLC (Owned by Dow) | Microposit S1818 Positive Photoresist (code 10018357) | |
| Quintel Contact Aligner | Neutronix Quintel Corp | NXQ 7500 Mask Aligner | |
| Reactive Ion Etching Tool | Oxford Instruments | Plasmalab System 100 Reactive Ion Etcher | |
| R2A Broth | TEKnova | R0005 | |
| Bovine Serum Albumin | Sigma | A9647 | |
| Multimode Plate Reader | Perkin Elmer | Enspire, 2300-0000 | |
| Fluorescent Microscope | Nikon | Eclipse Ti-U | |
| Automated Stage | Prior | ProScan III | |
| CCD camera | Nikon | DS-QiMc | |
| Stage-top environmental control chamber | In Vivo Scientific | STEV ECU-HOC | |
| Phosphate Buffered Saline | ThermoFisher Scientific | 14190144 | |
| UltraPure Agarose | ThermoFisher Scientific | 16500500 | |
| 25 x 75 mm No. 1.5 coverslip | Nexterion | High performance #1.5H coverslips | |
| Fluorescence Reference Slides | Ted Pella | 2273 | |
| Physical Stylus Profilometer | KLA Tencor | P-6 | |
| lab wipes | Kimberly Clark | Kimipe KIMTECH SCIENCE Brand, 34155 | |
| commercial software | Nikon | NIS Elements | |
| Zeiss 710 Confocal Microscope | Zeiss | ||
| filter cubes | Nikon | Nikon FITC (96311), Nikon Texas Red(96313) |
Referências
- Zhou, J., Deng, Y., et al. Stochasticity, succession, and environmental perturbations in a fluidic ecosystem. Proc Natl Acad Sci. 111, E836-E845 (2014).
- Valm, A. M., Welch, J. L. M., et al. Systems-level analysis of microbial community organization through combinatorial labeling and spectral imaging. Proc Natl Acad Sci USA. 108 (10), 4152-4157 (2011).
- Satoh, H., Miura, Y., Tsushima, I., Okabe, S. Layered structure of bacterial and archaeal communities and their in situ activities in anaerobic granules. Appl Environ Microbiol. 73 (22), 7300-7307 (2007).
- Kim, H. J., Boedicker, J. Q., Choi, J. W., Ismagilov, R. F. Defined spatial structure stabilizes a synthetic multispecies bacterial community. Proc Natl Acad Sci USA. 105 (47), 18188-18193 (2008).
- Nunan, N., Wu, K., Young, I. M., Crawford, J. W., Ritz, K. Spatial distribution of bacterial communities and their relationships with the micro-architecture of soil. FEMS Microbiol Ecol. 44, 203-215 (2003).
- Grundmann, G. L. Spatial scales of soil bacterial diversity - The size of a clone. FEMS Microbiol Ecol. 48, 119-127 (2004).
- Langenheder, S., Lindstrom, E. S., Tranvik, L. J. Structure and Function of Bacterial Communities Emerging from Different Sources under Identical Conditions. Appl Environ Microbiol. 72 (1), 212-220 (2006).
- Camp, J. G., Kanther, M., Semova, I., Rawls, J. F. Patterns and Scales in Gastrointestinal Microbial Ecology. Gastroenterology. 136 (6), 1989-2002 (2009).
- Renner, L. D., Weibel, D. B. Physicochemical regulation of biofilm formation. MRS Bull. 36 (5), 347-355 (2011).
- Wessel, A. K., Hmelo, L., Parsek, M. R., Whiteley, M. Going local: technologies for exploring bacterial microenvironments. Nat Rev Microbiol. 11 (5), 337-348 (2013).
- Stacy, A., McNally, L., Darch, S. E., Brown, S. P., Whiteley, M. The biogeography of polymicrobial infection. Nat Rev Microbiol. 14 (2), 93-105 (2015).
- Hansen, R. R., Shubert, K. R., Morrell-Falvey, J. L., Lokitz, B. S., Doktycz, M. J., Retterer, S. T. Microstructured block copolymer surfaces for control of microbe adhesion and aggregation. Biosensors. 4 (1), 63-75 (2014).
- Hansen, R. R., Hinestrosa, J. P., et al. Lectin-functionalized poly(glycidyl methacrylate)- block -poly(vinyldimethyl azlactone) surface scaffolds for high avidity microbial capture. Biomacromolecules. 14 (10), 3742-3748 (2013).
- Timm, C. M., Hansen, R. R., Doktycz, M. J., Retterer, S. T., Pelletier, D. A. Microstencils to generate defined, multi-species patterns of bacteria. Biomicrofluidics. 9 (6), (2015).
- Keymer, J. E., Galajda, P., Muldoon, C., Park, S., Austin, R. H. Bacterial metapopulations in nanofabricated landscapes. Proc Natl Acad Sci USA. 103 (46), 17290-17295 (2006).
- Zhang, Q., Lambert, G., et al. Acceleration of Emergence of Bacterial Antibiotic Resistance in Connected Microenvironments. Science. 333 (6050), 1764-1767 (2011).
- Friedlander, R. S., Vlamakis, H., Kim, P., Khan, M., Kolter, R., Aizenberg, J. Bacterial flagella explore microscale hummocks and hollows to increase adhesion. Proc Natl Acad Sci USA. 110 (14), 5624-5629 (2013).
- Zhou, J., Liu, W., et al. Stochastic Assembly Leads to Alternative Communities with Distinct Functions in a Bioreactor Microbial Community. MBio. 4 (2), 1-8 (2013).
- van Vliet, S., Hol, F. J., Weenink, T., Galajda, P., Keymer, J. E. The effects of chemical interactions and culture history on the colonization of structured habitats by competing bacterial populations. BMC Microbiol. 14 (1), 116 (2014).
- Niepa, T. H. R., Hou, L., et al. Microbial Nanoculture as an Artificial Microniche. Sci Rep. 6, 30578 (2016).
- Hansen, R. H., Timm, A. C., et al. Stochastic Assembly of Bacteria in Microwell Arrays Reveals the Importance of Confinement in Community Development. PLoS ONE. 11 (5), e0155080 (2016).
- Hood, R. D., Singh, P., et al. A Type VI Secretion System of Pseudomonas aeruginosa Targets a Toxin to Bacteria. Cell Host Microbe. 7 (1), 25-37 (2010).
- LeRoux, M., Ja De Leon, ., et al. Quantitative single-cell characterization of bacterial interactions reveals type VI secretion is a double-edged sword. Proc Natl Acad Sci. 109 (48), 19804-19809 (2012).
- Whitney, J. C., Beck, C. M., et al. Genetically distinct pathways guide effector export through the type VI secretion system. Mol Microbiol. 92 (3), 529-542 (2014).
- Warrick, J. W., Timm, A., Swick, A., Yin, J. Tools for Single-Cell Kinetic Analysis of Virus-Host Interactions. PLoS ONE. 11 (1), e0145081 (2016).
- Zwietering, M. H., Jongenburger, I., Rombouts, F. M., Van't Riet, K. Modeling of the Bacterial Growth Curve. Appl Environ Microbiol. 56 (6), 1875-1881 (1990).
- Halsted, M., Wilmoth, J. L., et al. Development of transparent microwell arrays for optical monitoring and dissection of microbial communities. J Vac Sci Technol B Nanotechnol Microelectron. 34 (6), 06KI03 (2016).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados