
このコンテンツを視聴するには、JoVE 購読が必要です。 サインイン又は無料トライアルを申し込む。
Method Article
マイクロウェルアレイプラットフォーム内での微生物コミュニティ開発の組み立てと追跡
要約
微生物群集の発達は、環境構造、メンバーの豊富さ、形質および相互作用を含む複数の要因の組み合わせに依存する。このプロトコルは、ニッチサイズや閉じ込めなどの重要な要素を近似できるフェムトリットルウェルに含まれる数千のコミュニティを同時に追跡するための合成の微細加工環境を記述しています。
要約
微生物群集の発達は、コミュニティメンバーの空間分布および活動を劇的に変えることができる複雑な決定論的および確率論的因子の組み合わせに依存する。我々は、数千の細菌群を並行して迅速にアセンブルおよび追跡するために使用できるマイクロウェルアレイプラットフォームを開発しました。このプロトコルは、プラットフォームの有用性を強調し、プラットフォーム内の配列のアンサンブル内の単純な2メンバーのコミュニティの開発を光学的に監視するための使用方法を説明します。このデモでは、 緑膿菌の 2つの突然変異体を使用しています。これは、タイプVIの分泌病原性を研究するために開発された一連の突然変異体の一部です。 mCherryまたはGFP遺伝子のいずれかの染色体挿入物は、各マイクロウェル内のコミュニティメンバーの存在および位置をモニターするために使用され得る異なる発光波長を有する蛍光タンパク質の構成的発現を促進する。このプロトコルは、アレイのウェルに細菌の混合物を組み立て、時間経過に伴う各メンバー集団の相対的成長を測定するための経時的蛍光イメージングおよび定量的イメージ分析を使用する。マイクロウェルプラットフォームの播種およびアセンブリ、アレイ内の微生物群集の定量分析に必要なイメージング手順、およびすべて説明した微生物種間の相互作用を明らかにするために使用できる方法。
概要
微生物群は、環境の構造や細胞死、分裂、タンパク質濃度、オルガネラ数、突然変異1に関連する確率論的プロセスのような決定論的要因の両方によって形成される。自然環境の中で、これらの影響がコミュニティの構成と活動に与える個々の影響を解析することは、ほとんど不可能です。自然の構造によって隠され、化学的および生物学的環境に埋もれて、コミュニティのメンバーを特定し、自然環境内で時空間分布をさらに解決することは非常に困難です。それにもかかわらず、最近の努力は、地域の機能に関する空間的組織の重要性を強調し、進行中の研究2,3,4でメンバーの豊富さと組織の両方を考慮する必要性を指摘している。
それ( 例えば、土壌構造、植物の根、海洋粒子、または腸の微絨毛)、酸素の存在または不存在、およびその存在が、病原性種はすべて、微生物群集の組成、構造、および機能に影響を及ぼす5,6,7,8,9,10,11。それにもかかわらず、これらの要因を捕らえることを怠った伝統的な文化の技法が引き続き普及している。コミュニティ構成( 例えば、共依存種の存在)、物理的付着、シグナル伝達分子濃度、および直接的細胞 - 細胞接触は、微生物共同体を形成するためのすべての重要な因子であり、c予防的培養条件。これらの特性は、バルク液体培養または寒天プレート上で複製することは困難である。しかし、自然環境の主要な物理的および化学的特徴の複製を可能にするマイクロ流体、マイクロパターン形成、およびナノ製造技術の可能性は、多くの研究者が相互作用を研究する細菌群集を構築することを可能にした12,13,14。自然条件4,15,16,17,18,19,20を模倣する。
このプロトコールは、マイクロウェルアレイデバイスを作製する方法を記載し、そして、機能的にするために使用され得る詳細な実験手順を提供する単一ウェルのウェル内で細菌を増殖させることができます。この研究はまた、蛍光レポータータンパク質を産生するように改変された細菌が、時間の経過とともにウェル内の細菌増殖をモニターするためにどのように使用され得るかを実証する。同様のアレイを以前に提示し、 マイクロウェル中の緑膿菌( P.緑膿菌)の単一種コロニーの成長を追跡することが可能であることを示した。ウェルサイズと播種密度を調整することにより、何千もの増殖実験の開始条件を並行して変化させることができ、初期接種条件がバクテリアの増殖能力にどのように影響するかを決定することができる21 。現在の研究では、複数のアレイの同時比較を可能にし、より堅牢な実験プロトコールを使用することにより、前の研究に基づいて構築されたわずかに改変されたマイクロウェルアレイのバージョンを使用する。この作品で使用されているアレイには、複数のサブアレイ、または配列ensem3つの異なるピッチ( すなわち 、ウェル直径の2倍、3倍、および4倍)で配置された、直径15〜100μmの範囲の異なるサイズのウェルを含む。アレイはシリコン内にエッチングされ、シリコン注入アレイに封入された細菌の増殖は、培地注入アガロースゲルで被覆されたカバースリップでアレイを密封することによって可能になる。タイプ6分泌系を研究するために設計されたP.aeruginosa変異体がこのデモンストレーションで使用される。
ここで提示された結果は、マイクロウェルアレイ内のマルチメンバーコミュニティを分析し、化学環境の制御と探索を行っている間、研究者がバイオウムの豊富さと組織をインサイチュで監視できる究極の目標に向かっています。これは、最終的に、コミュニティの発展と継承を支配する「ルール」についての洞察を提供するはずです。
プロトコル
1.シリコンマイクロウェルアレイ製造
- パリレンコーティング
- 市販のパリレンコーティングシステムを製造者の仕様および指示(設定:気化器設定点= 160℃;ファーネス設定点= 650℃)に従ってシリコンウェハ上に1-1.5μmのパリレンNの間に堆積させる。
注:約6gのパリレンNをチャンバーに装填すると、厚さ1〜1.5μmのコーティングが得られる。
- 市販のパリレンコーティングシステムを製造者の仕様および指示(設定:気化器設定点= 160℃;ファーネス設定点= 650℃)に従ってシリコンウェハ上に1-1.5μmのパリレンNの間に堆積させる。
- フォトリソグラフィー
- 接着促進剤、20%ヘキサメチルジシラザン(HMDS)、および80%プロピレングリコールモノメチルエーテルアセテート(PGMEA)( 表の表を参照)でパリレンNコートウェーハを3000rpmで45秒間スピンコートする。接着促進剤を含む2mLの移送ピペットを満たし、それをウェーハ全体に振りかける。ウェーハを乾燥させる前に約10秒間置いてください。
- 2 mLのトランスファーピペットに陽性( 表の表を参照 )、ウェーハの中心にフォトレジストを分配します。 3000rpmで45秒間回転させて、厚さ約1.5μmのレジストコーティングを得る。
- 115°Cでホットプレート上で1分間サンプルをソフトベークする。
- コンタクト・アライナーとフォトマスクを目的のウェル・パターンで使用して、サンプルを紫外線に曝す。パターン化されたフォトマスクを通してスピンコートされたウェハを6秒間露出させ、365nmで測定した約60〜80mJ / cm 2の線量を与える。
- パターンを現像液に浸して(2%未満の水酸化テトラメチルアンモニウム水酸化物;材料の表を参照 )、2分間現像する 。 DI水ですすぎ、清潔で乾燥した窒素で乾燥する。
注記:UVにさらされたフォトレジストの領域は現像中に消去する必要があります。
- 反応性イオンエッチング
- 露出したパリレンを除去するために酸素プラズマエッチングを使用するシリコン基板に至るまで。
注:パリレンのエッチング速度を変更するためにレシピを調整することができます。パリレン厚さが1〜5μmの場合は、反応性イオンエッチング(RIE)ツールで60 mTorr、20℃、100 sccm O 2、10 W RF、2,000 W ICPのレシピを使用します。露出したパリレン層をエッチングして除去した後、パターニングされた領域( すなわち 、露出したシリコン)は光沢を帯びて銀色に見えるべきである。 - 深いRIE(DRIE; 例えば、 Bosch DRIE)エッチングプロセスを用いてシリコンにエッチングする。
注:エッチング速度と継続時間によってウェルの深さが決まります。 Boschプロセスの1サイクル(3秒の堆積ステップ:20mTorr、15℃、140sccmのC 4 F 8、10WのRF、および1750WのICP、続いて10sのエッチングプロセス:20mTorr、15℃ 、120sccm SF 6,8W RF、および1,750W ICP)は、約1μmのエッチング深さに相当する。このデモンストレーションに使用される井戸は、3〜3.5μmの深さの範囲です。 - V物理的プロフィルメトリーを用いてエッチング深さをエージングする。
- サンプルを物理的なプロフィルメーターにロードします(材料表を参照 )。
- サンプルの真空を入れ、手動ロードボタンを押します。
- "Focus"ボタンを押してシステムをサンプルにフォーカスします。ビュー画面に測定のための適切な機能を配置します。
- サンプルをスキャンします。プロファイルを水平にし、フィーチャの深さを測定します。
- エッチング速度を記録し、その後のエッチング時間を調節して所望の深さを達成する。
注記:測定には、シリコンウェルの深さ、堆積されたパリレンの厚さ、およびフォトレジストの厚さが含まれる。正確な井戸深さを達成するためには、手順全体を通して各層の厚さを確認する必要があります。
- 露出したパリレンを除去するために酸素プラズマエッチングを使用するシリコン基板に至るまで。
細菌培養および播種( 図1a )
- Luriaブロス(LB)グリセロールストックからの寒天プレートおよび2週間以内に使用する。 LB寒天プレートから所望の株のコロニーを選び、P.aeruginosaの一晩培養を開始する。 R2A培地で220 rpmで振盪しながら、一晩培養物を37℃で約18時間インキュベートする。
注:コロニーは、突然変異および蛍光レポーター遺伝子が確実に保持されるように、プレーティングの2週間以内に選別する必要があります。すべてのP.緑膿菌の作業は、BSL-2条件下で行う必要があります。 - ダイアモンドスクライブを使用して、シリコンウエハを異なるサイズのアンサンブルとピッチウェル配列を含む個々のチップに分割します。各チップに研究のためのウェルサイズとピッチの完全な補完が含まれていることを確認します。

図1:作製および細胞シード形成手順。 ( a )マイクロウェルアレイaパリレン(i)の薄層で被覆されたシリコンウェーハに再エッチングされる。ウェルを濡らし、および/または表面を機能化するために、タンパク質溶液をアレイの上に液滴として加える(ii)。タンパク質溶液を除去し、ウェハを乾燥させ、所望の細菌を含有する新しい溶液を添加する(iii)。インキュベーション時間後に細菌溶液を除去し、ウェルを乾燥させ、ウェルおよび表面に細菌を残す(iv)。表面関連細菌は、パリレンリフトオフで除去され、マイクロウェルにきれいに播種された細菌を残し、ウェルを水和(v)に保つのに役立つ2%グリセロール培地のために依然として生存可能である。次いで、シリコンチップをアガロースゲルコートガラスカバースリップ上にアレイ側を下にして配置し、マイクロウェル内でバクテリア増殖を供給する(vi)。 ( b )単一のシリコンデバイス上のサブアレイのレイアウト。各サブアレイは、同一のウェルのセットを含む。すべてのサブaのマイクロウェルの直径rrayは直径5〜100μmの範囲であり、ボトムパネル概略図で白から暗灰色で示される井戸直径ピッチの2倍、3倍または4倍に編成される。ウェルの深さが浅い場合(<10μm)、5および10μmの井戸直径は、これらの非常に小さな井戸にコロニーを形成する細胞がないため、一般的に有用であることはめったにない。この研究では、直径15〜100μmのウェルからのデータのみを分析した。 この図の拡大版を見るには、ここをクリックしてください。
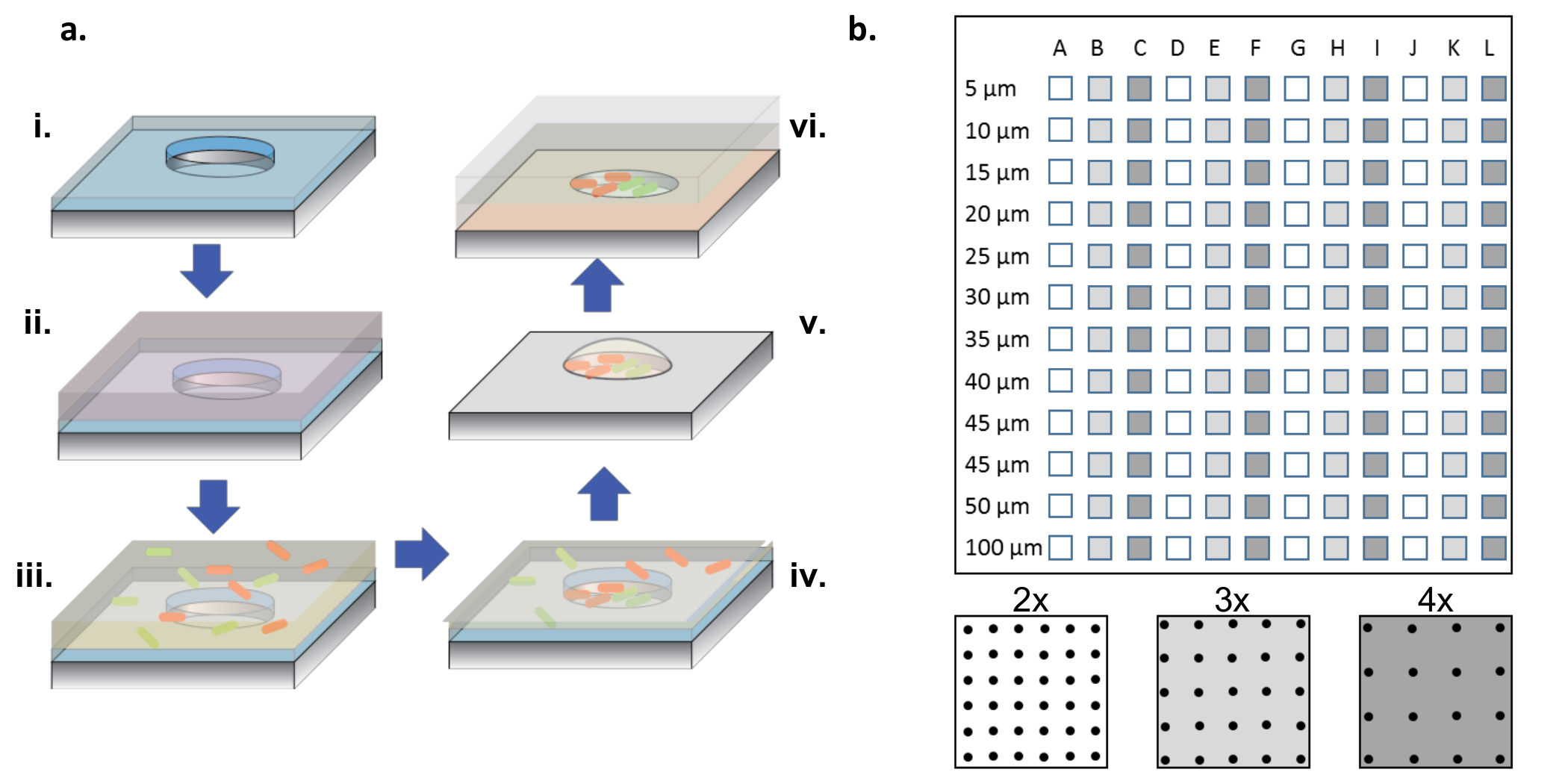{kind=link}
注: 図1bに示すように、完全なチップは、3つの異なるピッチ( すなわち、 2倍、3倍、および4倍の直径)が4回繰り返された、直径が5〜100μmのウェルのサブアレイを含む。
- 500μg/ mLウシ血清アルブミン(BSA)の150μL滴をPBS溶液に入れるマイクロウェルを濡らすためにアレイの上に置く。 BSA溶液をRTで湿ったチャンバー内でチップ上に1時間インキュベートする。
- 空のピペットチップボックスの底にリン酸緩衝化生理食塩水(PBS)を充填してチャンバーを作成する。
注:マイクロウェルの表面を機能化するために、BSAの代わりに特定のレクチンなどの他の物質を使用することができます。
- 空のピペットチップボックスの底にリン酸緩衝化生理食塩水(PBS)を充填してチャンバーを作成する。
- シリコンチップをBSA溶液でインキュベートしながら、2,500 rpm(平均950 xgに相当)で5分間遠心し、2%グリセロールを含む新鮮なR2A培地500μLに再懸濁します。
- 600nmでUV-vis分光計を使用して培養物のODを決定する。それを2%グリセロールR2A培地を用いて0.02のODに調整する。
注:グリセロールは、パリレンリフトオフ中にウェルが乾燥するのを防ぐのに役立ちます。
- 600nmでUV-vis分光計を使用して培養物のODを決定する。それを2%グリセロールR2A培地を用いて0.02のODに調整する。
- インキュベーション後、BSA溶液を除去し、液滴を除去して交換することによりPBSで3回リンスするシリコンマイクロウェルアレイを覆っている。窒素下で乾燥させる。
- 湿ったチャンバーに置かれた乾燥アレイのそれぞれに0.02 OD培養液150μLを加える。細菌を井戸壁に付着させるために、4℃で1時間インキュベートする。
注:インキュベーションには冷凍は必要ありません。 4℃のインキュベーション時間は、画像化が始まる前に細菌の増殖を防ぐために使用され、成長前のコミュニティの空間的構成を視覚化することができる。室温でのインキュベーションも使用することができる。どちらのプロトコールでも同様の増殖曲線が得られる。
3.顕微鏡のセットアップ
- シリコンチップ上で細菌のインキュベーションを開始する前に、ステージトップの環境制御チャンバー(材料の表を参照)をオンにし、湿度(〜100%)と温度(30〜32 ℃、ステップ3.2参照)は、サンプルを添加する前に平衡化することができる。
- サンプルホルダーを水平にして、チャンバー内の湿度を露点まで上昇させるために、PBSを浸した実験用ワイパー( 表の表を参照 )を使用してサンプル周りに置いてください。チャンバーの温度を30℃に、チャンバーの蓋の温度を32℃に設定して、結像面での結露を減らします。
注:スライドホルダーは、約1cmの厚さのガスケット付きの生細胞チャンバーに適合します。試料ホルダーは、試料ホルダーの上に置かれた気泡レベルの助けを借りて水平にされる。サンプルホルダはわずかに傾けられ、ガスケット内に密閉されたまま水平に保たれます。 - 培養物がシリコンチップ上でインキュベートしている間、イメージングの前に少なくとも30分間、水銀ランプの電源スイッチを手動でオンにする。手動でカメラと自動顕微鏡ステージの電源を入れます。顕微鏡と周辺機器を制御するソフトウェアを開き、機器がソフトウェアによって認識されていることを確認します。
注:倍率はNA = 0.3で10倍です。
- マイクロ波は、液体状態に達するまで、約60秒間前にアガロース溶液( すなわち 、R2A培地中の2%アガロース)を調製した。
- 75 mm×22 mmの#1.5ガラスカバースリップをエタノールで湿らせ、2 x 3インチ(50 x 75 mm)ガラススライド上に縦に中央に置く.2つのPDMSスペーサー(厚さ約1 mm)カバースリップの長い縁に沿って移動させ、カバースリップの約1mmがスライドの縁に張り出すようにガラスカバースリップを移動させる。
- カバーガラスとスライドの間に液体寒天を「サンドイッチ」するために、ガラスカバースリップの上に5mLの液体アガロース溶液を注ぎ、それを完全にカバーし、2×3インチのガラススライドをアセンブリの上に置きます。
注:これは、アガロースの深さを制御し、PDMSスペーサーと同じ厚さのカバーガラスと硬化アガロースの合計厚さを作る。 - アガロース溶液が凝固し始めるまでスライドガラス - カバーガラス - アガロースゲル - スライドスライドサンドイッチをセットし、それを冷蔵庫に移してください。 15分後、過剰の固体アガロースを除去し、ガラスカバースリップの周囲を切断する。これを清潔な皿に移し、使用するまで冷蔵庫に置きます。
アガロースコーティングされたカバースリップおよびイメージングによるウェルのシール
- 細菌のインキュベーション時間が完了した後、以下のように、冷蔵庫からアガロースコーティングされたカバースリップを除去し、シリコンチップを調製する。
- シリコンチップを超純水で一度に1つずつ10秒ずつ浸します。余分な液体のほとんどがチップの端から排出されるまで、ラボのワイプまたはティッシュの端にセットします。
- 各シリコンチップのエッジの長さに合わせてテープを切断します。シリコンを覆っているパリレン上にテープを置き、パリレンコーティングをすばやく剥がします。
- Immedマイクロチップアレイ側がアガロース被覆されたカバーガラスのアガロース被覆側に面する(かつ接触する)ように、各剥離チップを逆に反転させ、各チップを配置する。ウェルの外側の細菌の増殖を防ぐために、チップがアガロースに接触した後にチップを移動または移動させないように注意してください。
- アセンブリーされたマイクロウェルアレイ/アガロースカバースリップを、ステージトップ環境制御チャンバーのスライドホルダーに入れます。
- 関心のあるアレイの位置を特定するには、周囲光または有向ライト( 例:懐中電灯)を使用します。これらの位置を保存するために、自動ステージを制御する商用ソフトウェアを使用します(材料表を参照)。位置が保存された後、周囲光または有向光をオフにします。
- 商用ソフトウェアでは、 "ND Acquisition"パネルを開きます。
注記:このパネルには、特定のディレクトリへの自動保存のためのメニューとプログラマブルなイメージ取得が含まれています。これらの実験のために「時間」「XY」「λ」のメニューが使用されます。 - ソフトウェア内の位置を保存するには、「XYメニュー」をクリックし、保存する必要のある各位置の左側にある空のボックスをチェックします。また、 "Include Z"ボタンをクリックしてください。
- 適切な蛍光フィルターキューブを使用して、所望の波長および10倍の倍率で画像を取得する(表の表を参照)。
- 制御ソフトウェアと保存されたアレイ位置を使用して、保存された各位置に移動し、ウェルに集中します。保存されたリストの各XY位置をクリックし、緑色蛍光タンパク質(GFP)フィルターを使用してフォーカスを調整します。 z位置を示す矢印をクリックして新しいz位置を保存します。
注:このプロセスには時間がかかることがあります。ゲインを上げるための予防措置を講じること、および光退色を防ぐために中性濃度フィルターを使用して光強度を下げることを検討してください。 - アレイの表面に焦点を合わせたときのz軸の位置の違いに注目して、各波長の焦点面間のz軸の距離を求めます。混合赤/緑バクテリア集団を含むアレイから2-3の位置を選択し、赤色蛍光タンパク質(RFP)フィルターを用いて焦点を合わせる。
- GFPとRFP蛍光フィルターを使用して焦点面間の距離を差し引き、「λ」メニューの下に焦点面調整を追加します。
注:たとえば、アレイがGFPチャネルのz位置50μmにフォーカスされているように見え、同じアレイが55μmのRFPチャネルに焦点が合っているように見える場合は、RFP光学構成の隣に+5を「λ "メニュー。
- GFPとRFP蛍光フィルターを使用して焦点面間の距離を差し引き、「λ」メニューの下に焦点面調整を追加します。
- タイムラプス画像取得を開始します。
注:ここに示されている実験では、RFPおよびGFP画像を、30分間隔で、各アレイ位置について、多次元画像取得を、市販のソフトウェアカメラ、シャッター、フィルターホイール、および電動ステージ。- 「時間」メニューの下で、「間隔」を30分に、「実験の継続時間」を24時間に設定します。 [今すぐ実行]をクリックします。
注: "Time"、 "XY"、 "λ"の各チェックボックスをオンにすると、プログラムを実行するとステージが各位置( つまり保存されているXYZ位置)に移動し、1波長の画像を撮影し、 ( つまり、ラムダまたは波長制御)を考慮し、第2の画像を撮り、次の配列位置(多点)に移動し、これを30分間隔で(時間経過で)ループする。
- 「時間」メニューの下で、「間隔」を30分に、「実験の継続時間」を24時間に設定します。 [今すぐ実行]をクリックします。
- 制御ソフトウェアと保存されたアレイ位置を使用して、保存された各位置に移動し、ウェルに集中します。保存されたリストの各XY位置をクリックし、緑色蛍光タンパク質(GFP)フィルターを使用してフォーカスを調整します。 z位置を示す矢印をクリックして新しいz位置を保存します。
- 照明制御画像を取得する。
注: "ND Acquisition"、 "Time"、 "XY"メニューを使用して、それぞれ25倍の4つの位置を撮影します。- すべての光源をオフにして標準スライドの「画像」。これらの画像はカメラのノイズをキャプチャします。時間の経過中に使用された最も長い露光時間を使用する(ステップ5.3.3)。
- 所定の実験条件で不均一な照明を捕らえるために、いくつかの異なる場所で標準的なスライド( すなわち、一様なRFPまたはGFP強度)を画像化することによって、一連の100 "照明フィールド"画像を撮る。飽和に達することなく信号を最大化する露光時間を選択します。
6.分析
- 画像解析ソフトウェア(ImageJなど)を使用して画像スタックを処理します。
- 市販のソフトウェアを使用して、取得した画像をtiffファイル形式に変換します。 「ファイル」>「読み込み」>「画像シーケンス」をクリックして、画像解析ソフトウェアに画像をアップロードします。
- 「暗視野」と「照明フィールド」のすべての画像を平均して「補正画像」を作成します。サブトラック"Process"> "Image Calculator"を選択して、平均の "照射フィールド"画像から平均 "暗視野"画像を生成します。 2つのイメージ "Image1"と "Image2"を選択し、 "Operation"フィールドで "Subtract"を選択します。 [OK]をクリックします。
- 平均化するには、補正(または暗視野)画像を読み込み、「画像」>「スタック」>「Zプロジェクト」>「平均投影」をクリックします。
- 必要に応じて画像登録を行います。次に、「プロセス」>「バックグラウンドを差し引く」をクリックして、バックグラウンドの減算を実行します。 「半径」フィールドに半径( たとえば 125)を入力し、「滑り放物線」を選択します。
- "Process"> "Calculator Plus"を使用して照度補正を行います。以下のパラメータを選択します:operation、divide; i1、井戸像。 i2、補正画像。 k1、補正画像平均。 k2、0をクリックします。「Cre新しいウィンドウを食べた。
注:このデータセットは登録を必要としませんでしたが、他の作業ではImageJ Plugin StackRegが「変換」変換で使用されました。背景減算の場合は、すべての画像セットに同じ滑落放物線半径を使用します。例えば、画像化された最大のウェルが100のピクセル半径を有する場合、各画像セットに対して100より大きい半径( 例えば、 125)を使用する。
- マイクロウェル内の各株の増殖を決定する。
- ImageJ「MicroArray」プラグインを使用して、目的のアレイの各マイクロウェル周辺の関心領域(ROI)を選択します。
- [MAP]メニューの[Reset Grid]をクリックします。行、列、および直径を指定します(アレイ上のウェルのサイズと数に基づいて、 図1bを参照)。 「ROI形状」メニューから「円」を選択します。
- "Alt"キーを押しながらマウスの左上のROIを選択して移動する彼はROI配列。 「シフト」キーを押しながら左下のROIを選択してアレイのサイズを変更します。 「シフト」キーを押しながら、アレイの右側からROIを選択しますが、コーナーではなく、ROIの間隔を変更します。
- 上記のコマンドを使用して、イメージ内のウェルにROIアレイを合わせます。 「Measure RT」をクリックします。
注記:プラグインは、各ROIから必要な測定値をエクスポートします。 3つのROIサイズを使用して、ウェルの周囲に同心円状のリングを作成し、バックグラウンドシグナル( すなわち、外側リングから差し引かれた中間リングからのシグナル)および蛍光測定値( すなわち内側リングからのシグナル)を収集する。
- スプレッドシートソフトウェアでデータを収集し、バックグラウンドシグナルを計算します。さらに分析するためにカスタムスクリプトソフトウェアにインポートします。
- ImageJ「MicroArray」プラグインを使用して、目的のアレイの各マイクロウェル周辺の関心領域(ROI)を選択します。
- データの編成と分析
- データをインポートしてデータを整理するすべての回で次の順序で行列にImageJで収集されます:列1、サブ配列番号。列2、井戸列。カラム3、ウェルカラム;列4、平均強度;列5、バックグラウンド強度。列6は平均強度 - バックグラウンド強度である。
- mCherryとGFPの取得結果を別々のマトリックスに分けます。各サブアレイと各色の結果をセル配列の異なるセルに格納します。
注記:この構成では、イメージデータと測定結果の間を行き来し、データをクリーンアップし、データが正確にデータを表すことを保証します。
- mCherryとGFPの取得結果を別々のマトリックスに分けます。各サブアレイと各色の結果をセル配列の異なるセルに格納します。
- P.緑膿菌の自己蛍光を調整する。
注:GFPとmCherry株の共培養に関する実験では、mCherryのみのチップを分析して、mCherryと緑色自己蛍光の関係を確認する必要があります。- すべてのmCherryΔmからのmCherry対GFPシグナルをプロットするΔretS&#916; tse / i1-6ウェルを用いて、mCherryシグナルとGFPチャネルにおける自己蛍光との間の関係を決定した。共培養物から自己蛍光シグナルを差し引く。
- 軌跡をプロットし、修正されたロジスティック方程式を各軌道に当てはめて、スプレッドシートソフトウェアまたはカスタムスクリプトソフトウェアの最小二乗法を用いてパラメータを抽出する。
- GFPとmCherryの軌道パラメータの間の相関を探します。
- データをインポートしてデータを整理するすべての回で次の順序で行列にImageJで収集されます:列1、サブ配列番号。列2、井戸列。カラム3、ウェルカラム;列4、平均強度;列5、バックグラウンド強度。列6は平均強度 - バックグラウンド強度である。
結果
ここに提示された実験プラットフォームは、細菌群集のハイスループットおよびハイコンテント研究のために設計されています。このデザインにより、さまざまなサイズのウェルで成長する数千のコミュニティを同時に分析することができます。このマイクロウェルアレイ設計により、初期の播種密度、井戸の大きさ、および化学的環境への最終的なコミュニティ組?...
ディスカッション
この記事では、マイクロウェルアレイデバイスと、細菌のコミュニティ開発のハイスループットと高含量の生細胞イメージングベースの解析を可能にするための実験プロトコルを紹介しました。ここでのデモの焦点は、接触仲介型VI型分泌が地域社会の発展に及ぼす影響を研究することであったが、広範囲の微生物群集と微生物 - 微生物相互作用の研究に柔軟に対応するように設計された。?...
開示事項
著者は何も開示することはない。
謝辞
マイクロウェルアレイは、米国エネルギー省基礎エネルギー科学局のナノフェーズ材料科学センターのユーザー施設部門のセンターで製作され、特性評価されました。この研究の資金援助は、オークリッジ国立研究所所長の研究開発基金を通じて提供された。著者らはまた、これらの研究で使用されたP.緑膿菌株の供給についてJ. Mougous Laboratory(Washington、Seattle、WA)に感謝したい 。
資料
| Name | Company | Catalog Number | Comments |
| Parylene N | Specialty Coating Systems | CAS NO.:1633-22-3 | |
| Parylene coater | Specialty Coating Systems | Labcoter 2 Parylene Deposition Unit PDS2010 | |
| Silicon Wafer | WRS Materials | 100mm diameter, 500-550μm thickness, Prime, 10-20 resistivity, N/Phos<100>, | |
| adhesion promoter | Shin-Etsu Microsci | MicroPrime P20 adhesion promoter | |
| postive tone photoresist | Rohm and Haas Electronics Materials LLC (Owned by Dow) | Microposit S1818 Positive Photoresist (code 10018357) | |
| Quintel Contact Aligner | Neutronix Quintel Corp | NXQ 7500 Mask Aligner | |
| Reactive Ion Etching Tool | Oxford Instruments | Plasmalab System 100 Reactive Ion Etcher | |
| R2A Broth | TEKnova | R0005 | |
| Bovine Serum Albumin | Sigma | A9647 | |
| Multimode Plate Reader | Perkin Elmer | Enspire, 2300-0000 | |
| Fluorescent Microscope | Nikon | Eclipse Ti-U | |
| Automated Stage | Prior | ProScan III | |
| CCD camera | Nikon | DS-QiMc | |
| Stage-top environmental control chamber | In Vivo Scientific | STEV ECU-HOC | |
| Phosphate Buffered Saline | ThermoFisher Scientific | 14190144 | |
| UltraPure Agarose | ThermoFisher Scientific | 16500500 | |
| 25 x 75 mm No. 1.5 coverslip | Nexterion | High performance #1.5H coverslips | |
| Fluorescence Reference Slides | Ted Pella | 2273 | |
| Physical Stylus Profilometer | KLA Tencor | P-6 | |
| lab wipes | Kimberly Clark | Kimipe KIMTECH SCIENCE Brand, 34155 | |
| commercial software | Nikon | NIS Elements | |
| Zeiss 710 Confocal Microscope | Zeiss | ||
| filter cubes | Nikon | Nikon FITC (96311), Nikon Texas Red(96313) |
参考文献
- Zhou, J., Deng, Y., et al. Stochasticity, succession, and environmental perturbations in a fluidic ecosystem. Proc Natl Acad Sci. 111, E836-E845 (2014).
- Valm, A. M., Welch, J. L. M., et al. Systems-level analysis of microbial community organization through combinatorial labeling and spectral imaging. Proc Natl Acad Sci USA. 108 (10), 4152-4157 (2011).
- Satoh, H., Miura, Y., Tsushima, I., Okabe, S. Layered structure of bacterial and archaeal communities and their in situ activities in anaerobic granules. Appl Environ Microbiol. 73 (22), 7300-7307 (2007).
- Kim, H. J., Boedicker, J. Q., Choi, J. W., Ismagilov, R. F. Defined spatial structure stabilizes a synthetic multispecies bacterial community. Proc Natl Acad Sci USA. 105 (47), 18188-18193 (2008).
- Nunan, N., Wu, K., Young, I. M., Crawford, J. W., Ritz, K. Spatial distribution of bacterial communities and their relationships with the micro-architecture of soil. FEMS Microbiol Ecol. 44, 203-215 (2003).
- Grundmann, G. L. Spatial scales of soil bacterial diversity - The size of a clone. FEMS Microbiol Ecol. 48, 119-127 (2004).
- Langenheder, S., Lindstrom, E. S., Tranvik, L. J. Structure and Function of Bacterial Communities Emerging from Different Sources under Identical Conditions. Appl Environ Microbiol. 72 (1), 212-220 (2006).
- Camp, J. G., Kanther, M., Semova, I., Rawls, J. F. Patterns and Scales in Gastrointestinal Microbial Ecology. Gastroenterology. 136 (6), 1989-2002 (2009).
- Renner, L. D., Weibel, D. B. Physicochemical regulation of biofilm formation. MRS Bull. 36 (5), 347-355 (2011).
- Wessel, A. K., Hmelo, L., Parsek, M. R., Whiteley, M. Going local: technologies for exploring bacterial microenvironments. Nat Rev Microbiol. 11 (5), 337-348 (2013).
- Stacy, A., McNally, L., Darch, S. E., Brown, S. P., Whiteley, M. The biogeography of polymicrobial infection. Nat Rev Microbiol. 14 (2), 93-105 (2015).
- Hansen, R. R., Shubert, K. R., Morrell-Falvey, J. L., Lokitz, B. S., Doktycz, M. J., Retterer, S. T. Microstructured block copolymer surfaces for control of microbe adhesion and aggregation. Biosensors. 4 (1), 63-75 (2014).
- Hansen, R. R., Hinestrosa, J. P., et al. Lectin-functionalized poly(glycidyl methacrylate)- block -poly(vinyldimethyl azlactone) surface scaffolds for high avidity microbial capture. Biomacromolecules. 14 (10), 3742-3748 (2013).
- Timm, C. M., Hansen, R. R., Doktycz, M. J., Retterer, S. T., Pelletier, D. A. Microstencils to generate defined, multi-species patterns of bacteria. Biomicrofluidics. 9 (6), (2015).
- Keymer, J. E., Galajda, P., Muldoon, C., Park, S., Austin, R. H. Bacterial metapopulations in nanofabricated landscapes. Proc Natl Acad Sci USA. 103 (46), 17290-17295 (2006).
- Zhang, Q., Lambert, G., et al. Acceleration of Emergence of Bacterial Antibiotic Resistance in Connected Microenvironments. Science. 333 (6050), 1764-1767 (2011).
- Friedlander, R. S., Vlamakis, H., Kim, P., Khan, M., Kolter, R., Aizenberg, J. Bacterial flagella explore microscale hummocks and hollows to increase adhesion. Proc Natl Acad Sci USA. 110 (14), 5624-5629 (2013).
- Zhou, J., Liu, W., et al. Stochastic Assembly Leads to Alternative Communities with Distinct Functions in a Bioreactor Microbial Community. MBio. 4 (2), 1-8 (2013).
- van Vliet, S., Hol, F. J., Weenink, T., Galajda, P., Keymer, J. E. The effects of chemical interactions and culture history on the colonization of structured habitats by competing bacterial populations. BMC Microbiol. 14 (1), 116 (2014).
- Niepa, T. H. R., Hou, L., et al. Microbial Nanoculture as an Artificial Microniche. Sci Rep. 6, 30578 (2016).
- Hansen, R. H., Timm, A. C., et al. Stochastic Assembly of Bacteria in Microwell Arrays Reveals the Importance of Confinement in Community Development. PLoS ONE. 11 (5), e0155080 (2016).
- Hood, R. D., Singh, P., et al. A Type VI Secretion System of Pseudomonas aeruginosa Targets a Toxin to Bacteria. Cell Host Microbe. 7 (1), 25-37 (2010).
- LeRoux, M., Ja De Leon, ., et al. Quantitative single-cell characterization of bacterial interactions reveals type VI secretion is a double-edged sword. Proc Natl Acad Sci. 109 (48), 19804-19809 (2012).
- Whitney, J. C., Beck, C. M., et al. Genetically distinct pathways guide effector export through the type VI secretion system. Mol Microbiol. 92 (3), 529-542 (2014).
- Warrick, J. W., Timm, A., Swick, A., Yin, J. Tools for Single-Cell Kinetic Analysis of Virus-Host Interactions. PLoS ONE. 11 (1), e0145081 (2016).
- Zwietering, M. H., Jongenburger, I., Rombouts, F. M., Van't Riet, K. Modeling of the Bacterial Growth Curve. Appl Environ Microbiol. 56 (6), 1875-1881 (1990).
- Halsted, M., Wilmoth, J. L., et al. Development of transparent microwell arrays for optical monitoring and dissection of microbial communities. J Vac Sci Technol B Nanotechnol Microelectron. 34 (6), 06KI03 (2016).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved