
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Zusammenbau und Verfolgung der mikrobiellen Gemeinschaftsentwicklung innerhalb einer Microwell Array Plattform
In diesem Artikel
Zusammenfassung
Die Entwicklung der mikrobiellen Gemeinschaften hängt von einer Kombination von Faktoren ab, einschließlich Umweltarchitektur, Mitgliedshäufigkeit, Züge und Wechselwirkungen. Dieses Protokoll beschreibt eine synthetische, mikrogefertigte Umgebung für die gleichzeitige Verfolgung von Tausenden von Gemeinschaften, die in Femtoliter-Vertiefungen enthalten sind, wo Schlüsselfaktoren wie Nischengröße und Einschluss angenähert werden können.
Zusammenfassung
Die Entwicklung der mikrobiellen Gemeinschaften hängt von einer Kombination von komplexen deterministischen und stochastischen Faktoren ab, die die räumliche Verteilung und die Aktivitäten der Gemeinschaftsmitglieder drastisch verändern können. Wir haben eine microwell-Array-Plattform entwickelt, mit der Tausende von Bakteriengemeinschaften parallel versammelt und verfolgt werden können. Dieses Protokoll unterstreicht den Nutzen der Plattform und beschreibt seine Verwendung zur optischen Überwachung der Entwicklung einfacher, zweiköpfiger Gemeinschaften innerhalb eines Ensembles von Arrays innerhalb der Plattform. Diese Demonstration verwendet zwei Mutanten von Pseudomonas aeruginosa , Teil einer Reihe von Mutanten entwickelt, um Typ VI Sekretion Pathogenität zu studieren. Chromosomale Insertionen von entweder mCherry- oder GFP-Genen erleichtern die konstitutive Expression von fluoreszierenden Proteinen mit unterschiedlichen Emissionswellenlängen, die verwendet werden können, um die Häufigkeit und die Lage des Gemeinschaftsmitglieds innerhalb jeder Mikrovertiefung zu überwachen. Dieses Protokoll beschreibt ein detailliertes methoD zum Zusammenbau von Bakteriengemischen in die Vertiefungen des Arrays und unter Verwendung von zeitraffer Fluoreszenzbildgebung und quantitativer Bildanalyse, um das relative Wachstum jeder Mitgliedspopulation über die Zeit zu messen. Die Aussaat und Montage der Mikrotiterplattform, die bildgebenden Verfahren, die für die quantitative Analyse von mikrobiellen Gemeinschaften innerhalb des Arrays notwendig sind, und die Methoden, die verwendet werden können, um Wechselwirkungen zwischen mikrobiellen Speziesbereichen zu diskutieren, die alle diskutiert werden.
Einleitung
Die mikrobiellen Gemeinschaften sind sowohl durch deterministische Faktoren wie die Struktur der Umwelt und stochastische Prozesse geprägt, die mit dem Zelltod, der Teilung, der Proteinkonzentration, der Anzahl der Organellen und der Mutation verbunden sind. Innerhalb der natürlichen Umgebung kann es fast unmöglich sein, die individuellen Auswirkungen dieser Einflüsse auf die Zusammensetzung und die Aktivität der Gemeinschaft zu analysieren. Von natürlichen Strukturen verdeckt und in einem chemischen und biologischen Milieu begraben, die Mitglieder der Gemeinschaft zu identifizieren und ihre räumlich-zeitliche Verteilung in der natürlichen Umgebung zu lösen, ist äußerst anspruchsvoll. Dennoch haben die jüngsten Bemühungen die Bedeutung der räumlichen Organisation auf die Gemeinschaftsfunktion unterstrichen und auf die Notwendigkeit hingewiesen, sowohl die Mitgliedshäufigkeit als auch die Organisation in den laufenden Studien 2 , 3 , 4 zu berücksichtigen.
EsIst klar, dass die lokale chemische Umgebung ( dh die Verfügbarkeit von Nährstoffen und sekundären Metaboliten), die physikalische Struktur ( zB Bodenarchitektur, Pflanzenwurzeln, Ozeanpartikel oder die intestinalen Mikrovilli), die Anwesenheit oder Abwesenheit von Sauerstoff und die Einführung von Pathogene Arten alle beeinflussen die Zusammensetzung, die Architektur und die Funktion der mikrobiellen Gemeinschaften 5 , 6 , 7 , 8 , 9 , 10 , 11 . Nichtsdestotrotz dominieren traditionelle Techniken für Kulturen, die diese Faktoren nicht erfassen. Gemeinschaftszusammensetzung ( z. B. das Vorhandensein von Co-abhängigen Spezies), die physikalische Bindung, die Signalmolekülkonzentration und der direkte Zellzellkontakt sind alle wichtigen Faktoren für die Gestaltung einer mikrobiellen Gemeinschaft und können in c verloren gehenOnventionelle Kulturbedingungen. Diese Eigenschaften sind in einer Bulk-Flüssigkultur oder auf einer Agarplatte schwer zu replizieren. Die Verfügbarkeit von mikrofluidischen, Mikrostrukturen und Nanofabrikationstechniken, die die Replikation der wichtigsten physikalischen und chemischen Eigenschaften natürlicher Umgebungen ermöglichen, hat es vielen Forschern ermöglicht, bakterielle Gemeinschaften zu bauen, um ihre Wechselwirkungen 12 , 13 , 14 zu untersuchen und synthetische Umgebungen zu entwickeln Imitieren natürliche Bedingungen 4 , 15 , 16 , 17 , 18 , 19 , 20 .
Dieses Protokoll beschreibt eine Methode zur Herstellung einer Mikrowellen-Array-Vorrichtung und liefert detaillierte experimentelle Verfahren, die verwendet werden können, um Th zu funktionalisierenE Wells in der Array und Bakterien zu wachsen, sowohl als Single-Spezies-Kolonien und in Multi-Mitgliedsgemeinschaften. Diese Arbeit zeigt auch, wie Bakterien modifiziert, um fluoreszierende Reporter-Proteine zu produzieren, können verwendet werden, um das Bakterienwachstum in den Brunnen im Laufe der Zeit zu überwachen. Ein ähnliches Array wurde zuvor vorgestellt und zeigte, dass es möglich ist, das Wachstum von Einzelkolonien von Pseudomonas aeruginosa ( P. aeruginosa) in Mikrovertiefungen zu verfolgen. Durch die Modulation von Well-Size und Seeding-Dichte können die Ausgangsbedingungen von Tausenden von Wachstumsexperimenten parallel variiert werden, um zu bestimmen, wie die anfänglichen Inokulationsbedingungen die Fähigkeit der Bakterien beeinflussen, 21 zu wachsen. Die aktuelle Arbeit verwendet eine leicht modifizierte Version des microwell-Arrays, die auf der vorherigen Arbeit aufbaut, indem sie den gleichzeitigen Vergleich von mehreren Arrays ermöglicht und ein robusteres experimentelles Protokoll verwendet. Das Array, das in dieser Arbeit verwendet wird, enthält mehrere Subarrays oder Array ensemBles, die Brunnen unterschiedlicher Größe enthalten, die von 15 - 100 μm im Durchmesser reichen, die in drei verschiedenen Tonhöhen angeordnet sind ( dh 2x, 3x und 4x der Bohrlochdurchmesser). Die Arrays werden in Silizium geätzt, und das Wachstum der in den Silizium-Arrays ausgesetzten Bakterien wird durch Versiegeln der Arrays mit einem Deckglas ermöglicht, das mit einem Medium-infundierten Agarosegel beschichtet worden ist. P. aeruginosa- Mutanten, die entworfen wurden, um das Typ-VI-Sekretionssystem zu studieren, werden in dieser Demonstration verwendet.
Die hier vorgestellten Ergebnisse bauen auf das ultimative Ziel, Multimember-Communities innerhalb von Mikrowellen-Arrays zu analysieren, so dass Forscher die Fülle und Organisation von Bakterien in situ überwachen können, während sie die chemische Umgebung kontrollieren und untersuchen. Dies sollte letztlich Einblicke in die "Regeln" geben, die die Gemeinschaftsentwicklung und die Nachfolge bestimmen.
Protokoll
1. Silizium-Microwell-Array-Fertigung
- Parylenbeschichtung
- Ablagerung zwischen 1-1,5 μm Parylen N auf Silicium-Wafern unter Verwendung eines handelsüblichen Parylenbeschichtungssystems nach den Spezifikationen und Anweisungen des Herstellers (Einstellungen: Verdampfer-Sollwert = 160 ° C, Ofen-Sollwert = 650 ° C).
HINWEIS: Ungefähr 6 g Parylen N, das in eine Kammer geladen wird, ergeben Beschichtungen von 1-1,5 μm Dicke.
- Ablagerung zwischen 1-1,5 μm Parylen N auf Silicium-Wafern unter Verwendung eines handelsüblichen Parylenbeschichtungssystems nach den Spezifikationen und Anweisungen des Herstellers (Einstellungen: Verdampfer-Sollwert = 160 ° C, Ofen-Sollwert = 650 ° C).
- Photolithographie
- Die Parylen-N-beschichteten Wafer mit Haftvermittler, 20% Hexamethyldisilazan (HMDS) und 80% Propylenglykolmonomethyletheracetat (PGMEA) (siehe Tabelle der Materialien ) bei 3000 U / min für 45 s. Füllen Sie eine 2-ml-Transferpipette mit Haftvermittler und besprühen Sie sie über den gesamten Wafer. Lassen Sie den Wafer für ca. 10 s sitzen, bevor Sie ihn trocken drehen.
- Füllen Sie eine 2 mL Transferpipette mit positivem zuNe Photoresist (siehe Tabelle der Materialien ) und geben den Photoresist in der Mitte des Wafers ab. Drehen Sie bei 3.000 U / min für 45 s, um eine Resistbeschichtung zu ergeben, die etwa 1,5 μm dick ist.
- Soft-back die Proben auf einer Kochplatte bei 115 ° C für 1 min.
- Verwenden Sie einen Kontaktausrichter und eine Photomaske mit dem gewünschten Bohrlochmuster, um die Probe ultraviolettem Licht auszusetzen. Den spinbeschichteten Wafer für 6 s durch die gemusterte Photomaske auflösen, wobei eine ungefähre Dosis von 60-80 mJ / cm 2 bei 365 nm gemessen wird.
- Entwickeln Sie das Muster, indem Sie die Probe im Entwickler (<3% Tetramethylammoniumhydroxid in Wasser, siehe Tabelle der Materialien ) für 2 min eintauchen . Mit Wasser abspülen und mit sauberem, trockenem Stickstoff trocknen.
HINWEIS: Die UV-bezogenen Bereiche des Photoresists sollten während der Entwicklung gelöscht werden.
- Reaktives Ionenätzen
- Verwenden Sie ein Sauerstoff-Plasma-Ätzen, um das exponierte Parylen zu entfernenBis hin zum Siliziumsubstrat.
HINWEIS: Das Rezept kann moduliert werden, um die Ätzrate des Parylens zu verändern. Für Parylen-Dicken zwischen 1 und 5 μm verwenden Sie ein Rezept mit 60 mTorr, 20 ° C, 100 sccm O 2 , 10 W RF und 2.000 W ICP auf einem Reaktiv-Ionen-Ätzgerät (RIE). Nach dem Ätzen und Entfernen der exponierten Parylenschicht sollte der gemusterte Bereich ( dh das freiliegende Silizium) glänzend und silber aussehen. - Verwenden Sie einen tiefen RIE (DRIE, zB Bosch DRIE) Ätzprozess, um in das Silizium zu ätzen.
HINWEIS: Die Ätzrate und die Dauer bestimmen die Bohrlochtiefe. Ein vollständiger Zyklus des Bosch-Prozesses (ein 3-s-Abscheidungsschritt: 20 mTorr, 15 ° C, 140 sccm C 4 F 8 , 10 W RF und 1.750 W ICP, gefolgt von einem 10 s Ätzprozess: 20 mTorr, 15 ° C , 120 sccm SF 6 , 8 W RF und 1.750 W ICP) entspricht etwa 1 μm Ätztiefe. Die Brunnen, die in dieser Demonstration verwendet werden, reichen von 3 - 3,5 μm tief. - V.Erzwingen die Ätztiefe mit physikalischer Profilometrie.
- Legen Sie die Probe in ein physikalisches Profilometer (siehe Tabelle der Materialien ).
- Schalten Sie das Probenvakuum ein und drücken Sie die manuelle Lasttaste.
- Fokussieren Sie das System auf die Probe, indem Sie die Taste "Focus" drücken. Positionieren Sie eine geeignete Funktion für die Messung auf dem Bildschirm.
- Scannen Sie die Probe. Nähern Sie das Profil und messen Sie die Funktionstiefe.
- Aufzeichnen der Ätzrate und Modulierung der nachfolgenden Ätzzeiten, um die gewünschte Tiefe zu erreichen.
HINWEIS: Die Messungen umfassen die Tiefe der Silizium-Vertiefung, die Dicke des abgeschiedenen Parylens und die Dicke des Photoresists. Die Überprüfung der Dicke jeder Schicht während des gesamten Verfahrens ist notwendig, um eine genaue Bohrtiefe zu erreichen.
- Verwenden Sie ein Sauerstoff-Plasma-Ätzen, um das exponierte Parylen zu entfernenBis hin zum Siliziumsubstrat.
2. Bakterienkultur und -saat (Abbildung 1a )
- Starten Sie Kolonien auf Luria Brühe (LB) Agarplatten aus Glycerinbeständen und Gebrauch innerhalb von zwei Wochen. Wählen Sie Kolonien der gewünschten Stämme aus LB-Agarplatten und beginnen Sie über Nachtkulturen von P. aeruginosa. Inkubieren Sie die Übernachtkulturen für ca. 18 h bei 37 ° C unter Schütteln bei 220 U / min in R2A-Medium.
ANMERKUNG: Kolonien sollten innerhalb von zwei Wochen nach der Beschichtung ausgewählt werden, um sicherzustellen, dass die Mutationen und fluoreszierenden Reportergene beibehalten werden. Alle P. aeruginosa Arbeit sollte unter BSL-2 Bedingungen durchgeführt werden. - Verwenden Sie einen Diamantenschreiber, um den Siliziumwafer in einzelne Chips zu schneiden, die die Ensembles unterschiedlicher Größen und Pitch-Well-Arrays enthalten. Stellen Sie sicher, dass jeder Chip die volle Ergänzung von Brunnengrößen und Tonhöhen für die Studie enthält.

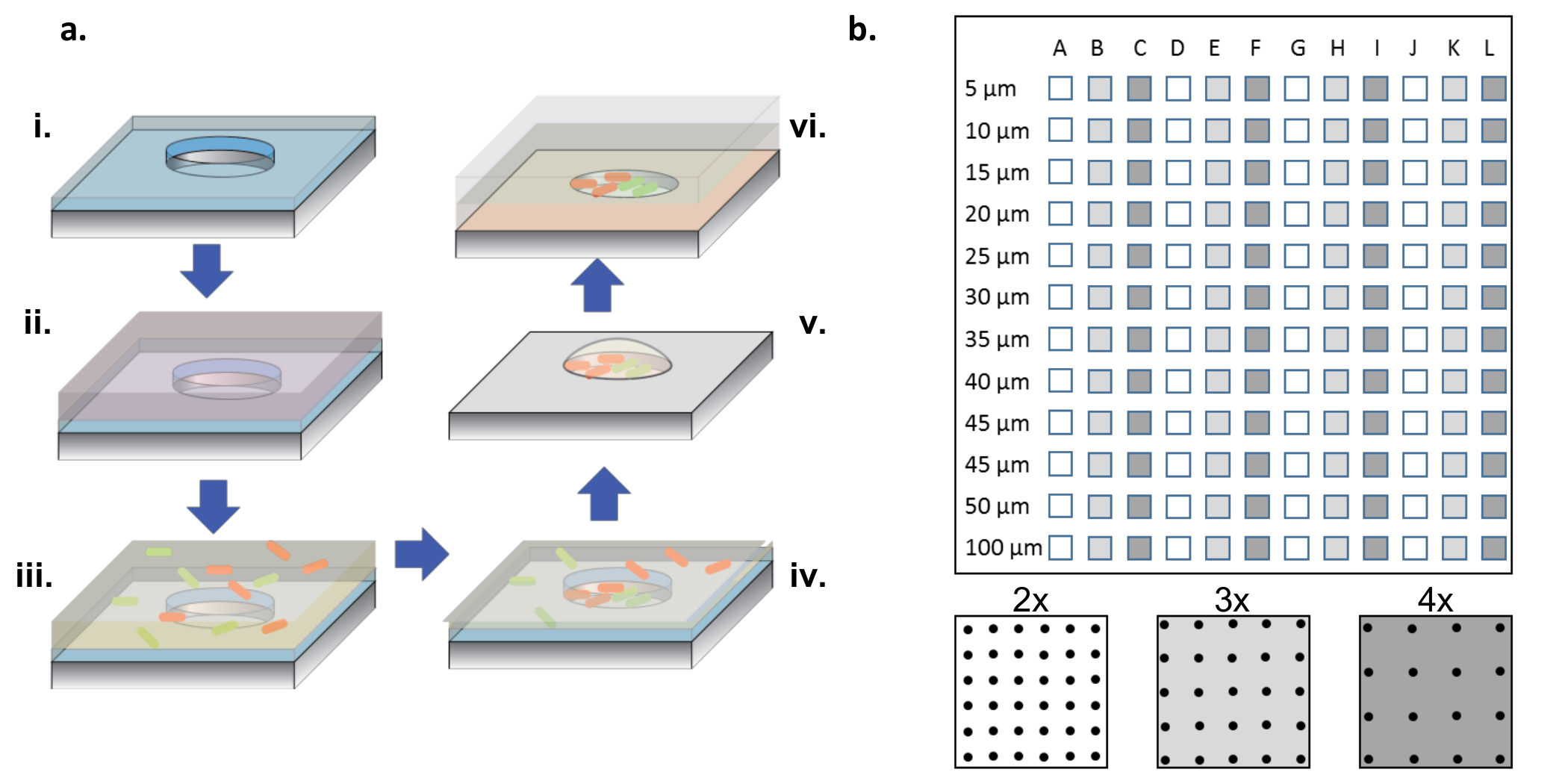
Abbildung 1: Fertigungs- und Zellseeding-Verfahren. ( A ) Microwell Arrays aIn Silicium-Wafer geätzt, die mit einer dünnen Schicht aus Parylen (i) beschichtet sind. Um die Wells zu benetzen und / oder die Oberfläche zu funktionalisieren, wird eine Proteinlösung in einem Tröpfchen oben auf den Arrays (ii) zugegeben. Die Proteinlösung wird entfernt, die Wafer getrocknet und eine neue Lösung, die die gewünschten Bakterien enthält, zugegeben (iii). Die Bakterienlösung wird nach einer Inkubationszeit entfernt und die Wafer trocknen gelassen und hinterlassen Bakterien in den Vertiefungen und auf der Oberfläche (iv). Die oberflächenassoziierten Bakterien werden mit dem Parylen-Abheben entfernt, wobei die Bakterien in den Mikrovertiefungen sauber gesät werden und aufgrund des 2% Glycerin-Mediums noch lebensfähig sind, was dazu beiträgt, die Vertiefungen hydratisiert zu halten (v). Die Silizium-Chips werden dann Arrayseite nach unten auf ein mit Agarose gelbeschichtetes Glasdeckel verlegt, welches das Bakterienwachstum in den Mikrovertiefungen (vi) zuführt. ( B ) Anordnung von Sub-Arrays auf einem einzigen Silizium-Gerät. Jedes Sub-Array enthält einen Satz identischer Wells. Der Durchmesser der Mikrovertiefungen über alle Sub-aRrays im Durchmesser von 5-100 μm und sind bei 2x, 3x oder 4x der Well-Durchmesser-Tonhöhe organisiert, die mit den weißen bis dunkelgrauen Farben auf dem Bodenplatten-Schema bezeichnet wird. Wenn die Bohrtiefen flach (<10 μm) sind, sind die 5 und 10 μm Bohrlochdurchmesser selten nützlich, im allgemeinen wegen des Mangels an Zellen, die diese sehr kleinen Vertiefungen kolonisieren. In dieser Arbeit wurden nur die Daten aus Brunnen mit 15-100 μm Durchmesser analysiert. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}
HINWEIS: Wie in Abbildung 1b gezeigt , enthält ein kompletter Chip Sub-Arrays von Wells mit Durchmessern von 5 bis 100 μm, wobei drei verschiedene Tonhöhen ( dh 2x, 3x und 4x der Durchmesser) viermal wiederholt werden.
- Legen Sie ein 150 μl Tröpfchen von 500 μg / ml Rinderserumalbumin (BSA) in PBS-LösungOben auf dem Array, um die microwells zu benetzen. Inkubieren Sie die BSA-Lösung für 1 h auf dem Chip bei RT in einer feuchten Kammer.
- Erstellen Sie die Kammer, indem Sie den Boden eines leeren Pipettenspitzenkastens mit Phosphat-gepufferter Kochsalzlösung (PBS) füllen.
ANMERKUNG: Andere Substanzen, wie z. B. spezifische Lektine, können anstelle von BSA verwendet werden, um die Oberfläche der Mikrovertiefungen zu funktionalisieren.
- Erstellen Sie die Kammer, indem Sie den Boden eines leeren Pipettenspitzenkastens mit Phosphat-gepufferter Kochsalzlösung (PBS) füllen.
- Bei der Inkubation von Siliciumchips mit BSA-Lösung werden die Kulturen bei 2500 U / min (entsprechend einem Durchschnitt von 950 xg) für 5 min zentrifugiert und dann in 500 μl frischem R2A-Medium mit 2% Glycerin resuspendiert.
- Bestimmen Sie die OD der Kultur mit einem UV-Vis-Spektrometer bei 600 nm. Stellen Sie es auf eine OD von 0,02 mit 2% Glycerin R2A Medium.
HINWEIS: Das Glycerin hilft, die Brunnen beim Austrocknen beim Parylenabheben zu verhindern.
- Bestimmen Sie die OD der Kultur mit einem UV-Vis-Spektrometer bei 600 nm. Stellen Sie es auf eine OD von 0,02 mit 2% Glycerin R2A Medium.
- Nach der Inkubation die BSA-Lösung entfernen und 3x mit PBS abspülen, indem man den flüssigen Tropfen entfernt und ersetztT, die das Silizium-Mikrowellen-Array abdecken. Trocken unter Stickstoff.
- Füge 150 μl 0,02 OD-Kulturen zu jeder der trockenen Arrays hinzu, die in einer feuchten Kammer angeordnet sind. Inkubieren für 1 h bei 4 ° C, damit die Bakterien an den Brunnenwänden haften können.
HINWEIS: Für die Inkubation ist keine Kühlung erforderlich. Die Inkubationszeit von 4 ° C kann verwendet werden, um das Wachstum von Bakterien zu verhindern, bevor die Bildgebung beginnt, so dass man die räumliche Organisation der Gemeinschaften vor dem Wachstum visualisieren kann. Raumtemperatur-Inkubation kann auch verwendet werden. Beide Protokolle führen zu ähnlichen Wachstumskurven.
3. Mikroskopaufstellung
- Bevor Sie die Bakterieninkubation auf den Siliziumchips starten, schalten Sie die Bühnenüberwachungskammer ein (siehe Tabelle der Materialien) und stellen Sie die Einstellungen auf dem Schaltkasten so ein, dass die Feuchtigkeit (~ 100%) und die Temperatur (30-32 ° C, siehe Schritt 3.2) können vor dem Hinzufügen von Proben äquilibrieren.
- Den Probenhalter einrasten und einsteckenTerior um die Probe mit PBS- getränkten Labortüchern (siehe Tabelle der Materialien ), um die Feuchtigkeit in der Kammer auf den Taupunkt zu erhöhen. Stellen Sie die Temperatur der Kammer auf 30 ° C und die des Kammerdeckels auf 32 ° C, um die Kondensation auf der Abbildungsebene zu reduzieren.
HINWEIS: Der Schiebehalter passt in die lebende Zellenkammer mit einer etwa 1 cm dicken Dichtung. Der Probenhalter wird mit Hilfe einer Blasenhöhe geebnet, die auf den Probenhalter gelegt wird. Der Probenhalter kann leicht gekippt werden und in der Dichtung auf dem Niveau versiegelt bleiben. - Während die Kulturen auf den Silizium-Chips inkubieren, schalten Sie den Netzschalter für die Quecksilberlampe manuell mindestens 30 Minuten vor der Bildgebung ein. Manuelles Einschalten der Kamera und automatisierte Mikroskopstufe. Öffnen Sie die Software zur Steuerung des Mikroskops und der Peripheriegeräte und stellen Sie sicher, dass das Gerät von der Software erkannt wird.
HINWEIS: Vergrößerung ist 10X mit NA = 0,3.
- Mikrowelle zuvor hergestellte Agarose-Lösungen ( dh 2% Agarose in R2A-Medium), bis ein flüssiger Zustand erreicht ist, ca. 60 s.
- Benetzen Sie die Rückseite eines 75 mm x 22 mm, # 1,5 Glasdeckel mit Ethanol und legen Sie es in Längsrichtung, zentriert, über eine 2 x 3 "(50 x 75 mm) Glasrutsche. Platzieren Sie zwei PDMS-Abstandshalter (Dicke von ~ 1 mm) Entlang der langen Ränder des Deckglases und verschieben Sie den Glasdeckel, so dass etwa 1 mm des Deckglases über dem Rand des Rutschens liegt.
- Gießen Sie 5 ml flüssige Agarose-Lösung auf das Glasdeckel, gerade genug, um es vollständig zu bedecken, und legen Sie einen zweiten 2 x 3 "Glasschieber auf die Oberseite der Montage, um den flüssigen Agar zwischen dem Deckglas und der Folie zu" sandwich ".
HINWEIS: Dies steuert die Tiefe der Agarose, so dass die Gesamtdicke des Deckglases und der gehärteten Agarose in der Dicke der PDMS-Abstandshalter gleich ist. - Lassen Sie das Glas-Gleit-Deckglas-Agarose-Gel-Glas-Dia-Sandwich so lange einstellen, bis die Agarose-Lösung zu erstarren beginnt. Dann überträgt es in einen kühlschrank. Nach 15 min die überschüssige feste Agarose entfernen und um das Glasdeckel schneiden. Bewegen Sie diese zu einem sauberen Teller und legen Sie es in einen Kühlschrank bis zum Gebrauch.
5. Die Wells mit einem Agarose-beschichteten Deckglas und Imaging abdichten
- Nachdem die Bakterien-Inkubationszeit abgeschlossen ist, entfernen Sie das mit Agarose beschichtete Deckglas aus dem Kühlschrank und bereiten die Silizium-Chips wie folgt vor.
- Dip die Silizium-Chips in Reinstwasser, eine zu einer Zeit, für jeweils 10 s. Setzen Sie sie an ihren Kanten auf ein Labor-Wischen oder Gewebe, bis die meisten der überschüssigen Flüssigkeit aus den Kanten der Chips abgelassen hat.
- Schneiden Sie ein Stück Klebeband, um die Kantenlänge jedes Siliziumchips anzupassen. Legen Sie das Band auf das Parylen, das das Silikon bedeckt, und verwenden Sie es, um schnell die Parylenbeschichtung wegzuschälen.
- ImmedStecke jeden einzelnen geschliffenen Chip ein und platziere jeden Chip so, dass die Microwell-Array-Seite mit der Agarose-beschichteten Seite eines mit Agarose beschichteten Deckglases in Berührung kommt. Achten Sie darauf, den Chip nicht zu bewegen oder zu verschieben, nachdem er die Agarose berührt hat, um das Wachstum von Bakterien außerhalb der Brunnen zu verhindern.
- Legen Sie das zusammengesetzte Mikrotiter-Array / Agarose-Deckglas in den Schieberhalter der Bühnen-Top-Umgebungs-Kontrollkammer auf dem Miscroskop.
- Verwenden Sie Umgebungslicht oder gerichtetes Licht ( zB eine Taschenlampe), um Arrays von Interesse zu lokalisieren. Verwenden Sie die kommerzielle Software, die die automatisierte Bühne steuert, um diese Positionen zu speichern (siehe Tabelle der Materialien). Schalten Sie das Umgebungs- oder gerichtete Licht aus, nachdem die Positionen gespeichert sind.
- In der kommerziellen Software öffnen Sie das "ND Acquisition" Panel.
HINWEIS: Dieses Panel enthält ein Menü zum automatischen Speichern in ein bestimmtes Verzeichnis sowie eine programmierbare Bilderfassung. Für diese Experimente, Werden die "Time", "XY" und "λ" Menüs verwendet. - Um die Standorte in der Software zu speichern, klicke auf das "XY-Menü" und sehe dann eine leere Box auf der linken Seite für jede Position, die gespeichert werden soll. Klicken Sie auch auf die Schaltfläche "Include Z".
- Erfassen von Bildern über die Zeit bei den gewünschten Wellenlängen und 10 Vergrößerung mit den entsprechenden Fluoreszenzfilterwürfeln (siehe Tabelle der Materialien).
- Verwenden Sie die Steuerungssoftware und die gespeicherten Arraypositionen, um zu jedem gespeicherten Ort zu gelangen und sich auf die Brunnen zu konzentrieren. Klicken Sie auf jede XY-Position in der gespeicherten Liste und stellen Sie den Fokus mit dem Green Flourescence Protein (GFP) -Filter ein. Speichern Sie die neue z-Position, indem Sie auf den Pfeil klicken, der auf den z-Standort zeigt.
HINWEIS: Dieser Vorgang kann zeitaufwändig sein. Berücksichtigen Sie die Vorsicht, die Verstärkung zu erhöhen und den Neutral-Dichte-Filter zu verwenden, um die Lichtintensität zu reduzieren, um das Photobleichen zu verhindern. - Bestimmen Sie den Abstand der z-Achse zwischen den Fokalebenen für jede Wellenlänge, indem Sie die Differenz in der z-Achsenposition notieren, wenn sie auf die Oberfläche des Arrays fokussiert sind. Wählen Sie 2-3 Stellen aus dem Array mit der gemischten rot / grünen Bakterienpopulation und konzentrieren Sie sich mit dem Red Fluorescence Protein (RFP) Filter.
- Subtrahieren Sie den Abstand zwischen den Brennebenen mit den GFP- und RFP-Fluoreszenzfiltern und fügen Sie die Blendenebeneneinstellung unter dem Menü "λ" hinzu.
HINWEIS: Wenn zum Beispiel das Array im GFP-Kanal in einer Z-Position von 50 μm fokussiert ist und das gleiche Array im RFP-Kanal bei 55 μm fokussiert ist, füge +5 neben der RFP-Optik in der "λ" hinzu "Menü.
- Subtrahieren Sie den Abstand zwischen den Brennebenen mit den GFP- und RFP-Fluoreszenzfiltern und fügen Sie die Blendenebeneneinstellung unter dem Menü "λ" hinzu.
- Beginnen Sie mit der Zeitraffer-Bildaufnahme.
HINWEIS: Für die hier gezeigten Experimente wurden RFP- und GFP-Bilder für jede Array-Position in 30-minütigen Intervallen unter Verwendung einer mehrdimensionalen Bildaufnahme über eine kommerzielle Software erfasst, die steuertDie Kamera, den Verschluss, das Filterrad und die motorisierte Bühne.- Setzen Sie das "Intervall" auf 30 min und die "Dauer des Experiments" bis 24 h unter dem Menü "Zeit". Klicken Sie auf "Jetzt ausführen".
HINWEIS: Wenn die Felder "Time", "XY" und "λ" aktiviert sind, verschiebt das Programm die Bühne, um jeden Ort ( dh die gespeicherten XYZ-Stellen) zu fotografieren, ein Bild in einer Wellenlänge zu machen, die z-Position zu bewegen Um fokale Ebenendifferenzen ( dh Lambda- oder Wellenlängensteuerung) zu berücksichtigen, das zweite Bild zu nehmen, auf die nächste Array-Position (Multipoint) zu gehen und diese in 30-Minuten-Intervallen (Zeitraffer) zu schleifen.
- Setzen Sie das "Intervall" auf 30 min und die "Dauer des Experiments" bis 24 h unter dem Menü "Zeit". Klicken Sie auf "Jetzt ausführen".
- Verwenden Sie die Steuerungssoftware und die gespeicherten Arraypositionen, um zu jedem gespeicherten Ort zu gelangen und sich auf die Brunnen zu konzentrieren. Klicken Sie auf jede XY-Position in der gespeicherten Liste und stellen Sie den Fokus mit dem Green Flourescence Protein (GFP) -Filter ein. Speichern Sie die neue z-Position, indem Sie auf den Pfeil klicken, der auf den z-Standort zeigt.
- Erfassen von Beleuchtungssteuerungsbildern.
HINWEIS: Verwenden Sie die Menüs "ND Acquisition", "Time" und "XY", um Bilder von 4 Standorten zu machen, jeweils 25x.- Nehmen Sie eine Reihe von 100 "Dunkelfeld" -Bilder, indem Sie alle Lichtquellen ausschalten und nehmenEin "Bild" einer Standardfolie. Diese Bilder erfassen das Kameralärm. Verwenden Sie die längste Belichtungszeit, die während des Zeitraffens verwendet wird (Schritt 5.3.3).
- Nehmen Sie eine Reihe von 100 "Beleuchtungsfeld" -Bilder, indem Sie eine Standardfolie ( dh einheitliche RFP- oder GFP-Intensität) an wenigen verschiedenen Stellen abbilden, um die ungleichmäßige Beleuchtung bei den gegebenen experimentellen Bedingungen zu erfassen. Wählen Sie eine Belichtungszeit, die das Signal maximiert, ohne die Sättigung zu erreichen.
6. Analyse
- Verarbeiten Sie die Bildstapel mit einer Bildanalyse-Software ( zB ImageJ).
- Konvertieren Sie die erworbenen Bilder in das TIFF-Dateiformat mit der kommerziellen Software. Laden Sie Bilder in die Bildanalyse-Software, indem Sie auf "Datei"> "Importieren"> "Bildsequenz" klicken.
- Erstellen Sie ein "Korrekturbild", indem Sie alle Bilder "Dunkelfeld" und "Beleuchtungsfeld" SubtracT das durchschnittliche "Dunkelfeld" -Bild aus dem durchschnittlichen "Beleuchtungsfeld" -Bild, indem du "Prozess"> "Bildrechner" wählst. Wählen Sie die beiden Bilder "Image1" und "Image2" und dann "Subtrahieren" im Feld "Operation". OK klicken."
- Für die Mittelung, laden Sie die Korrektur- (oder Dunkelfeld-) Bilder, klicken Sie auf "Bild"> "Stapel"> "Z-Projekt"> "Durchschnittliche Projektion".
- Führen Sie ggf. die Bildregistrierung durch. Dann führen Sie die Hintergrund-Subtraktion durch, indem Sie auf "Process"> "Subtract Background" klicken. Geben Sie im Feld "Radius" einen Radius ( zB 125) ein und wählen Sie "Schiebeparaboloid".
- Führen Sie die Beleuchtungskorrektur mit "Process"> "Calculator Plus" durch. Wählen Sie die folgenden Parameter: Operation, divide; I1, gut Bild; I2, Korrekturbild; K1, Korrekturbildmittel; Und k2, 0. Klicken Sie auf "CreAß neues Fenster. "
HINWEIS: Dieser Datensatz erforderte keine Registrierung, aber bei anderen Arbeiten wurde das ImageJ Plugin StackReg mit der Transformation "Übersetzung" verwendet. Für die Hintergrund-Subtraktion verwenden Sie für jeden Bildsatz denselben gleitenden Paraboloid-Radius. Zum Beispiel, wenn die größten gut abgebildeten Brunnen einen Pixelradius von 100 haben , verwenden Sie für jeden Bildsatz einen Radius größer als 100 ( zB 125).
- Bestimmen Sie das Wachstum jedes Stammes in den Mikrovertiefungen.
- Wählen Sie die Bereiche von Interesse (ROIs) um jede microwell in den gewünschten Arrays mit dem ImageJ "MicroArray" Plugin.
- Klicken Sie im Menü "MAP" auf "Raster zurücksetzen". Geben Sie Zeilen, Spalten und Durchmesser an (basierend auf der Größe und der Anzahl auf dem Array, siehe Abbildung 1b ). Wählen Sie "Kreis" aus dem Menü "ROI Form".
- Halten Sie die "Alt" -Taste gedrückt, während Sie den obersten linken ROI mit der Maus auswählen, um t zu bewegenEr ROI Array. Halten Sie die "Shift" -Taste gedrückt, während Sie den unteren ROI auswählen, um die Größe des Arrays zu ändern. Halten Sie die "Shift" -Taste gedrückt, während Sie einen ROI von der rechten Seite des Arrays auswählen, aber nicht an den Ecken, um den Abstand der ROIs zu ändern.
- Verwenden Sie die obigen Befehle, um das ROI-Array über die Wells in einem Bild zu platzieren. Klicken Sie auf "RT messen".
HINWEIS: Das Plugin exportiert die gewünschten Messungen von jedem ROI. Verwenden Sie drei ROI-Größen, um konzentrische Ringe um die Vertiefungen zu erzeugen, um lokal das Hintergrundsignal zu sammeln ( dh das Signal vom mittleren Ring, das vom äußeren Ring subtrahiert wird) und Fluoreszenzmessungen ( dh das Signal vom inneren Ring).
- Sammeln Sie die Daten in einer Tabellenkalkulationssoftware und berechnen Sie das Hintergrundsignal. Importieren Sie es in eine benutzerdefinierte Scripting-Software für weitere Analysen.
- Wählen Sie die Bereiche von Interesse (ROIs) um jede microwell in den gewünschten Arrays mit dem ImageJ "MicroArray" Plugin.
- Datenorganisation und -analyse
- Importieren Sie die Daten und organisieren Sie die DatenGesammelt in ImageJ in eine Matrix in der folgenden Reihenfolge für alle Zeiten: Spalte 1, Sub-Array-Nummer; Spalte 2, Brunnenreihe; Spalte 3, Brunnensäule; Spalte 4, mittlere Intensität; Spalte 5, Hintergrundintensität; Und Spalte 6, mittlere Intensität - Hintergrundintensität.
- Trennen Sie die Ergebnisse der mCherry- und GFP-Akquisition in verschiedene Matrizen. Speichern Sie die Ergebnisse aus jedem Sub-Array und jede Farbe in einer anderen Zelle in einem Zellen-Array.
HINWEIS: Diese Organisation macht es einfacher, zwischen Bilddaten und Messergebnissen hin und her zu gehen, die Daten zu reinigen und sicherzustellen, dass die Messungen die Daten genau darstellen.
- Trennen Sie die Ergebnisse der mCherry- und GFP-Akquisition in verschiedene Matrizen. Speichern Sie die Ergebnisse aus jedem Sub-Array und jede Farbe in einer anderen Zelle in einem Zellen-Array.
- Anpassung für die Autofluoreszenz von P. aeruginosa .
HINWEIS: In Experimenten, die die Co-Kultur von GFP- und mCherry-Stämmen beinhalten, sollte ein mCherry-only-Chip analysiert werden, um die Beziehung zwischen mCherry und grüner Autofluoreszenz zu ermitteln.- Zeichnen Sie das mCherry-versus-GFP-Signal von allen mCherry ΔretS &# 916; tse / i1-6 Wells zu allen Zeitpunkten, um die Beziehung zwischen dem mCherry-Signal und der Autofluoreszenz im GFP-Kanal zu bestimmen. Subtrahieren Sie das Autofluoreszenzsignal von den Co-Kulturen.
- Zeichnen Sie die Trajektorien und passen Sie eine modifizierte Logistikgleichung an jede Trajektorie an, um Parameter zu extrahieren, die Kleinste Quadrate verwenden, die entweder in einer Tabellenkalkulationssoftware oder einer benutzerdefinierten Scripting-Software passen.
- Suchen Sie nach Korrelationen zwischen und zwischen GFP- und mCherry-Trajektorienparametern.
- Importieren Sie die Daten und organisieren Sie die DatenGesammelt in ImageJ in eine Matrix in der folgenden Reihenfolge für alle Zeiten: Spalte 1, Sub-Array-Nummer; Spalte 2, Brunnenreihe; Spalte 3, Brunnensäule; Spalte 4, mittlere Intensität; Spalte 5, Hintergrundintensität; Und Spalte 6, mittlere Intensität - Hintergrundintensität.
Ergebnisse
Die hier vorgestellte experimentelle Plattform ist für Hochdurchsatz- und Hochinhaltensstudien von Bakteriengemeinschaften ausgelegt. Das Design ermöglicht es Tausende von Gemeinden, die in Brunnen verschiedener Größen wachsen, gleichzeitig analysiert werden. Mit diesem Microwell-Array-Design kann die Abhängigkeit der endgültigen Gemeinschaftszusammensetzung auf den anfänglichen Seeding-Dichten, der Well-Größe und der chemischen Umgebung bestimmt werden. Diese Arbeit zeigt das W...
Diskussion
Dieser Artikel präsentierte ein microwell-Array-Gerät und experimentelle Protokolle entwickelt, um High-Throughput-und High-Content-Live-Zelle Imaging-basierte Analyse der bakteriellen Community-Entwicklung zu ermöglichen. Während der Schwerpunkt der Demonstration hier war, um die Effekte der kontaktvermittelten Typ-VI-Sekretion auf die Entwicklung der Gemeinschaft zu untersuchen, wurden die Arrays so konzipiert, dass sie flexibel sind und die Untersuchung einer breiten Palette von mikrobiellen Gemeinschaften und Mi...
Offenlegungen
Die Autoren haben nichts zu offenbaren.
Danksagungen
Microwell Arrays wurden hergestellt und charakterisiert im Zentrum für Nanophase Materials Sciences User Facilities Division, Office of Basic Energy Sciences, US Department of Energy. Die finanzielle Unterstützung für diese Arbeit wurde durch den Forschungs- und Entwicklungsfonds des Oak Ridge National Laboratory Director Die Autoren danken auch dem J. Mougous Laboratory (University of Washington, Seattle, WA) für die Lieferung von P. aeruginosa- Stämmen, die in diesen Studien verwendet wurden.
Materialien
| Name | Company | Catalog Number | Comments |
| Parylene N | Specialty Coating Systems | CAS NO.:1633-22-3 | |
| Parylene coater | Specialty Coating Systems | Labcoter 2 Parylene Deposition Unit PDS2010 | |
| Silicon Wafer | WRS Materials | 100mm diameter, 500-550μm thickness, Prime, 10-20 resistivity, N/Phos<100>, | |
| adhesion promoter | Shin-Etsu Microsci | MicroPrime P20 adhesion promoter | |
| postive tone photoresist | Rohm and Haas Electronics Materials LLC (Owned by Dow) | Microposit S1818 Positive Photoresist (code 10018357) | |
| Quintel Contact Aligner | Neutronix Quintel Corp | NXQ 7500 Mask Aligner | |
| Reactive Ion Etching Tool | Oxford Instruments | Plasmalab System 100 Reactive Ion Etcher | |
| R2A Broth | TEKnova | R0005 | |
| Bovine Serum Albumin | Sigma | A9647 | |
| Multimode Plate Reader | Perkin Elmer | Enspire, 2300-0000 | |
| Fluorescent Microscope | Nikon | Eclipse Ti-U | |
| Automated Stage | Prior | ProScan III | |
| CCD camera | Nikon | DS-QiMc | |
| Stage-top environmental control chamber | In Vivo Scientific | STEV ECU-HOC | |
| Phosphate Buffered Saline | ThermoFisher Scientific | 14190144 | |
| UltraPure Agarose | ThermoFisher Scientific | 16500500 | |
| 25 x 75 mm No. 1.5 coverslip | Nexterion | High performance #1.5H coverslips | |
| Fluorescence Reference Slides | Ted Pella | 2273 | |
| Physical Stylus Profilometer | KLA Tencor | P-6 | |
| lab wipes | Kimberly Clark | Kimipe KIMTECH SCIENCE Brand, 34155 | |
| commercial software | Nikon | NIS Elements | |
| Zeiss 710 Confocal Microscope | Zeiss | ||
| filter cubes | Nikon | Nikon FITC (96311), Nikon Texas Red(96313) |
Referenzen
- Zhou, J., Deng, Y., et al. Stochasticity, succession, and environmental perturbations in a fluidic ecosystem. Proc Natl Acad Sci. 111, E836-E845 (2014).
- Valm, A. M., Welch, J. L. M., et al. Systems-level analysis of microbial community organization through combinatorial labeling and spectral imaging. Proc Natl Acad Sci USA. 108 (10), 4152-4157 (2011).
- Satoh, H., Miura, Y., Tsushima, I., Okabe, S. Layered structure of bacterial and archaeal communities and their in situ activities in anaerobic granules. Appl Environ Microbiol. 73 (22), 7300-7307 (2007).
- Kim, H. J., Boedicker, J. Q., Choi, J. W., Ismagilov, R. F. Defined spatial structure stabilizes a synthetic multispecies bacterial community. Proc Natl Acad Sci USA. 105 (47), 18188-18193 (2008).
- Nunan, N., Wu, K., Young, I. M., Crawford, J. W., Ritz, K. Spatial distribution of bacterial communities and their relationships with the micro-architecture of soil. FEMS Microbiol Ecol. 44, 203-215 (2003).
- Grundmann, G. L. Spatial scales of soil bacterial diversity - The size of a clone. FEMS Microbiol Ecol. 48, 119-127 (2004).
- Langenheder, S., Lindstrom, E. S., Tranvik, L. J. Structure and Function of Bacterial Communities Emerging from Different Sources under Identical Conditions. Appl Environ Microbiol. 72 (1), 212-220 (2006).
- Camp, J. G., Kanther, M., Semova, I., Rawls, J. F. Patterns and Scales in Gastrointestinal Microbial Ecology. Gastroenterology. 136 (6), 1989-2002 (2009).
- Renner, L. D., Weibel, D. B. Physicochemical regulation of biofilm formation. MRS Bull. 36 (5), 347-355 (2011).
- Wessel, A. K., Hmelo, L., Parsek, M. R., Whiteley, M. Going local: technologies for exploring bacterial microenvironments. Nat Rev Microbiol. 11 (5), 337-348 (2013).
- Stacy, A., McNally, L., Darch, S. E., Brown, S. P., Whiteley, M. The biogeography of polymicrobial infection. Nat Rev Microbiol. 14 (2), 93-105 (2015).
- Hansen, R. R., Shubert, K. R., Morrell-Falvey, J. L., Lokitz, B. S., Doktycz, M. J., Retterer, S. T. Microstructured block copolymer surfaces for control of microbe adhesion and aggregation. Biosensors. 4 (1), 63-75 (2014).
- Hansen, R. R., Hinestrosa, J. P., et al. Lectin-functionalized poly(glycidyl methacrylate)- block -poly(vinyldimethyl azlactone) surface scaffolds for high avidity microbial capture. Biomacromolecules. 14 (10), 3742-3748 (2013).
- Timm, C. M., Hansen, R. R., Doktycz, M. J., Retterer, S. T., Pelletier, D. A. Microstencils to generate defined, multi-species patterns of bacteria. Biomicrofluidics. 9 (6), (2015).
- Keymer, J. E., Galajda, P., Muldoon, C., Park, S., Austin, R. H. Bacterial metapopulations in nanofabricated landscapes. Proc Natl Acad Sci USA. 103 (46), 17290-17295 (2006).
- Zhang, Q., Lambert, G., et al. Acceleration of Emergence of Bacterial Antibiotic Resistance in Connected Microenvironments. Science. 333 (6050), 1764-1767 (2011).
- Friedlander, R. S., Vlamakis, H., Kim, P., Khan, M., Kolter, R., Aizenberg, J. Bacterial flagella explore microscale hummocks and hollows to increase adhesion. Proc Natl Acad Sci USA. 110 (14), 5624-5629 (2013).
- Zhou, J., Liu, W., et al. Stochastic Assembly Leads to Alternative Communities with Distinct Functions in a Bioreactor Microbial Community. MBio. 4 (2), 1-8 (2013).
- van Vliet, S., Hol, F. J., Weenink, T., Galajda, P., Keymer, J. E. The effects of chemical interactions and culture history on the colonization of structured habitats by competing bacterial populations. BMC Microbiol. 14 (1), 116 (2014).
- Niepa, T. H. R., Hou, L., et al. Microbial Nanoculture as an Artificial Microniche. Sci Rep. 6, 30578 (2016).
- Hansen, R. H., Timm, A. C., et al. Stochastic Assembly of Bacteria in Microwell Arrays Reveals the Importance of Confinement in Community Development. PLoS ONE. 11 (5), e0155080 (2016).
- Hood, R. D., Singh, P., et al. A Type VI Secretion System of Pseudomonas aeruginosa Targets a Toxin to Bacteria. Cell Host Microbe. 7 (1), 25-37 (2010).
- LeRoux, M., Ja De Leon, ., et al. Quantitative single-cell characterization of bacterial interactions reveals type VI secretion is a double-edged sword. Proc Natl Acad Sci. 109 (48), 19804-19809 (2012).
- Whitney, J. C., Beck, C. M., et al. Genetically distinct pathways guide effector export through the type VI secretion system. Mol Microbiol. 92 (3), 529-542 (2014).
- Warrick, J. W., Timm, A., Swick, A., Yin, J. Tools for Single-Cell Kinetic Analysis of Virus-Host Interactions. PLoS ONE. 11 (1), e0145081 (2016).
- Zwietering, M. H., Jongenburger, I., Rombouts, F. M., Van't Riet, K. Modeling of the Bacterial Growth Curve. Appl Environ Microbiol. 56 (6), 1875-1881 (1990).
- Halsted, M., Wilmoth, J. L., et al. Development of transparent microwell arrays for optical monitoring and dissection of microbial communities. J Vac Sci Technol B Nanotechnol Microelectron. 34 (6), 06KI03 (2016).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten