
Для просмотра этого контента требуется подписка на Jove Войдите в систему или начните бесплатную пробную версию.
Method Article
Сборка и отслеживание развития микробного сообщества в рамках платформы массивов микролистов
В этой статье
Резюме
Развитие микробных сообществ зависит от сочетания факторов, включая экологическую архитектуру, численность членов, черты и взаимодействия. В этом протоколе описывается синтетическая микроструктурированная среда для одновременного отслеживания тысяч сообществ, содержащихся в ямах фемтолитера, где могут быть аппроксимированы ключевые факторы, такие как размер ниши и ограничение.
Аннотация
Развитие микробных сообществ зависит от сочетания сложных детерминированных и стохастических факторов, которые могут кардинально изменить пространственное распределение и деятельность членов сообщества. Мы разработали платформу массива microwell, которая может использоваться для быстрой сборки и отслеживания тысяч бактериальных сообществ параллельно. В этом протоколе подчеркивается полезность платформы и описывается ее использование для оптического мониторинга развития простых сообществ из двух членов в составе массивов в рамках платформы. Эта демонстрация использует два мутанта Pseudomonas aeruginosa , часть серии мутантов, разработанных для изучения патогенности секреции типа VI. Хромосомные вставки генов mCherry или GFP облегчают конститутивную экспрессию флуоресцентных белков с четкими длинами волн излучения, которые могут использоваться для мониторинга численности и местоположения членов сообщества в каждом микролухе. Этот протокол описывает подробный методD для сборки смесей бактерий в лунки массива и использования временной флуоресцентной визуализации и количественного анализа изображений для измерения относительного роста популяции каждого члена с течением времени. Посев и сборка платформы microwell, процедуры визуализации, необходимые для количественного анализа микробных сообществ внутри массива, и методы, которые могут быть использованы для выявления взаимодействий между областью микробных видов, обсуждались.
Введение
Микробные сообщества формируются как детерминированными факторами, такими как структура окружающей среды, так и стохастическими процессами, которые связаны с гибелью клеток, делением, концентрацией белка, количеством органелл и мутацией 1 . В естественной среде практически невозможно проанализировать индивидуальное воздействие этих влияний на состав и деятельность сообщества. Замаскированные естественными структурами и погребенные в химической и биологической среде, выявление членов сообщества и дальнейшее разрешение их пространственно-временного распределения в естественной среде чрезвычайно сложны. Тем не менее, недавние усилия подчеркнули важность пространственной организации для функционирования сообщества и указывают на необходимость учета как численности, так и организации в текущих исследованиях 2 , 3 , 4 .
ЭтоЯсно, что локальная химическая среда ( то есть доступность питательных веществ и вторичных метаболитов), физическая структура ( например, почвенная архитектура, корни растений, частицы океана или кишечные микроворсинки), наличие или отсутствие кислорода и введение Патогенные виды влияют на состав, архитектуру и функцию микробных сообществ 5 , 6 , 7 , 8 , 9 , 10 , 11 . Тем не менее традиционные методы для культур, которые пренебрегают этим фактором, продолжают преобладать. Состав сообщества ( например, наличие со-зависимых видов), физическое прикрепление, концентрация сигнальной молекулы и прямой контакт клеток-клеток являются важными факторами формирования микробного сообщества и могут быть потеряны в cОбщепринятые условия культивирования. Эти свойства трудно реплицировать в объемной жидкой культуре или на агаровой пластине. Тем не менее, наличие микрофлюидных, микронапряжений и технологий нанообработки, которые позволяют реплицировать ключевые физические и химические свойства природных сред, позволило многим исследователям построить сообщества бактерий для изучения их взаимодействий 12 , 13 , 14 и разработать синтетические среды, которые Имитировать природные условия 4 , 15 , 16 , 17 , 18 , 19 , 20 .
В этом протоколе описан способ изготовления устройства массива микроволн и подробные экспериментальные процедуры, которые могут быть использованы для функционализацииE колодцев в массиве и выращивать бактерии, как колонии одного вида, так и в сообществах с несколькими членами. Эта работа также демонстрирует, как бактерии, модифицированные для получения флуоресцентных репортерных белков, могут использоваться для мониторинга роста бактерий в лунках с течением времени. Аналогичный массив был представлен ранее и показал, что можно отслеживать рост одновидовых колоний Pseudomonas aeruginosa ( P. aeruginosa) в микролунках. Путем модуляции плотности и плотности посева начальные условия тысяч экспериментов по росту могут варьироваться параллельно, чтобы определить, как исходные условия инокуляции влияют на способность бактерий расти 21 . Текущая работа использует слегка измененную версию массива микроячейки, которая основывается на предыдущей работе, позволяя одновременное сравнение нескольких массивов и используя более надежный экспериментальный протокол. Массив, используемый в этой работе, содержит несколько подмассивов или массивСодержащие лунки разного размера, от 15 до 100 мкм в диаметре, которые расположены на трех разных смолах ( т.е. 2x, 3x и 4x диаметр скважины). Массивы выгравированы в кремнии, а рост бактерий, засеянных в кремниевых массивах, обеспечивается запечатыванием массивов с покровным стеклом, покрытым агарозным гелем с средней интенсивностью. В этой демонстрации используются мутанты P. aeruginosa, предназначенные для изучения системы секреции типа VI.
Представленные здесь результаты направлены на достижение конечной цели анализа сообществ многочленов в массивах матриц микроволн, что позволяет исследователям контролировать численность и организацию бактерий in situ , контролируя и исследуя химическую среду. Это должно в конечном итоге дать представление о «правилах», которые регулируют развитие и преемственность сообщества.
протокол
1. Изготовление матрицы из микроволнового кремния
- Париленовое покрытие
- Депозит между 1-1,5 мкм парилена N на кремниевых пластинах с использованием имеющейся в продаже системы покрытия парилена в соответствии с техническими условиями и инструкциями производителя (настройки: заданное значение испарителя = 160 ° C, заданное значение печи = 650 ° C).
ПРИМЕЧАНИЕ. Приблизительно 6 г парилена N, загруженного в камеру, дает покрытия толщиной 1-1,5 мкм.
- Депозит между 1-1,5 мкм парилена N на кремниевых пластинах с использованием имеющейся в продаже системы покрытия парилена в соответствии с техническими условиями и инструкциями производителя (настройки: заданное значение испарителя = 160 ° C, заданное значение печи = 650 ° C).
- фотолитография
- Спиновое покрытие париленовых N-покрытых пластин с адгезионным промотором, 20% гексаметилдисилазаном (HMDS) и 80% ацетатом монометилового эфира пропиленгликоля (PGMEA) (см. Таблицу материалов ) при 3000 об / мин в течение 45 с. Заполните 2 мл переносной пипетки промотором адгезии и посыпьте ее по всей пластине. Дайте вафельной плите сидеть в течение приблизительно 10 с, прежде чем высушить ее.
- Заполните 2 мл пипетки с положительнымNe фоторезист (см. Таблицу материалов ) и распределите фоторезист в центре пластины. Спин при 3000 об / мин в течение 45 с, чтобы получить покрытие сопротивлением, которое составляет приблизительно 1,5 мкм.
- Мягко выпекать образцы на конфорке при 115 ° C в течение 1 мин.
- Используйте контактный выравниватель и фотомаску с желаемым рисунком лунки, чтобы подвергнуть образец ультрафиолетовому излучению. Экспонируйте спин-покрытую пластину через узорчатую фотомаску в течение 6 с, давая приблизительную дозу 60-80 мДж / см 2, измеренную при 365 нм.
- Разработайте образец, погрузив образец в проявитель (<3% гидроксид тетраметиламмония в воде, см. Таблицу материалов ) в течение 2 мин. Промыть водой DI и сушить чистым сухим азотом.
ПРИМЕЧАНИЕ. Области фоторезиста, подвергающиеся воздействию УФ, должны быть очищены во время разработки.
- Реактивное ионное травление
- Используйте плазменную травление кислородной плазмы для удаления обнаженного париленаВплоть до кремниевой подложки.
ПРИМЕЧАНИЕ. Рецепт можно модулировать, чтобы изменить скорость травления парилена. Для толщины парилена от 1 до 5 мкм используйте рецепт с 60 мТорр, 20 ° C, 100 сccm O 2 , 10 W RF и 2000 Вт ICP на инструменте реактивного ионного травления (RIE). После травления и удаления открытого слоя парилена узорчатая область ( то есть облученный кремний) должна выглядеть блестящей и серебристой. - Используйте глубокий процесс RIE (DRIE, например, Bosch DRIE) для травления в кремнии.
ПРИМЕЧАНИЕ. Скорость и продолжительность травления определяют глубину скважины. Один полный цикл процесса Bosch (шаг осаждения 3 с: 20 мТорр, 15 ° С, 140 см 3 C 4 F 8 , 10 Вт RF и 1750 Вт ICP с последующим 10-летним процессом травления: 20 мТорр, 15 ° C , 120 sccm SF 6 , 8 W RF и 1 750 W ICP) соответствует приблизительно 1 мкм глубины травления. Колодцы, используемые в этой демонстрации, составляют от 3 до 3,5 мкм. - ВУточнить глубину травления, используя физическую профилометрию.
- Загрузите образец в физический профилометр (см. Таблицу материалов ).
- Включите вакуум образца и нажмите кнопку ручной загрузки.
- Сфокусируйте систему на образце, нажав кнопку «Фокус». Расположите соответствующую функцию для измерения на экране просмотра.
- Сканирование образца. Выровняйте профиль и измерьте глубину функции.
- Запишите скорость травления и модулируйте последующие времена травления для достижения желаемой глубины.
ПРИМЕЧАНИЕ. Измерения будут включать глубину кремниевой скважины, толщину осажденного парилена и толщину фоторезиста. Проверка толщины каждого слоя в течение всей процедуры необходима для достижения точной глубины скважины.
- Используйте плазменную травление кислородной плазмы для удаления обнаженного париленаВплоть до кремниевой подложки.
2. Бактериальная культура и сеяние ( рис. 1а )
- Начинайте колонии на Лурийском бульоне (LB) агаровые пластины из глицериновых запасов и использовать в течение двух недель. Выбирайте колонии желаемых штаммов из пластин агара LB и начинайте ночные культуры P. aeruginosa. Инкубируйте ночные культуры в течение приблизительно 18 ч при 37 ° С при встряхивании со скоростью 220 об / мин в среде R2A.
ПРИМЕЧАНИЕ. Колонии следует собирать в течение двух недель после нанесения покрытия, чтобы гарантировать, что мутации и флуоресцентные репортерные гены сохранены. Все работы P. aeruginosa должны выполняться в условиях BSL-2. - Используйте алмазный писец, чтобы разрезать кремниевую пластину на отдельные чипы, содержащие ансамбли разных размеров и массивы высоты тона. Убедитесь, что каждый чип содержит полный набор размеров и смол для исследования.

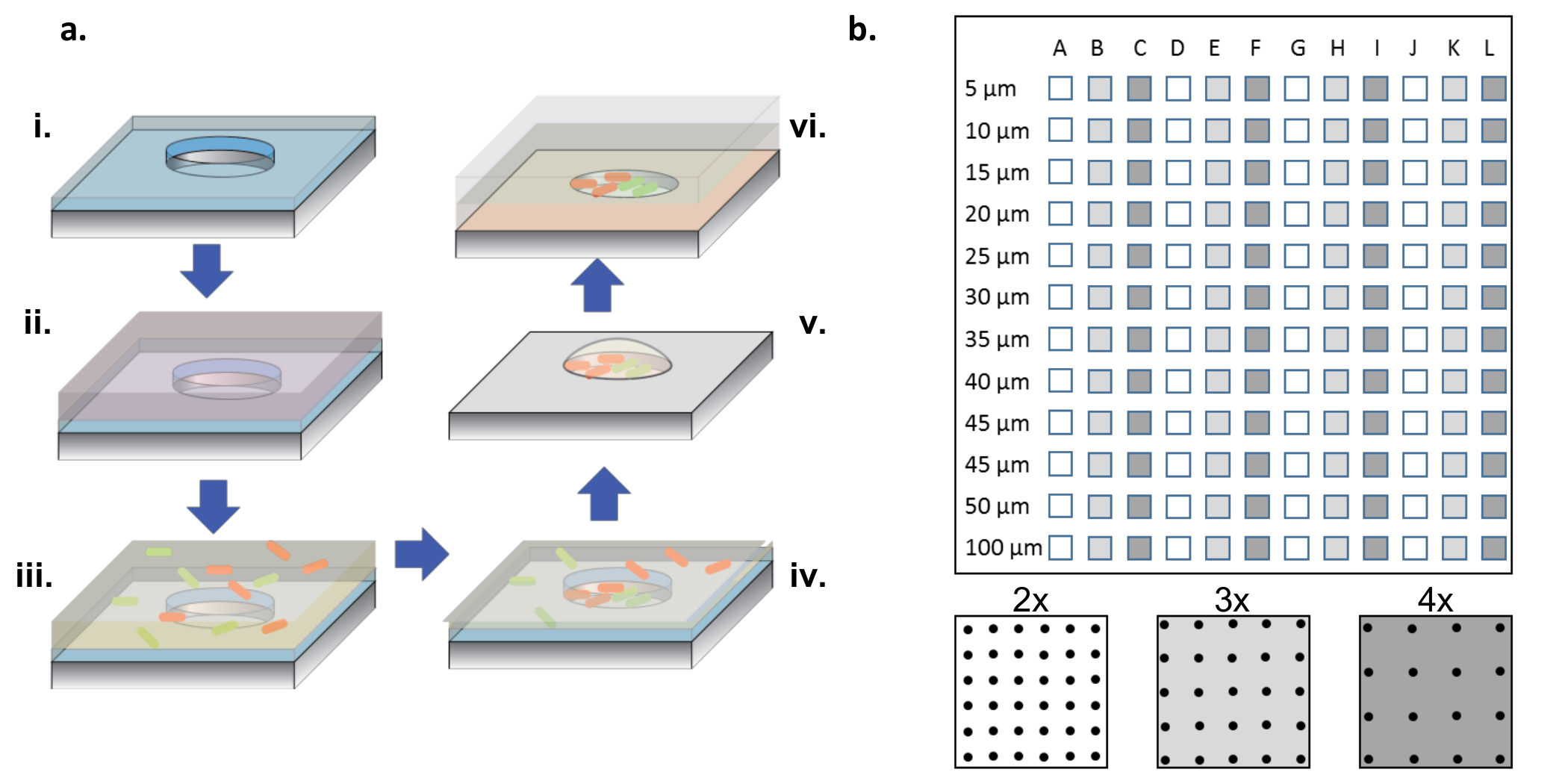
Рисунок 1: Процесс изготовления и сортировки ячеек. ( A ) Массивы MicrowellРектифицированных в кремниевые пластины, покрытые тонким слоем парилена (i). Для смачивания лунок и / или функционализации поверхности раствор белка добавляется в каплю поверх массивов (ii). Раствор белка удаляют, вафли сушат и добавляют новый раствор, содержащий искомые бактерии (iii). Бактериальный раствор удаляют после инкубационного периода, и вафли дают высохнуть, оставляя бактерии в лунках и на поверхности (iv). Поверхностно-ассоциированные бактерии удаляются с выделением парилена, оставляя позади бактерии, засеянные чисто в микролунках, и все еще жизнеспособны из-за 2% -ной глицериновой среды, что помогает удерживать лунки гидратированными (v). Затем кремниевые чипы размещают на стороне массива на покрытом агарозным гелеобразным стеклянным покровным стеклом, который питает бактериальный рост в микролунах (vi). ( Б ) Макет поддиапазонов на одном кремниевом устройстве. Каждый суб-массив содержит набор одинаковых скважин. Диаметр микролунков по всем суб-аДиапазоны в диапазоне от 5 до 100 мкм и организованы в 2x, 3x или 4x шаг диаметра скважины, который обозначен белым или темно-серым цветом на нижней панели. Когда глубина скважины неглубокая (<10 мкм), диаметры отверстий 5 и 10 мкм редко используются, как правило, из-за отсутствия клеток, колонизирующих эти очень маленькие лунки. В этой работе были проанализированы только данные из скважин диаметром 15-100 мкм. Нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
ПРИМЕЧАНИЕ. Как показано на рисунке 1b , полный чип содержит подвалы колодцев с диаметром от 5 до 100 мкм с тремя разными смолами ( т.е. 2x, 3x и 4x диаметр), повторяющимися 4 раза.
- Поместите 150 мкл капли 500 мкг / мл альбумина бычьей сыворотки (BSA) в раствор PBSНа верхней части массива, чтобы намочить микролунки. Инкубируйте раствор BSA в течение 1 часа на чипе при комнатной температуре во влажной камере.
- Создайте камеру, заполнив дно пустого наконечника пипетки с помощью фосфатного буферного раствора (PBS).
ПРИМЕЧАНИЕ. Другие вещества, такие как конкретные лектины, могут использоваться вместо BSA для функционализации поверхности микролунов.
- Создайте камеру, заполнив дно пустого наконечника пипетки с помощью фосфатного буферного раствора (PBS).
- При инкубации кремниевых чипов с раствором BSA центрифугируйте культуры при 2500 об / мин (что соответствует среднему значению 950 × g) в течение 5 мин, а затем ресуспендируют их в 500 мкл свежей среды R2A с 2% глицерином.
- Определите OD культуры с использованием УФ-пористого спектрометра при 600 нм. Отрегулируйте его до OD 0,02, используя 2% глицерин R2A.
ПРИМЕЧАНИЕ. Глицерин помогает предотвратить высыхание колодцев во время отрыва парилена.
- Определите OD культуры с использованием УФ-пористого спектрометра при 600 нм. Отрегулируйте его до OD 0,02, используя 2% глицерин R2A.
- После инкубации удалите раствор BSA и промойте 3 раза с помощью PBS, удалив и заменив каплю жидкостиT, покрывающий массив кремниевых микроячейков. Высушивают в атмосфере азота.
- Добавьте 150 мкл 0.02 OD культур в каждый сухой массив, расположенный во влажной камере. Инкубируйте в течение 1 часа при 4 ° C, чтобы бактерии могли прилипать к стенкам колодца.
ПРИМЕЧАНИЕ. Для инкубации охлаждения не требуется. Время инкубации 4 ° C можно использовать для предотвращения роста бактерий до начала изображения, чтобы можно было визуализировать пространственную организацию сообществ до роста. Также можно использовать инкубацию при комнатной температуре. Оба протокола приводят к аналогичным кривым роста.
3. Настройка микроскопа
- Перед началом бактериальной инкубации на кремниевых чипах включите верхнюю камеру контроля окружающей среды (см. Таблицу материалов) и отрегулируйте настройки на блоке управления так, чтобы влажность (~ 100%) и температура (30-32 ° C, см. Шаг 3.2) может уравновешиваться перед добавлением образцов.
- Выровняйте держатель образца иTerior вокруг образца с пропитанными PBS лабораторными салфетками (см. Таблицу материалов ) для увеличения влажности в камере до точки росы. Установите температуру камеры на 30 ° C и температуру крышки камеры до 32 ° C, чтобы уменьшить конденсацию на плоскости формирования изображения.
ПРИМЕЧАНИЕ. Держатель слайда входит в камеру с жидкими ячейками с прокладкой толщиной около 1 см. Держатель образца выравнивается с помощью уровня пузырьков, который помещается поверх держателя образца. Держатель образца может слегка наклоняться и оставаться герметичным в прокладке до уровня. - В то время как культуры инкубируются на кремниевых чипах, вручную включите выключатель питания для ртутной лампы по крайней мере за 30 минут до изображения. Вручную включите камеру и станцию микроскопа. Откройте программное обеспечение, используемое для управления микроскопом и периферийным оборудованием, и убедитесь, что оборудование распознано программным обеспечением.
ПРИМЕЧАНИЕ. Увеличение составляет 10X с NA = 0,3.
- Микроволновые предварительно приготовленные растворы агарозы ( т.е. 2% агарозы в среде R2A) до достижения жидкого состояния примерно 60 с.
- Намочите заднюю часть стеклянного покровного стекла размером 75 мм x 22 мм, 1,5 дюйма с этанолом и поместите его по длине, по центру, через стеклянный слайд размером 2 х 3 дюйма (50 х 75 мм). Поместите две прокладки PDMS (толщина ~ 1 мм) По длинным краям покровного стекла и сдвиньте покровное стекло, чтобы примерно 1 мм покровного стекла нависало над краем слайда.
- Налейте 5 мл жидкого раствора агарозы поверх покровного стекла, достаточно, чтобы полностью покрыть его, и поместите второй стеклянный слайд 2 x 3 дюйма сверху сборки в «сэндвич» жидкого агара между покровным и скользким.
ПРИМЕЧАНИЕ. Это контролирует глубину агарозы, при этом общая толщина покровного стекла и упрочненной агарозы равна толщине прокладок PDMS. - Дать стеклянному слайд-покровно-агарозному гелеобразному стеклянному сэндвичу установить до тех пор, пока раствор агарозы не начнет затвердевать; Затем переведите его в холодильник. Через 15 мин удалите избыток твердой агарозы и обрезайте покровное стекло. Переместите это на чистую посуду и поместите ее в холодильник до использования.
5. Уплотнение колодцев покрытием с агарозным покрытием и визуализацией
- После завершения периода инкубации бактерий удалите покрытое агарозой покровное стекло из холодильника и подготовьте кремниевую стружку следующим образом.
- Окуните кремниевые чипы в сверхчистую воду, по одной за раз, по 10 с каждый. Установите их по краям на протирание или ткань в лаборатории до тех пор, пока большая часть избыточной жидкости не будет слита с краев стружки.
- Отрежьте кусок ленты, чтобы она соответствовала длине края каждого кремниевого чипа. Поместите ленту на парилену, покрывающую кремний, и используйте ее для быстрого отслаивания париленового покрытия.
- IMMEDIally инвертируйте каждый очищенный чип и помещайте каждый чип таким образом, чтобы сторона сопел микроисточника обращена (и контактирует) с покрытой агарозой стороной покрытого агарозой покровного стекла. Старайтесь не перемещать или перемещать чип после того, как он касается агарозы, чтобы предотвратить рост бактерий за пределами колодцев.
- Поместите собранное покровное покрытие микролинии / агарозы в держатель слайдов камеры контроля окружающей среды на микроскопе.
- Используйте окружающий свет или направленный свет ( например, фонарик), чтобы найти интересующие массивы. Используйте коммерческое программное обеспечение, управляющее автоматическим этапом, чтобы сохранить эти позиции (см. Таблицу материалов). Выключите окружающий или направленный свет после сохранения позиций.
- В коммерческом программном обеспечении откройте панель «ND Acquisition».
ПРИМЕЧАНИЕ. Эта панель содержит меню для автоматического сохранения в конкретном каталоге, а также программируемое получение изображения. Для этих экспериментов, Используются меню «Время», «XY» и «λ». - Чтобы сохранить места в программном обеспечении, щелкните «XY menu», а затем проверьте пустое поле с левой стороны для каждой позиции, которую необходимо сохранить. Также нажмите кнопку «Включить Z».
- Приобретайте изображения со временем на требуемых длинах волн и 10 увеличения с использованием соответствующих кубиков фильтра флуоресценции (см. Таблицу материалов).
- Используйте программное обеспечение управления и сохраненные позиции массива для перемещения в каждое сохраненное место и для фокусировки на скважинах. Нажмите на каждое место XY в сохраненном списке и отрегулируйте фокус с помощью фильтра Green Flourescence Protein (GFP). Сохраните новую позицию z, щелкнув стрелку, указывающую на местоположение z.
ПРИМЕЧАНИЕ. Этот процесс может занять много времени. Попробуйте принять меры предосторожности для увеличения коэффициента усиления и использования фильтра нейтральной плотности, чтобы уменьшить интенсивность света, чтобы предотвратить фотообесцвечивание. - Определите расстояние по оси z между фокальными плоскостями для каждой длины волны, отметив разницу в поведении оси z при фокусировке на поверхности массива. Выберите 2-3 места из массива со смешанной популяцией красных / зеленых бактерий и сфокусируйтесь с помощью фильтра Red Fluorescence Protein (RFP).
- Вычтите расстояние между фокальными плоскостями с помощью флуоресцентных фильтров GFP и RFP и добавьте настройку фокальной плоскости в меню «λ».
ПРИМЕЧАНИЕ. Например, если массив выглядит сфокусированным в канале GFP в z-месте 50 мкм, и тот же массив отображается сфокусированным в канале RFP на уровне 55 мкм, добавьте +5 рядом с оптической конфигурацией RFP в «λ ".
- Вычтите расстояние между фокальными плоскостями с помощью флуоресцентных фильтров GFP и RFP и добавьте настройку фокальной плоскости в меню «λ».
- Начните поиск изображений с задержкой по времени.
ПРИМЕЧАНИЕ. Для экспериментов, показанных здесь, изображения RFP и GFP были получены для каждой позиции массива с интервалом 30 минут с использованием многомерного захвата изображения с помощью коммерческого программного обеспечения, которое контролируетКамера, затвор, колесо фильтров и моторизованная ступень.- Установите «интервал» на 30 минут, а «продолжительность эксперимента» - на 24 часа в меню «Время». Нажмите «Запустить сейчас».
ПРИМЕЧАНИЕ. Когда флажки «Время», «XY» и «λ» проверены, запуск программы приведет к перемещению сцены для изображения каждого местоположения ( т.е. сохраненных местоположений XYZ), возьмите изображение на одной длине волны, переместите z-позицию Для учета различий фокальной плоскости ( т. Е. Управления длиной лямбда или длины волны), возьмите второе изображение, перейдите к следующему местоположению массива (многоточечный) и зациклируйте его с интервалом 30 минут (с течением времени).
- Установите «интервал» на 30 минут, а «продолжительность эксперимента» - на 24 часа в меню «Время». Нажмите «Запустить сейчас».
- Используйте программное обеспечение управления и сохраненные позиции массива для перемещения в каждое сохраненное место и для фокусировки на скважинах. Нажмите на каждое место XY в сохраненном списке и отрегулируйте фокус с помощью фильтра Green Flourescence Protein (GFP). Сохраните новую позицию z, щелкнув стрелку, указывающую на местоположение z.
- Приобретите изображения управления освещением.
ПРИМЕЧАНИЕ. Используйте меню «ND Acquisition», «Time» и «XY» для получения изображений из 4-х мест, по 25 раз.- Возьмите серию из 100 изображений «темного поля», отключив все источники света и«Образ» стандартного слайда. Эти изображения будут снимать шум камеры. Используйте наибольшее время экспозиции, используемое во время временной шкалы (шаг 5.3.3).
- Возьмите серию изображений «поля освещенности 100» путем визуализации стандартного слайда ( т. Е. Равномерной интенсивности RFP или GFP) в нескольких разных местах для захвата неравномерного освещения в заданных условиях эксперимента. Выберите время экспозиции, которое максимизирует сигнал без насыщения.
6. Анализ
- Обработка стеков изображений с помощью программного обеспечения для анализа изображений ( например, ImageJ).
- Конвертируйте полученные изображения в формат файла tiff с помощью коммерческого программного обеспечения. Загрузите изображения в программное обеспечение для анализа изображений, нажав «Файл»> «Импорт»> «Последовательность изображений».
- Создайте «Корректирующее изображение», усреднив все изображения «темного поля» и «поля освещенности». SubtracT среднее изображение «темного поля» из среднего изображения «поля освещенности», выбрав «Процесс»> «Калькулятор изображений». Выберите два изображения: «Image1» и «Image2», а затем «Subtract» в поле «Operation». Нажмите «ОК».
- Для усреднения загрузите изображения коррекции (или темного поля), нажмите «Изображение»> «Стеки»> «Проект Z»> «Средняя проекция».
- При необходимости выполните регистрацию изображений. Затем выполните вычитание фона, нажав «Процесс»> «Вычесть фон». Введите радиус ( например, 125) в поле «радиус» и выберите «скользящий параболоид».
- Выполните коррекцию освещенности с помощью «Процесс»> «Калькулятор плюс». Выберите следующие параметры: операция, деление; I1, образ скважины; I2, коррекционное изображение; K1, среднее значение коррекции; И k2, 0. Нажмите «CreСъели новое окно ».
ПРИМЕЧАНИЕ. Этот набор данных не требовал регистрации, но в другой работе ImageJ Plugin StackReg использовался с преобразованием «Перевод». Для вычитания фона используйте один и тот же скользящий радиус параболоида для каждого набора изображений. Например, если изображения с наибольшими лунками имеют радиус пикселя 100, используйте радиус больше 100 ( например, 125) для каждого набора изображений.
- Определите рост каждого штамма в микролунках.
- Выберите области интереса (ROI) вокруг каждого микрошара в желаемых массивах с помощью модуля ImageJ «MicroArray».
- В меню «MAP» нажмите «Сбросить сетку». Укажите строки, столбцы и диаметр (в зависимости от размера и количества скважины в массиве, см . Рисунок 1b ). Выберите «круг» в меню «Форма ROI».
- Удерживая клавишу «Alt», выбирая верхний левый ROI с помощью мыши, чтобы переместить tОн ROI. Удерживая клавишу «shift», выбирая нижний левый ROI, чтобы изменить размер массива. Удерживайте клавишу «shift» при выборе ROI с правой стороны массива, но не в углах, чтобы изменить интервал ROI.
- Используйте приведенные выше команды для соответствия массиву ROI над лунками изображения. Нажмите «Measure RT».
ПРИМЕЧАНИЕ. Плагин будет экспортировать требуемые измерения из каждого ROI. Используйте три размера ROI, создавая концентрические кольца вокруг колодцев, чтобы собрать локальный фоновый сигнал ( т. Е. Сигнал от среднего кольца, вычитаемого из внешнего кольца) и измерения флуоресценции ( т. Е. Сигнал от внутреннего кольца).
- Соберите данные в программном обеспечении для работы с электронными таблицами и вычислите фоновый сигнал. Импортируйте его в специальное программное обеспечение для создания сценариев для дальнейшего анализа.
- Выберите области интереса (ROI) вокруг каждого микрошара в желаемых массивах с помощью модуля ImageJ «MicroArray».
- Организация и анализ данных
- Импорт данных и организация данныхСобранные в ImageJ в матрицу в следующем порядке для всех времен: столбец 1, номер поддиапазона; Столбец 2, ряд скважин; Столбец 3, колонка скважины; Столбец 4, средняя интенсивность; Столбец 5, интенсивность фона; И столбец 6, средняя интенсивность - интенсивность фона.
- Отделите результаты получения mCherry и GFP в разных матрицах. Сохраняйте результаты из каждого подматрица и каждого цвета в другой ячейке в массиве ячеек.
ПРИМЕЧАНИЕ. Эта организация упрощает перемещение между данными изображения и результатами измерений, очисткой данных и гарантированием того, что измерения точно представляют данные.
- Отделите результаты получения mCherry и GFP в разных матрицах. Сохраняйте результаты из каждого подматрица и каждого цвета в другой ячейке в массиве ячеек.
- Отрегулируйте для автофлуоресценции P. aeruginosa .
ПРИМЕЧАНИЕ. В экспериментах, связанных с совместной культурой штаммов GFP и mCherry, необходимо проанализировать только чип mCherry, чтобы выяснить взаимосвязь между mCherry и зеленой автофлуоресценцией.- Постройте сигнал mCherry-versus-GFP от всех mCherry ΔretS &# 916; tse / i1-6 лунок во всех временных точках для определения взаимосвязи между сигналом mCherry и автофлуоресценцией в канале GFP. Вычтите сигнал аутофлуоресценции из сокультур.
- Постройте траектории и установите модифицированное логистическое уравнение на каждую траекторию для извлечения параметров с использованием меток наименьших квадратов в программном обеспечении электронной таблицы или пользовательском скриптовом программном обеспечении.
- Найдите корреляции между параметрами траектории GFP и mCherry.
- Импорт данных и организация данныхСобранные в ImageJ в матрицу в следующем порядке для всех времен: столбец 1, номер поддиапазона; Столбец 2, ряд скважин; Столбец 3, колонка скважины; Столбец 4, средняя интенсивность; Столбец 5, интенсивность фона; И столбец 6, средняя интенсивность - интенсивность фона.
Результаты
Представленная здесь экспериментальная платформа предназначена для высокопроизводительных и высококонцентрированных исследований бактериальных сообществ. Этот проект позволяет одновременно анализировать тысячи сообществ, растущих в лунках разного размера. При ...
Обсуждение
В этой статье представлено устройство с микроволновыми решетками и экспериментальные протоколы, предназначенные для высокопроизводительного и высокоточного анализа изображений на основе живых клеток на основе бактериальных сообществ. В то время как в центре внимания демонстрации ?...
Раскрытие информации
Авторам нечего раскрывать.
Благодарности
Массивы Microwell были сфабрикованы и охарактеризованы в Отделе пользовательских услуг Центра нанофазных материалов, Управление по основным энергетическим наукам Министерства энергетики США. Финансовая поддержка этой работы была оказана через Фонд исследований и развития директора Национальной библиотеки Оук-Ридж. Авторы также хотели бы поблагодарить лабораторию J. Mougous (Вашингтонский университет, Сиэтл, штат Вашингтон) за поставку штаммов P. aeruginosa, используемых в этих исследованиях .
Материалы
| Name | Company | Catalog Number | Comments |
| Parylene N | Specialty Coating Systems | CAS NO.:1633-22-3 | |
| Parylene coater | Specialty Coating Systems | Labcoter 2 Parylene Deposition Unit PDS2010 | |
| Silicon Wafer | WRS Materials | 100mm diameter, 500-550μm thickness, Prime, 10-20 resistivity, N/Phos<100>, | |
| adhesion promoter | Shin-Etsu Microsci | MicroPrime P20 adhesion promoter | |
| postive tone photoresist | Rohm and Haas Electronics Materials LLC (Owned by Dow) | Microposit S1818 Positive Photoresist (code 10018357) | |
| Quintel Contact Aligner | Neutronix Quintel Corp | NXQ 7500 Mask Aligner | |
| Reactive Ion Etching Tool | Oxford Instruments | Plasmalab System 100 Reactive Ion Etcher | |
| R2A Broth | TEKnova | R0005 | |
| Bovine Serum Albumin | Sigma | A9647 | |
| Multimode Plate Reader | Perkin Elmer | Enspire, 2300-0000 | |
| Fluorescent Microscope | Nikon | Eclipse Ti-U | |
| Automated Stage | Prior | ProScan III | |
| CCD camera | Nikon | DS-QiMc | |
| Stage-top environmental control chamber | In Vivo Scientific | STEV ECU-HOC | |
| Phosphate Buffered Saline | ThermoFisher Scientific | 14190144 | |
| UltraPure Agarose | ThermoFisher Scientific | 16500500 | |
| 25 x 75 mm No. 1.5 coverslip | Nexterion | High performance #1.5H coverslips | |
| Fluorescence Reference Slides | Ted Pella | 2273 | |
| Physical Stylus Profilometer | KLA Tencor | P-6 | |
| lab wipes | Kimberly Clark | Kimipe KIMTECH SCIENCE Brand, 34155 | |
| commercial software | Nikon | NIS Elements | |
| Zeiss 710 Confocal Microscope | Zeiss | ||
| filter cubes | Nikon | Nikon FITC (96311), Nikon Texas Red(96313) |
Ссылки
- Zhou, J., Deng, Y., et al. Stochasticity, succession, and environmental perturbations in a fluidic ecosystem. Proc Natl Acad Sci. 111, E836-E845 (2014).
- Valm, A. M., Welch, J. L. M., et al. Systems-level analysis of microbial community organization through combinatorial labeling and spectral imaging. Proc Natl Acad Sci USA. 108 (10), 4152-4157 (2011).
- Satoh, H., Miura, Y., Tsushima, I., Okabe, S. Layered structure of bacterial and archaeal communities and their in situ activities in anaerobic granules. Appl Environ Microbiol. 73 (22), 7300-7307 (2007).
- Kim, H. J., Boedicker, J. Q., Choi, J. W., Ismagilov, R. F. Defined spatial structure stabilizes a synthetic multispecies bacterial community. Proc Natl Acad Sci USA. 105 (47), 18188-18193 (2008).
- Nunan, N., Wu, K., Young, I. M., Crawford, J. W., Ritz, K. Spatial distribution of bacterial communities and their relationships with the micro-architecture of soil. FEMS Microbiol Ecol. 44, 203-215 (2003).
- Grundmann, G. L. Spatial scales of soil bacterial diversity - The size of a clone. FEMS Microbiol Ecol. 48, 119-127 (2004).
- Langenheder, S., Lindstrom, E. S., Tranvik, L. J. Structure and Function of Bacterial Communities Emerging from Different Sources under Identical Conditions. Appl Environ Microbiol. 72 (1), 212-220 (2006).
- Camp, J. G., Kanther, M., Semova, I., Rawls, J. F. Patterns and Scales in Gastrointestinal Microbial Ecology. Gastroenterology. 136 (6), 1989-2002 (2009).
- Renner, L. D., Weibel, D. B. Physicochemical regulation of biofilm formation. MRS Bull. 36 (5), 347-355 (2011).
- Wessel, A. K., Hmelo, L., Parsek, M. R., Whiteley, M. Going local: technologies for exploring bacterial microenvironments. Nat Rev Microbiol. 11 (5), 337-348 (2013).
- Stacy, A., McNally, L., Darch, S. E., Brown, S. P., Whiteley, M. The biogeography of polymicrobial infection. Nat Rev Microbiol. 14 (2), 93-105 (2015).
- Hansen, R. R., Shubert, K. R., Morrell-Falvey, J. L., Lokitz, B. S., Doktycz, M. J., Retterer, S. T. Microstructured block copolymer surfaces for control of microbe adhesion and aggregation. Biosensors. 4 (1), 63-75 (2014).
- Hansen, R. R., Hinestrosa, J. P., et al. Lectin-functionalized poly(glycidyl methacrylate)- block -poly(vinyldimethyl azlactone) surface scaffolds for high avidity microbial capture. Biomacromolecules. 14 (10), 3742-3748 (2013).
- Timm, C. M., Hansen, R. R., Doktycz, M. J., Retterer, S. T., Pelletier, D. A. Microstencils to generate defined, multi-species patterns of bacteria. Biomicrofluidics. 9 (6), (2015).
- Keymer, J. E., Galajda, P., Muldoon, C., Park, S., Austin, R. H. Bacterial metapopulations in nanofabricated landscapes. Proc Natl Acad Sci USA. 103 (46), 17290-17295 (2006).
- Zhang, Q., Lambert, G., et al. Acceleration of Emergence of Bacterial Antibiotic Resistance in Connected Microenvironments. Science. 333 (6050), 1764-1767 (2011).
- Friedlander, R. S., Vlamakis, H., Kim, P., Khan, M., Kolter, R., Aizenberg, J. Bacterial flagella explore microscale hummocks and hollows to increase adhesion. Proc Natl Acad Sci USA. 110 (14), 5624-5629 (2013).
- Zhou, J., Liu, W., et al. Stochastic Assembly Leads to Alternative Communities with Distinct Functions in a Bioreactor Microbial Community. MBio. 4 (2), 1-8 (2013).
- van Vliet, S., Hol, F. J., Weenink, T., Galajda, P., Keymer, J. E. The effects of chemical interactions and culture history on the colonization of structured habitats by competing bacterial populations. BMC Microbiol. 14 (1), 116 (2014).
- Niepa, T. H. R., Hou, L., et al. Microbial Nanoculture as an Artificial Microniche. Sci Rep. 6, 30578 (2016).
- Hansen, R. H., Timm, A. C., et al. Stochastic Assembly of Bacteria in Microwell Arrays Reveals the Importance of Confinement in Community Development. PLoS ONE. 11 (5), e0155080 (2016).
- Hood, R. D., Singh, P., et al. A Type VI Secretion System of Pseudomonas aeruginosa Targets a Toxin to Bacteria. Cell Host Microbe. 7 (1), 25-37 (2010).
- LeRoux, M., Ja De Leon, ., et al. Quantitative single-cell characterization of bacterial interactions reveals type VI secretion is a double-edged sword. Proc Natl Acad Sci. 109 (48), 19804-19809 (2012).
- Whitney, J. C., Beck, C. M., et al. Genetically distinct pathways guide effector export through the type VI secretion system. Mol Microbiol. 92 (3), 529-542 (2014).
- Warrick, J. W., Timm, A., Swick, A., Yin, J. Tools for Single-Cell Kinetic Analysis of Virus-Host Interactions. PLoS ONE. 11 (1), e0145081 (2016).
- Zwietering, M. H., Jongenburger, I., Rombouts, F. M., Van't Riet, K. Modeling of the Bacterial Growth Curve. Appl Environ Microbiol. 56 (6), 1875-1881 (1990).
- Halsted, M., Wilmoth, J. L., et al. Development of transparent microwell arrays for optical monitoring and dissection of microbial communities. J Vac Sci Technol B Nanotechnol Microelectron. 34 (6), 06KI03 (2016).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеСмотреть дополнительные статьи
This article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены