
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Assemblea e monitoraggio dello sviluppo microbico della comunità all'interno di una piattaforma di array Microwell
In questo articolo
Riepilogo
Lo sviluppo delle comunità microbiche dipende da una combinazione di fattori, tra cui l'architettura ambientale, l'abbondanza dei membri, i tratti e le interazioni. Questo protocollo descrive un ambiente sintetico e microfabbricato per il monitoraggio simultaneo di migliaia di comunità contenute nei pozzetti di femtoliter, in cui si possono approssimare fattori chiave come la dimensione e il confinamento di nicchia.
Abstract
Lo sviluppo delle comunità microbiche dipende da una combinazione di fattori deterministici e stochastici complessi che possono alterare drasticamente la distribuzione spaziale e le attività dei membri della comunità. Abbiamo sviluppato una piattaforma di array microwell che può essere utilizzata per assemblare e monitorare rapidamente migliaia di comunità batteriche in parallelo. Questo protocollo evidenzia l'utilità della piattaforma e descrive il suo utilizzo per monitorare ottimamente lo sviluppo di semplici comunità a due componenti all'interno di un insieme di array all'interno della piattaforma. Questa dimostrazione utilizza due mutanti di Pseudomonas aeruginosa , parte di una serie di mutanti sviluppati per studiare la patogenicità della secrezione di tipo VI. Gli inserti cromosomici di geni mCherry o GFP facilitano l'espressione costitutiva di proteine fluorescenti con distinte lunghezze d'onda di emissione che possono essere utilizzate per monitorare l'abbondanza e la posizione del membro della comunità all'interno di ciascuna microssacca. Questo protocollo descrive un metodo dettagliatoD per l'assemblaggio di miscele di batteri nei pozzetti dell'array e utilizzando l'imaging di fluorescenza a tempo intervallo e l'analisi quantitativa delle immagini per misurare la crescita relativa di ciascuna popolazione membro nel tempo. La semina e l'assemblaggio della piattaforma microwell, le procedure di imaging necessarie per l'analisi quantitativa delle comunità microbiche all'interno dell'array e i metodi che possono essere utilizzati per rivelare le interazioni tra le specie di specie microbiche tutte discusse.
Introduzione
Le comunità microbiche sono formate da fattori deterministici come la struttura dell'ambiente e processi stocastici associati a morte cellulare, divisione, concentrazione proteica, numero di organelli e mutazione 1 . All'interno dell'ambiente naturale, può essere quasi impossibile analizzare l'impatto individuale di tali influenze sulla composizione e l'attività della comunità. Oscurati da strutture naturali e sepolte in un ambiente chimico e biologico, identificare i membri della comunità e risolvere ulteriormente la loro distribuzione spaziale nell'ambiente naturale è estremamente impegnativo. Tuttavia, gli sforzi recenti hanno sottolineato l'importanza dell'organizzazione spaziale sulla funzione della comunità e sottolineano la necessità di tenere conto dell'abbondanza e dell'organizzazione dei membri negli studi in corso 2 , 3 , 4 .
essoÈ chiaro che l'ambiente chimico locale ( ossia la disponibilità di sostanze nutritive e metaboliti secondari), la struttura fisica ( ad esempio, l'architettura del suolo, le radici di piante, le particelle oceaniche oi microvilli intestinali), la presenza o l'assenza di ossigeno e l'introduzione di Specie patogene influenzano la composizione, l'architettura e la funzione delle comunità microbiche 5 , 6 , 7 , 8 , 9 , 10 , 11 . Tuttavia, le tecniche tradizionali per le culture che trascurano di catturare questi fattori continuano a prevalere. La composizione comunitaria ( ad esempio, la presenza di specie co-dipendenti), l'attaccamento fisico, la concentrazione di molecole di segnalazione e il contatto diretto delle cellule cellulari sono tutti fattori importanti per la formazione di una comunità microbica e possono essere persi in cCondizioni culturali convenzionali. Queste proprietà sono difficili da replicare in una coltura liquida o in una piastra agar. La disponibilità di tecniche microfluidiche, micropatternate e nanofabbricazione che consentono la replica delle principali caratteristiche fisiche e chimiche degli ambienti naturali ha comunque consentito a molti ricercatori di costruire comunità batteriche per studiare le loro interazioni 12 , 13 e 14 e sviluppare ambienti sintetici che Mimare le condizioni naturali 4 , 15 , 16 , 17 , 18 , 19 , 20 .
Questo protocollo descrive un metodo per la fabbricazione di un dispositivo di array microwell e fornisce procedure sperimentali dettagliate che possono essere utilizzate per funzionalizzare thI pozzi nell'array e la crescita dei batteri, sia come colonie di singola specie che in comunità multi-membro. Questo lavoro dimostra anche come i batteri modificati per produrre proteine reporter fluorescenti possono essere utilizzati per monitorare la crescita batterica nei pozzetti nel tempo. Un reticolo simile è stato presentato in precedenza e ha dimostrato che è possibile seguire la crescita delle colonie di singola specie di Pseudomonas aeruginosa ( P. aeruginosa) in microonde. Modulando la dimensione del pozzo e della densità di semina, le condizioni di partenza di migliaia di esperimenti di crescita possono essere variate in parallelo per determinare come le condizioni iniziali di inoculazione influenzano la capacità dei batteri di crescere 21 . Il lavoro corrente utilizza una versione leggermente modificata dell'array microwell che si basa sul lavoro precedente abilitando il confronto simultaneo di più array e utilizzando un protocollo sperimentale più robusto. L'array utilizzato in questo lavoro contiene più subarray, o array ensemChe contengono pozzetti di diverse dimensioni, che vanno da 15 a 100 μm di diametro, che sono disposti in tre differenti gradazioni ( ossia 2x, 3x e 4x il diametro del pozzo). Le matrici sono incisi in silicio e la crescita dei batteri seminati negli array di silicio è abilitata sigillando le matrici con una copertura rivestita con un gel agarosio a media infusione. I mutanti P. aeruginosa progettati per studiare il sistema di secrezione di tipo VI sono utilizzati in questa dimostrazione.
I risultati qui presentati si basano sull'obiettivo finale di analizzare le comunità di multipli all'interno delle matrici di microwell, consentendo ai ricercatori di monitorare l'abbondanza e l'organizzazione dei batteri in situ controllando e sondando l'ambiente chimico. Questo dovrebbe infine fornire approfondimenti sulle "regole" che governano lo sviluppo e la successione della comunità.
Protocollo
1. Fabbricazione Silicon Microwell-array
- Rivestimento di Parylene
- Depurare tra 1-1,5 μm di parilene N su piastre di silicio usando un sistema di rivestimento di parilene disponibile in commercio secondo le specifiche e le istruzioni del produttore (impostazioni: set point vaporizzatore = 160 ° C; punto di forno = 650 ° C).
NOTA: Circa 6 g di parilene N caricato in una camera porta i rivestimenti di spessore di 1-1,5 μm.
- Depurare tra 1-1,5 μm di parilene N su piastre di silicio usando un sistema di rivestimento di parilene disponibile in commercio secondo le specifiche e le istruzioni del produttore (impostazioni: set point vaporizzatore = 160 ° C; punto di forno = 650 ° C).
- fotolitografia
- Spin-coat le polveri N-rivestite di parilene con promotore di adesione, 20% di esametildisilazano (HMDS) e 80% di acetato di monometil etere di propilenglicole (PGMEA) (vedi tabella dei materiali ) a 3.000 giri / min per 45 s. Riempire una pipetta da 2 ml di trasferimento con il promotore di adesione e spargerla su tutta la cialda. Lasciare che la cialda si posiziona per circa 10 secondi prima di spinarla asciutta.
- Riempire una pipetta da 2 mL di trasferimento con positivo a(Vedere la tabella dei materiali ) e dispensare il fotoresist nel centro della fetta. Spinare a 3.000 giri / min per 45 s per ottenere un rivestimento resistente, di circa 1,5 μm di spessore.
- Infornare i campioni su una piastra a 115 ° C per 1 minuto.
- Utilizzare un allineatore di contatti e una fotomassa con il modello di pozzetto desiderato per esporre il campione alla luce ultravioletta. Esporre la vernice spin-rivestita attraverso il fotomontaggio modellato per 6 s, dando una dose approssimativa di 60-80 mJ / cm 2 misurata a 365 nm.
- Sviluppare il modello sottomarendo il campione nello sviluppatore (<3% tetrametilammonio idrossido in acqua, vedere la tabella dei materiali ) per 2 min. Sciacquare con acqua DI e asciugare con azoto pulito e asciutto.
NOTA: Le aree del fotoresist esposto a UV devono essere eliminate durante lo sviluppo.
- Incisione di ioni reattiva
- Utilizzare un getto di plasma di ossigeno per rimuovere il parilene espostoFino al substrato di silicio.
NOTA: la ricetta può essere modulata per modificare la velocità di etch del parilene. Per gli spessori del parilene compreso tra 1 e 5 μm, utilizzare una ricetta con 60 mTorr, 20 ° C, 100 sccm O 2 , 10 W RF e 2.000 W ICP su un attrezzo Reattivo Ion Etching (RIE). Dopo l'incisione e la rimozione dello strato di parilene esposto, l'area modellata ( cioè il silicio esposto) dovrebbe sembrare lucido e argento. - Utilizzare un processo di etching profondo RIE (DRIE, ad esempio, Bosch DRIE) da incidere nel silicio.
NOTA: La frequenza e la durata di etchè determineranno la profondità di profondità. Un ciclo completo del processo Bosch (passo di deposizione di 3 s: 20 mTorr, 15 ° C, 140 sccm C 4 F 8 , 10 W RF e 1,750 W ICP seguiti da un processo di etching da 10 s: 20 mTorr, 15 ° C , 120 sccm SF 6 , 8 W RF e 1.750 W ICP) corrisponde a circa 1 μm di profondità di etch. I pozzetti utilizzati in questo campo di dimostrazione variano da 3 a 3,5 μm di profondità. - VDeterminare la profondità dell'acqua utilizzando la profilometria fisica.
- Caricare il campione in un profilometro fisico (vedere la tabella dei materiali ).
- Accendere il vuoto del campione e premere il pulsante di caricamento manuale.
- Mettete a fuoco il sistema sul campione premendo il pulsante "Focus". Posizionare una funzione appropriata per la misura sulla schermata di visualizzazione.
- Eseguire la scansione del campione. Livellare il profilo e misurare la profondità delle funzioni.
- Registrare la velocità di etching e modulare i tempi di incisione successivi per ottenere la profondità desiderata.
NOTA: Le misure comprendono la profondità del pozzo di silicio, lo spessore del parilene depositato e lo spessore del fotoresist. Verificare lo spessore di ogni strato durante la procedura è necessario per ottenere una profondità accurata.
- Utilizzare un getto di plasma di ossigeno per rimuovere il parilene espostoFino al substrato di silicio.
2. Cultura batterica e semina ( Figura 1a )
- Avviare le colonie sul brodo Luria (LB) le piastre di agar da giacenze di glicole e utilizzare entro due settimane. Selezionare le colonie dei ceppi desiderati dalle piastre LB agar e avviare colture di P. aeruginosa durante la notte. Incubare le colture durante la notte per circa 18 ore a 37 ° C mentre si agita a 220 rpm nel mezzo R2A.
NOTA: Le colonie devono essere raccolte entro due settimane dalla placcatura per assicurare che le mutazioni ei geni del reporter fluorescenti siano mantenuti. Tutti i lavori di P. aeruginosa devono essere eseguiti in condizioni BSL-2. - Utilizzare uno scriba diamante per la sezione del wafer di silicio in singoli trucioli contenenti gli ensemble di diverse dimensioni e array di pozzetti. Assicurarsi che ogni chip contenga il pieno complemento di buche e campi per lo studio.

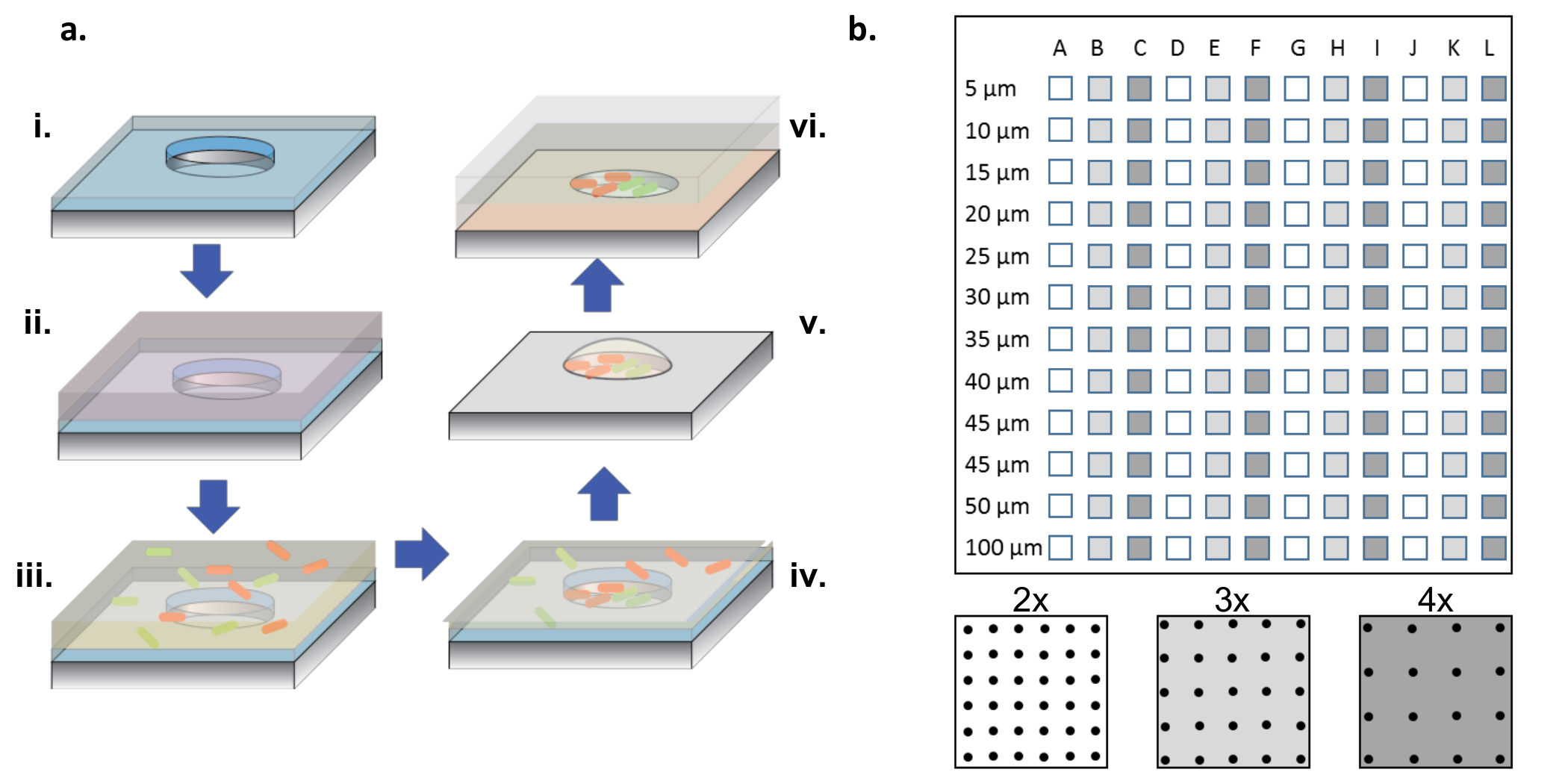
Figura 1: Fabbricazione e procedura di semina delle cellule. ( A ) Unità microwell aRicoperto in piastre di silicio rivestite con uno strato sottile di parilene (i). Per bagnare i pozzetti e / o funzionalizzare la superficie, si aggiunge una soluzione proteica in una gocciola in cima alle matrici (ii). La soluzione proteica viene rimossa, le cialde vengono essiccate e viene aggiunta una nuova soluzione contenente i batteri desiderati (iii). La soluzione batterica viene rimossa dopo un periodo di incubazione e le cialde sono lasciate asciugare, lasciando dietro i batteri nei pozzetti e sulla superficie (iv). I batteri associati alla superficie vengono rimossi con il sollevamento del parilene, lasciando i batteri seminati puliti nei microssi e ancora praticabili a causa del 2% di glicerolo, che aiuta a mantenere i pozzetti idratati (v). I chip di silicio vengono poi collocati in basso verso l'alto su un coperchio di vetro rivestito di gel agarosio, che alimenta la crescita batterica nei microssi (vi). ( B ) Layout di sotto-array su un unico dispositivo di silicio. Ogni sotto-array contiene un insieme di pozzetti identici. Il diametro delle microonde in tutti i sotto-aI raschi hanno un diametro compreso tra 5 e 100 μm e sono organizzati a 2x, 3x o 4x il passo del diametro del diametro, indicato dai colori bianco o grigio scuro sullo schema del pannello inferiore. Quando le profondità del pozzo sono superficiali (<10 μm), i diametri dei pozzetti da 5 e 10 μm sono raramente utili, generalmente a causa della mancanza di cellule che colonizzano questi pozzi molto piccoli. In questo lavoro sono stati analizzati solo i dati di pozzi con diametri da 15-100 μm. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
NOTA: Come illustrato nella figura 1b , un chip completo contiene sotto-array di pozzetti, con diametri compresi tra 5 e 100 μm, con tre diverse tonalità ( ossia 2x, 3x e 4x del diametro) ripetendo 4 volte.
- Inserire una goccia di 150 μl di 500 μg / mL di albumina bovina serum (BSA) in soluzione PBSSulla parte superiore dell'array per bagnare i microsangue. Incubare la soluzione BSA per 1 h sul chip a RT in una camera umida.
- Creare la camera riempendo la parte inferiore di una cassetta vuota di pipetta con Phosphate-Buffered Saline (PBS).
NOTA: al posto di BSA possono essere utilizzate altre sostanze, come lectine specifiche, per funzionalizzare la superficie delle microonde.
- Creare la camera riempendo la parte inferiore di una cassetta vuota di pipetta con Phosphate-Buffered Saline (PBS).
- Durante l'incubazione di patatine di silicio con soluzione BSA, centrifugare le colture a 2,500 giri / min (corrispondenti a una media di 950 xg) per 5 min e quindi riassorbirle in 500 μL di mezzo fresco R2A con glicerolo al 2%.
- Determinare l'OD della coltura usando uno spettrometro UV-vis a 600 nm. Adeguarlo ad un OD di 0,02 utilizzando il 2% di glicerolo R2A.
NOTA: Il glicerolo aiuta a impedire che i pozzetti si asciugano durante il sollevamento di parilene.
- Determinare l'OD della coltura usando uno spettrometro UV-vis a 600 nm. Adeguarlo ad un OD di 0,02 utilizzando il 2% di glicerolo R2A.
- Dopo l'incubazione, rimuovere la soluzione BSA e risciacquare 3x con PBS rimuovendo e sostituendo la goccia di liquidoT che copre l'array di microsfere del silicio. Asciugare sotto l'azoto.
- Aggiungere 150 μl di 0.02 colture OD ad ognuno degli archi asciutti posti in una camera umida. Incubare per 1 h a 4 ° C per permettere ai batteri di aderire alle pareti del pozzo.
NOTA: La refrigerazione non è necessaria per l'incubazione. Il tempo di incubazione di 4 ° C può essere utilizzato per impedire la crescita dei batteri prima dell'inizio dell'immagine in modo da poter visualizzare l'organizzazione spaziale delle comunità prima della crescita. È anche possibile utilizzare l'incubazione a temperatura ambiente. Entrambi i protocolli portano a curve di crescita simili.
3. Impostazione del microscopio
- Prima di avviare l'incubazione batterica sui circuiti integrati di silicio, accendere la camera di controllo ambientale (vedere la tabella dei materiali) e regolare le impostazioni sulla scatola di controllo in modo che l'umidità (~ 100%) e la temperatura (30-32 ° C, vedere la fase 3.2) possono equilibrare prima di aggiungere campioni.
- Livellare il supporto del campione e allineare(Vedere tabella dei materiali ) per aumentare l'umidità nella camera al punto di rugiada. Impostare la temperatura della camera a 30 ° C e quella del coperchio della camera a 32 ° C per ridurre la condensa sul piano di imaging.
NOTA: Il portalampada si inserisce nella camera a celle viva con una guarnizione di circa 1 cm di spessore. Il supporto di campionamento viene livellato con l'aiuto di un livello di bolla posto sulla parte superiore del supporto di campionamento. Il supporto del campione può essere inclinato leggermente e rimanere sigillato nella guarnizione per livellarsi. - Mentre le colture stanno incubando sui chip di silicio, azionare manualmente l'interruttore di alimentazione per la lampada a mercurio almeno 30 minuti prima dell'immagine. Accendere manualmente la fotocamera e la fase automatica del microscopio. Aprire il software utilizzato per controllare il microscopio e le periferiche e assicurarsi che l'attrezzatura sia riconosciuta dal software.
NOTA: l'ingrandimento è 10 volte con NA = 0,3.
- Le soluzioni agarosiche precedentemente preparate a microonde ( cioè il 2% di agarosio nel mezzo R2A) fino a raggiungere uno stato liquido di circa 60 sec.
- Bagnare la parte posteriore di una copertura in vetro di etichetta da 75 mm x 22 mm e coprire con l'etanolo e posizionarla in senso longitudinale, attraversando una scocca in vetro da 50 x 75 mm. Posizionare due distanziali PDMS (spessore di circa 1 mm) Lungo i bordi lunghi del coperchio e spostare la copertura in vetro in modo che circa 1 mm della copertura copre il bordo della slitta.
- Versare 5 ml di soluzione di agarosio liquido sulla parte superiore della copertura in vetro, abbastanza per coprire completamente e mettere una seconda vetrata di vetro da 2 x 3 "sulla parte superiore dell'assemblaggio per" panino "dell'agar liquido tra il coperchio e lo scivolo.
NOTA: questo controlla la profondità dell'agarosa, rendendo lo spessore totale della copertura e dell'agarosio temprato uguale nello spessore ai distanziatori PDMS. - Lasciare il sandwich di scorrimento vetro-slide-agarose in vetro-vetro per impostare fino a quando la soluzione agarosa inizia a solidificare; Quindi, trasferirlo in un frigorifero. Dopo 15 minuti, rimuovere l'agarosio solido in eccesso e tagliare intorno alla copertura in vetro. Spostare questo in un piatto pulito e metterlo in frigorifero fino all'uso.
5. Sigillare i pozzetti con una Coverslip e l'imaging rivestiti con agarosio
- Dopo che il periodo di incubazione dei batteri è completo, rimuovere la copertura rivestita di agarosio dal frigorifero e preparare i chip di silicio, come segue.
- Immergere i chip di silicio in acqua ultrapunta, uno alla volta, per 10 secondi ciascuno. Posizionarli sui bordi su una striscia o un tessuto da laboratorio finché la maggior parte del liquido in eccesso non è scomparsa dai bordi delle patatine.
- Tagliare un pezzo di nastro per abbinare la lunghezza del bordo di ogni chip di silicio. Posizionare il nastro sul parilene che copre il silicio e lo usa per sbucciare rapidamente il rivestimento di parilene.
- ImmedInvertire ciascuno i chip sbucciati e posizionare ciascun chip in modo che il lato dell'allargamento del microsfondo si affaccia (e faccia contatto con) il lato rivestito di agarosio di una copertura rivestita di agarosio. Fare attenzione a non spostare o spostare il circuito integrato dopo che tocca l'agarosio per impedire la crescita di batteri al di fuori dei pozzetti.
- Posizionare la copertura del microtelefono assemblato / agarosio nel supporto di scorrimento della camera di controllo ambientale superiore sul miscroscopio.
- Utilizzare la luce ambiente o la luce diretta ( ad esempio, una torcia) per individuare array di interesse. Utilizzare il software commerciale che controlla la fase automatizzata per salvare tali posizioni (vedere la tabella dei materiali). Spegnere la luce ambientale o diretta dopo che le posizioni vengono memorizzate.
- Nel software commerciale aprire il pannello "ND Acquisition".
NOTA: Questo pannello include un menu per il salvataggio automatico in una directory specifica, nonché l'acquisizione di immagini programmabili. Per questi esperimentiVengono utilizzati i menu "Time", "XY" e "λ". - Per salvare le posizioni nel software, fare clic sul menu "XY" e quindi selezionare una casella vuota a sinistra per ogni posizione che deve essere salvata. Inoltre, fai clic sul pulsante "Includi Z".
- Acquisisci le immagini nel tempo alle lunghezze d'onda desiderate e 10 ingrandimenti utilizzando i cubi appropriati di filtro fluorescenza (vedi tabella dei materiali).
- Utilizzare il software di controllo e le posizioni di array salvate per spostarsi in ogni posizione salvata e concentrarsi sui pozzetti. Fare clic su ciascuna posizione XY nell'elenco salvato e regolare la messa a fuoco usando il filtro Proteine Verde Flourescence (GFP). Salvare la nuova posizione z facendo clic sulla freccia che punta alla posizione z.
NOTA: Questo processo può richiedere molto tempo. Si consideri la precauzione di aumentare il guadagno e di utilizzare il filtro di densità neutra per ridurre l'intensità della luce per evitare la fotolibrazione. - Determinare la distanza dell'asse z tra i piani focali per ciascuna lunghezza d'onda rilevando la differenza nella posizione dell'asse z se focalizzata sulla superficie dell'array. Scegli 2-3 posizioni dall'array con la popolazione di batteri rossi / verdi misti e concentrarsi utilizzando il filtro Red Fluorescence Protein (RFP).
- Sottrarre la distanza tra i piani focali utilizzando i filtri di fluorescenza GFP e RFP e aggiungere la regolazione del piano focale sotto il menu "λ".
NOTA: Ad esempio, se l'array appare focalizzato nel canale GFP in una posizione z di 50 μm e la stessa matrice appare focalizzata nel canale RFP a 55 μm, aggiungere +5 accanto alla configurazione ottica RFP nella casella "λ "Menu.
- Sottrarre la distanza tra i piani focali utilizzando i filtri di fluorescenza GFP e RFP e aggiungere la regolazione del piano focale sotto il menu "λ".
- Inizia l'acquisizione di immagini a tempo trascorso.
NOTA: Per gli esperimenti mostrati qui, le immagini RFP e GFP sono state acquisite per ogni posizione di matrice a intervalli di 30 minuti utilizzando l'acquisizione di immagini multi-dimensione tramite un software commerciale che controllaLa macchina fotografica, l'otturatore, la rotella del filtro e la fase motorizzata.- Impostare "l'intervallo" a 30 minuti e la "durata dell'esperimento" a 24 ore sotto il menu "Ora". Fai clic su "Esegui ora".
NOTA: con le caselle "Time", "XY" e "λ" selezionate, eseguire il programma sposterà lo stadio all'immagine di ciascuna posizione ( cioè le posizioni XYZ salvate), prende un'immagine in una lunghezza d'onda, sposta la posizione z Per tenere conto delle differenze di piano focale ( ad esempio, controllo lambda o lunghezza d'onda), prendere la seconda immagine, passare alla prossima posizione di matrice (multipoint) e ripetere questo a intervalli di 30 minuti (time-lapse).
- Impostare "l'intervallo" a 30 minuti e la "durata dell'esperimento" a 24 ore sotto il menu "Ora". Fai clic su "Esegui ora".
- Utilizzare il software di controllo e le posizioni di array salvate per spostarsi in ogni posizione salvata e concentrarsi sui pozzetti. Fare clic su ciascuna posizione XY nell'elenco salvato e regolare la messa a fuoco usando il filtro Proteine Verde Flourescence (GFP). Salvare la nuova posizione z facendo clic sulla freccia che punta alla posizione z.
- Acquisisci le immagini di controllo dell'illuminazione.
NOTA: Utilizzare i menu "ND Acquisition", "Time" e "XY" per scattare immagini di 4 posizioni, ciascuna di 25x.- Prendi una serie di 100 immagini "darkfield" disattivando tutte le sorgenti luminose e prendendoUn'immagine di una diapositiva standard. Queste immagini catturano il rumore della fotocamera. Utilizzare il tempo di esposizione più lungo utilizzato durante il timelapse (passo 5.3.3).
- Prendete una serie di 100 "immagini di campo di illuminazione" immaginando una diapositiva standard ( ad esempio un'intensità RFP uniforme o GFP) in alcune posizioni diverse per catturare l'illuminazione irregolare nelle condizioni sperimentali date. Scegliere un tempo di esposizione che massimizza il segnale senza raggiungere la saturazione.
6. Analisi
- Processare le pile di immagini utilizzando un software di analisi delle immagini ( ad esempio, ImageJ).
- Convertire le immagini acquisite in formato TIFF utilizzando il software commerciale. Carica le immagini nel software di analisi delle immagini facendo clic su "File"> "Importa"> "Sequenza immagine".
- Crea un "Immagine di correzione", misurando tutte le immagini "darkfield" e "illumination field". sottrazioneT l'immagine media "darkfield" dall'immagine "campo di illuminazione" medio selezionando "Process"> "Calculator Image". Selezionare le due immagini, "Image1" e "Image2", e poi "Sottrarre" nel campo "Operation". Fai clic su "OK".
- Per la media, caricate le immagini di correzione (o scuro), fate clic su "Immagine"> "Stack"> "Progetto Z"> "Proiezione media".
- Se necessario, eseguire la registrazione delle immagini. Quindi eseguire la sottrazione di sfondo facendo clic su "Process"> "Sottrarre sfondo". Inserisci un raggio ( ad esempio, 125) nel campo "raggio" e seleziona "paraboloid scorrevole".
- Effettuare la correzione dell'illuminazione usando "Process"> "Calculator Plus". Scegli i seguenti parametri: operazione, divide; I1, ben immagine; I2, immagine di correzione; K1, media immagine di correzione; E k2, 0. Fare clic su "CreaMangiato Nuova Finestra ".
NOTA: Questo set di dati non richiede la registrazione, ma in altri lavori, il plugin ImageJ StackReg è stato utilizzato con la trasformazione "Traduzione". Per la sottrazione di sfondo, utilizzare lo stesso raggio parabolico scorrevole per ogni set di immagini. Ad esempio, se i più grandi pozzetti visualizzati hanno un raggio di pixel di 100, utilizzare un raggio maggiore di 100 ( ad esempio, 125) per ogni set di immagini.
- Determinare la crescita di ciascun ceppo nelle microonde.
- Selezionare le aree di interesse (ROI) attorno a ciascuna microwell negli array desiderati utilizzando il plugin ImageJ "MicroArray".
- Nel menu "MAP" fare clic su "Reset Grid". Specificare righe, colonne e diametro (in base alla dimensione e al numero dell'allineamento, vedere la Figura 1b ). Seleziona "cerchio" dal menu "ROI shape".
- Tenere premuto il tasto "Alt" mentre seleziona il ROI in alto a sinistra con il mouse per spostare tLui array di ROI. Tenere premuto il tasto "shift" mentre seleziona il ROI in basso a sinistra per modificare la dimensione dell'array. Tenere premuto il tasto "shift" mentre seleziona un ROI dal lato destro dell'array, ma non agli angoli, per modificare la spaziatura dei ROI.
- Utilizzare i comandi di cui sopra per adattare l'array ROI sui pozzetti di un'immagine. Fai clic su "Misura RT".
NOTA: Il plugin esporterà le misure desiderate da ciascun ROI. Utilizzare tre dimensioni ROI, creando anelli concentrici attorno ai pozzetti per raccogliere localmente il segnale di sfondo ( cioè il segnale dall'anello centrale sottratto dall'anello esterno) e le misurazioni della fluorescenza ( cioè il segnale dall'anello interno).
- Raccogliere i dati in un software di calcolo e calcolare il segnale di sfondo. Importarlo ad un software di script personalizzato per ulteriori analisi.
- Selezionare le aree di interesse (ROI) attorno a ciascuna microwell negli array desiderati utilizzando il plugin ImageJ "MicroArray".
- Organizzazione e analisi dei dati
- Importa i dati e organizza i datiRaccolti in ImageJ in una matrice nel seguente ordine per tutti i tempi: colonna 1, numero di sotto-array; Colonna 2, fila; Colonna 3, colonna colonna; Colonna 4, intensità media; Colonna 5, intensità di fondo; E colonna 6, intensità media - intensità di fondo.
- Separare i risultati dell'acquisizione di mCherry e GFP in diverse matrici. Memorizza i risultati di ogni sotto-array e ogni colore in una cella diversa in un array di celle.
NOTA: Questa organizzazione rende più semplice spostarsi avanti e indietro tra dati di immagine e risultati di misura, pulendo i dati e assicurando che le misure rappresentino esattamente i dati.
- Separare i risultati dell'acquisizione di mCherry e GFP in diverse matrici. Memorizza i risultati di ogni sotto-array e ogni colore in una cella diversa in un array di celle.
- Regolare l'autofluorescenza di P. aeruginosa .
NOTA: Negli esperimenti che coinvolgono la co-cultura di ceppi di GFP e mCherry, si dovrebbe analizzare un chip solo di mCherry per accertare la relazione tra mCherry e autofluorescenza verde.- Tracciare il segnale mCherry-versus-GFP da tutti i parametri mCherry ΔretS &# 916; tse / i1-6 in tutti i punti di tempo per determinare la relazione tra il segnale mCherry e l'autofluorescenza nel canale GFP. Sottrai il segnale di autofluorescenza dalle co-culture.
- Tracciare le traiettorie e adattare un'equazione logistica modificata a ciascuna traiettoria per estrarre i parametri utilizzando i minimi quadrati che si adattano sia a un software di fogli di calcolo che a un software di script personalizzato.
- Cercare correlazioni tra e tra i parametri del tracciamento di GFP e mCherry.
- Importa i dati e organizza i datiRaccolti in ImageJ in una matrice nel seguente ordine per tutti i tempi: colonna 1, numero di sotto-array; Colonna 2, fila; Colonna 3, colonna colonna; Colonna 4, intensità media; Colonna 5, intensità di fondo; E colonna 6, intensità media - intensità di fondo.
Risultati
La piattaforma sperimentale qui presentata è progettata per studi di alta percentuale e ad alto contenuto di comunità batteriche. Il progetto consente di analizzare contemporaneamente migliaia di comunità, che crescono nei pozzi di varie dimensioni. Con questo disegno di array microwell, è possibile determinare la dipendenza della composizione finale della comunità sulle densità iniziali di semina, dimensione e ambiente chimico. Questo lavoro dimostra la crescita di una comunità a...
Discussione
In questo articolo è stato presentato un dispositivo a matrice microwell e protocolli sperimentali progettati per consentire l'analisi basata su immagini basate su immagini viventi ad alta percentuale e ad alto contenuto di sviluppo della comunità batterica. Mentre il fuoco della dimostrazione è stato quello di studiare gli effetti della secrezione di tipo VI mediata dal contatto sullo sviluppo della comunità, gli array sono stati progettati per essere flessibili e ospitare lo studio di un'ampia gamma di com...
Divulgazioni
Gli autori non hanno niente da rivelare.
Riconoscimenti
Le matrici di Microwell sono state fabbricate e caratterizzate presso il Centro per le Divisioni Ufficio delle Scienze dei materiali di Nanophase, Ufficio delle Scienze Energetiche di base, Dipartimento di Energia Americana. Il sostegno finanziario a questo lavoro è stato fornito attraverso il fondo di Ricerca e Sviluppo del Direttore Nazionale del Laboratorio di Oak Ridge. Gli autori vorrebbero inoltre ringraziare il J. Mougous Laboratory (Università di Washington, Seattle, WA) per la fornitura di ceppi P. aeruginosa utilizzati in questi studi.
Materiali
| Name | Company | Catalog Number | Comments |
| Parylene N | Specialty Coating Systems | CAS NO.:1633-22-3 | |
| Parylene coater | Specialty Coating Systems | Labcoter 2 Parylene Deposition Unit PDS2010 | |
| Silicon Wafer | WRS Materials | 100mm diameter, 500-550μm thickness, Prime, 10-20 resistivity, N/Phos<100>, | |
| adhesion promoter | Shin-Etsu Microsci | MicroPrime P20 adhesion promoter | |
| postive tone photoresist | Rohm and Haas Electronics Materials LLC (Owned by Dow) | Microposit S1818 Positive Photoresist (code 10018357) | |
| Quintel Contact Aligner | Neutronix Quintel Corp | NXQ 7500 Mask Aligner | |
| Reactive Ion Etching Tool | Oxford Instruments | Plasmalab System 100 Reactive Ion Etcher | |
| R2A Broth | TEKnova | R0005 | |
| Bovine Serum Albumin | Sigma | A9647 | |
| Multimode Plate Reader | Perkin Elmer | Enspire, 2300-0000 | |
| Fluorescent Microscope | Nikon | Eclipse Ti-U | |
| Automated Stage | Prior | ProScan III | |
| CCD camera | Nikon | DS-QiMc | |
| Stage-top environmental control chamber | In Vivo Scientific | STEV ECU-HOC | |
| Phosphate Buffered Saline | ThermoFisher Scientific | 14190144 | |
| UltraPure Agarose | ThermoFisher Scientific | 16500500 | |
| 25 x 75 mm No. 1.5 coverslip | Nexterion | High performance #1.5H coverslips | |
| Fluorescence Reference Slides | Ted Pella | 2273 | |
| Physical Stylus Profilometer | KLA Tencor | P-6 | |
| lab wipes | Kimberly Clark | Kimipe KIMTECH SCIENCE Brand, 34155 | |
| commercial software | Nikon | NIS Elements | |
| Zeiss 710 Confocal Microscope | Zeiss | ||
| filter cubes | Nikon | Nikon FITC (96311), Nikon Texas Red(96313) |
Riferimenti
- Zhou, J., Deng, Y., et al. Stochasticity, succession, and environmental perturbations in a fluidic ecosystem. Proc Natl Acad Sci. 111, E836-E845 (2014).
- Valm, A. M., Welch, J. L. M., et al. Systems-level analysis of microbial community organization through combinatorial labeling and spectral imaging. Proc Natl Acad Sci USA. 108 (10), 4152-4157 (2011).
- Satoh, H., Miura, Y., Tsushima, I., Okabe, S. Layered structure of bacterial and archaeal communities and their in situ activities in anaerobic granules. Appl Environ Microbiol. 73 (22), 7300-7307 (2007).
- Kim, H. J., Boedicker, J. Q., Choi, J. W., Ismagilov, R. F. Defined spatial structure stabilizes a synthetic multispecies bacterial community. Proc Natl Acad Sci USA. 105 (47), 18188-18193 (2008).
- Nunan, N., Wu, K., Young, I. M., Crawford, J. W., Ritz, K. Spatial distribution of bacterial communities and their relationships with the micro-architecture of soil. FEMS Microbiol Ecol. 44, 203-215 (2003).
- Grundmann, G. L. Spatial scales of soil bacterial diversity - The size of a clone. FEMS Microbiol Ecol. 48, 119-127 (2004).
- Langenheder, S., Lindstrom, E. S., Tranvik, L. J. Structure and Function of Bacterial Communities Emerging from Different Sources under Identical Conditions. Appl Environ Microbiol. 72 (1), 212-220 (2006).
- Camp, J. G., Kanther, M., Semova, I., Rawls, J. F. Patterns and Scales in Gastrointestinal Microbial Ecology. Gastroenterology. 136 (6), 1989-2002 (2009).
- Renner, L. D., Weibel, D. B. Physicochemical regulation of biofilm formation. MRS Bull. 36 (5), 347-355 (2011).
- Wessel, A. K., Hmelo, L., Parsek, M. R., Whiteley, M. Going local: technologies for exploring bacterial microenvironments. Nat Rev Microbiol. 11 (5), 337-348 (2013).
- Stacy, A., McNally, L., Darch, S. E., Brown, S. P., Whiteley, M. The biogeography of polymicrobial infection. Nat Rev Microbiol. 14 (2), 93-105 (2015).
- Hansen, R. R., Shubert, K. R., Morrell-Falvey, J. L., Lokitz, B. S., Doktycz, M. J., Retterer, S. T. Microstructured block copolymer surfaces for control of microbe adhesion and aggregation. Biosensors. 4 (1), 63-75 (2014).
- Hansen, R. R., Hinestrosa, J. P., et al. Lectin-functionalized poly(glycidyl methacrylate)- block -poly(vinyldimethyl azlactone) surface scaffolds for high avidity microbial capture. Biomacromolecules. 14 (10), 3742-3748 (2013).
- Timm, C. M., Hansen, R. R., Doktycz, M. J., Retterer, S. T., Pelletier, D. A. Microstencils to generate defined, multi-species patterns of bacteria. Biomicrofluidics. 9 (6), (2015).
- Keymer, J. E., Galajda, P., Muldoon, C., Park, S., Austin, R. H. Bacterial metapopulations in nanofabricated landscapes. Proc Natl Acad Sci USA. 103 (46), 17290-17295 (2006).
- Zhang, Q., Lambert, G., et al. Acceleration of Emergence of Bacterial Antibiotic Resistance in Connected Microenvironments. Science. 333 (6050), 1764-1767 (2011).
- Friedlander, R. S., Vlamakis, H., Kim, P., Khan, M., Kolter, R., Aizenberg, J. Bacterial flagella explore microscale hummocks and hollows to increase adhesion. Proc Natl Acad Sci USA. 110 (14), 5624-5629 (2013).
- Zhou, J., Liu, W., et al. Stochastic Assembly Leads to Alternative Communities with Distinct Functions in a Bioreactor Microbial Community. MBio. 4 (2), 1-8 (2013).
- van Vliet, S., Hol, F. J., Weenink, T., Galajda, P., Keymer, J. E. The effects of chemical interactions and culture history on the colonization of structured habitats by competing bacterial populations. BMC Microbiol. 14 (1), 116 (2014).
- Niepa, T. H. R., Hou, L., et al. Microbial Nanoculture as an Artificial Microniche. Sci Rep. 6, 30578 (2016).
- Hansen, R. H., Timm, A. C., et al. Stochastic Assembly of Bacteria in Microwell Arrays Reveals the Importance of Confinement in Community Development. PLoS ONE. 11 (5), e0155080 (2016).
- Hood, R. D., Singh, P., et al. A Type VI Secretion System of Pseudomonas aeruginosa Targets a Toxin to Bacteria. Cell Host Microbe. 7 (1), 25-37 (2010).
- LeRoux, M., Ja De Leon, ., et al. Quantitative single-cell characterization of bacterial interactions reveals type VI secretion is a double-edged sword. Proc Natl Acad Sci. 109 (48), 19804-19809 (2012).
- Whitney, J. C., Beck, C. M., et al. Genetically distinct pathways guide effector export through the type VI secretion system. Mol Microbiol. 92 (3), 529-542 (2014).
- Warrick, J. W., Timm, A., Swick, A., Yin, J. Tools for Single-Cell Kinetic Analysis of Virus-Host Interactions. PLoS ONE. 11 (1), e0145081 (2016).
- Zwietering, M. H., Jongenburger, I., Rombouts, F. M., Van't Riet, K. Modeling of the Bacterial Growth Curve. Appl Environ Microbiol. 56 (6), 1875-1881 (1990).
- Halsted, M., Wilmoth, J. L., et al. Development of transparent microwell arrays for optical monitoring and dissection of microbial communities. J Vac Sci Technol B Nanotechnol Microelectron. 34 (6), 06KI03 (2016).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati