
Method Article
Secuenciación de ARNm de sangre entera utilizando secuenciación de nanoporos
En este artículo
Resumen
La secuenciación de nanoporos es una tecnología novedosa que permite una secuenciación rentable en ubicaciones remotas y entornos con escasos recursos. Aquí, presentamos un protocolo para la secuenciación de mRNAs de sangre entera que es compatible con tales condiciones.
Resumen
La secuenciación en ubicaciones remotas y entornos con escasos recursos presenta desafíos únicos. La secuenciación de nanoporos se puede utilizar con éxito en esas condiciones y se desplegó en África occidental durante la reciente epidemia de virus del Ebola, destacando esta posibilidad. Además de sus ventajas prácticas (bajo costo, facilidad de transporte y uso de equipos), esta tecnología también proporciona ventajas fundamentales sobre los enfoques de secuenciación de segunda generación, en particular la longitud de lectura muy larga, la capacidad de secuenciar directamente ARN y disponibilidad de datos en tiempo real. La precisión de lectura RAW es menor que con otras plataformas de secuenciación, lo que representa la principal limitación de esta tecnología; sin embargo, esto se puede mitigar parcialmente por la alta profundidad de lectura generada. Aquí, presentamos un protocolo compatible con el campo para la secuenciación de la codificación mRNAs para Niemann-Pick C1, que es el receptor celular para Ebolavirus. Este protocolo abarca la extracción de ARN de muestras de sangre animal, seguida de RT-PCR para el enriquecimiento objetivo, código de barras, preparación de la biblioteca, y la secuencia de ejecución en sí, y se puede adaptar fácilmente para su uso con otros objetivos de ADN o ARN.
Introducción
La secuenciación es una herramienta poderosa e importante en la investigación biológica y biomédica. Permite el análisis de genomas, variaciones genéticas y perfiles de expresión de ARN, y por lo tanto desempeña un papel importante en la investigación de enfermedades humanas y animales por igual1,2. La secuenciación de Sanger, uno de los métodos más antiguos disponibles para la secuenciación del ADN, todavía se utiliza rutinariamente hasta el día de hoy y ha sido una piedra angular de la biología molecular. En los últimos 50 años, esta tecnología se ha mejorado para lograr longitudes de lectura superiores a 1.000 NT y una precisión de hasta 99,999%1. Sin embargo, la secuenciación de Sanger también tiene limitaciones. Secuenciar un conjunto más grande de muestras o el análisis de genomas enteros con este método consume mucho tiempo y es costoso1,3. Los métodos de secuenciación de ADN de segunda generación (de próxima generación), como la pirosecuenciación 454 y la tecnología Illumina, nos han permitido reducir significativamente el costo y la carga de trabajo requeridos para la secuenciación en la última década, y han conducido a un tremendo aumento en el cantidad de información de la secuencia biológica disponible4. Sin embargo, las ejecuciones de secuenciación individuales utilizando estas tecnologías de segunda generación son costosas, y la secuenciación en condiciones de campo es desafiante, ya que el equipo necesario es voluminoso y frágil (similar a los dispositivos de secuenciación de Sanger), y a menudo tiene que ser calibrados y atendidos por personal especialmente capacitado. Además, para muchas de las tecnologías de segunda generación las longitudes de lectura son bastante limitadas, lo que a menudo hace que el análisis Bioinformático posterior de estos datos sea un desafío.
La secuenciación de tercera generación que utiliza dispositivos de secuenciación de nanoporos de tamaño de bolsillo (consulte la tabla de materiales) puede servir como una alternativa a estas plataformas de secuenciación establecidas. En estos dispositivos, una molécula de ADN o ARN de una sola cadena pasa a través de un nanoporo simultáneamente con una corriente iónica que luego es medida por un sensor (figura 1). A medida que la hebra atraviesa el nanoporo, se detecta la modulación de la corriente por los nucleótidos presentes en el poro en un momento dado, y se traduce de forma computacional hacia atrás en la secuencia de nucleótidos5. Debido a este principio operacional, la secuenciación de nanoporos permite tanto la generación de lecturas muy largas (cerca de 1 x 106 nucleótidos6) como el análisis de datos de secuenciación en tiempo real. El código de barras es posible adjuntando secuencias de nucleótidos definidas a los ácidos nucleicos de una muestra, lo que permite el análisis de múltiples muestras en una sola ejecución de secuenciación, aumentando así el rendimiento de la muestra y reduciendo los costos de muestra. Debido a su alta portabilidad y facilidad de uso, los dispositivos de secuenciación de nanoporos se han utilizado con éxito en el campo durante la reciente epidemia de enfermedad por el virus del Ebola en África occidental, destacando su idoneidad para un rápido despliegue en regiones remotas7 , 8.
Aquí describimos un protocolo detallado de campo compatible para la secuenciación de la codificación de mRNA para la proteína Niemann-Pick C1 (NPC1), que es el receptor de entrada obligado para filovirus como los Ebolavirus, y se ha demostrado que limita la susceptibilidad de las especies a estos virus9,10. El protocolo abarca la extracción de ARN entero a partir de muestras de sangre, la amplificación específica de NPC1 mRNA por RT-PCR, código de barras de muestras, preparación de la biblioteca y secuenciación con un dispositivo de secuenciación de nanoporos. El análisis de datos no puede discutirse debido a limitaciones de espacio, aunque se proporcionan algunas orientaciones básicas en los resultados representativos; sin embargo, el lector interesado se refiere a una publicación anterior11 para una descripción más detallada del flujo de trabajo que usamos, así como a publicaciones deotros 12,13,14 para obtener información detallada sobre las herramientas de análisis utilizadas en este flujo de trabajo.
Protocolo
Las muestras fueron recogidas siguiendo el protocolo de la Junta de revisión institucional de la Universidad de Njala (NUIRB). IRB00008861/FWA00018924.
1. extracción de ARN de muestras de sangre
- Recoger 3 mL de sangre entera de la especie a analizar en un tubo de recolección de sangre precargada con 6 mL de reactivo estabilizador de ADN/ARN (ver tabla de materiales) y mezclar invirtiendo 5 veces. Conservar la muestra de sangre hasta un mes a 4 ° c.
- Transfiera el contenido del tubo de recolección de sangre a un tubo de recogida de 50 ml, añada 120 μl de proteinasa K y mezcle con vórtex durante 5 s. Incubar la muestra durante 30 minutos a temperatura ambiente.
- Añadir 9 mL de isopropanol a la mezcla y vórtice durante 5 s.
- Coloque un reservorio en una columna de espín de purificación de ARN (consulte la tabla de materiales) y coloque el ensamblaje en un colector de vacío (consulte la tabla de materiales). Agregue la mezcla de muestra al depósito. Aplique una aspiradora hasta que todo el líquido haya pasado a través de la columna.
- Alternativamente, si no hay un colector de vacío disponible, la sangre se puede pasar a través de la columna en 700 μL de porciones por centrifugación repetida durante 30 s a 12.000 x g con el flujo descartado entre los pasos de centrifugación. Sin embargo, esto requerirá aproximadamente 26 pasos de centrifugación.
- Coloque la columna de espín de purificación de ARN en un tubo de recolección y añada 400 μL de tampón de preparación de ADN/ARN (ver tabla de materiales). Centrifugar a 12.000 x g durante 30 s y desechar el caudal.
- Añadir 400 μL de tampón de lavado de ADN/ARN (ver tabla de materiales) a la columna, centrifugar a 12.000 x g durante 30 s y desechar el flujo.
- Mezclar 5 μL de DNase I (1 U/μL) (ver tabla de materiales) con 75 μl de tampón de digestión de ADN (ver tabla de materiales) y añadir la mezcla en la columna. Incubar durante 15 min a temperatura ambiente.
- Añadir 400 μL de tampón de preparación de ADN/ARN a la columna y centrifugar a 12.000 x g durante 30 s. Deseche el flujo.
- Lave la columna con 700 μL de tampón de lavado de ADN/ARN y centrifugue a 12.000 x g durante 30 s. Deseche el flujo.
- Repita el paso 1,9 con 400 μL de tampón de lavado de ADN/ARN y centrifugue a 12.000 x g durante 2 min para eliminar todo el tampón de lavado residual y secar la columna. Cuando quite la columna del tubo de recolección, asegúrese de no humedecer la parte inferior de la columna con el búfer en el tubo de recolección.
-
Coloque la columna en un nuevo tubo de microcentrífuga de 1,5 mL y añada 70 μL de agua sin nucleasa. Incubar durante 1 min a temperatura ambiente y centrifugar durante 30 s a 12.000 x g. Conservar el ARN a-80 ° c hasta su uso posterior (o utilizarlo inmediatamente).
- Opcional: Para cuantificar el ARN, tomar una alícuota y determinar la concentración utilizando un espectrofotómetro UV (ver tabla de materiales).
2. transcripción inversa de NPC1 mRNA en cDNA
- En un tubo de reacción de 0,2 mL, añada 8 μL de ARN de plantilla (1 PG a 2,5 μg de ARN) y 1 μL de tampón de DNase de 10x y de enzima DNase (ver tabla de materiales). Incubar a 37 ° c durante 2 min. Posteriormente, centrifugar la reacción brevemente y colocarla sobre hielo.
- Añadir 4 μL de mezcla maestra de transcriptasa inversa (ver tabla de materiales) y 6 μl de agua sin nucleasa al tubo de reacción y mezclar suavemente. Incubar la reacción en un termociclador durante 10 min a 25 ° c (para el recocido de imprimación), seguida de 10 min a 50 ° c (para la transcripción inversa de ARN). Para inactivar la enzima, incubar durante 5 min a 85 ° c.
- Transfiera el cDNA a un nuevo tubo de microcentrífuga de 1,5 mL y Almacénese a-80 ° c hasta su uso posterior (o utilice inmediatamente).
3. amplificación del marco de lectura abierto NPC1
-
Paso de amplificación inicial
- Configure una PCR de touchdown de15,16 para amplificar el cDNA NPC1 con el primer set 1 (ver tabla 1), utilizando una polimerasa de ADN de alta fidelidad de inicio caliente (ver tabla de materiales) con el tampón de reacción apropiado en una reacción de 50 μl volumen con 1 μL de plantilla. Si es posible, configure la reacción sobre hielo o en un bloque frío de 4 ° c.
- Incubar la reacción en un termociclador con un paso de desnaturalización inicial de 30 s a 98 ° c, seguido de 10 ciclos con desnaturalización a 98 ° c durante 10 s, recocido de imprimación para 20 s a 65 ° c, bajando la temperatura en 0,5 ° c por ciclo, y alargamiento durante 1 min a 72 ° c. Posteriormente, ejecutar 20 ciclos adicionales con 10 s a 98 ° c, 20 s a 60 ° c y 1 min a 72 ° c, seguido de un paso final de elongación de 5 min a 72 ° c.
-
Purificación de PCR utilizando Perlas magnéticas
- Transferir 50 μL de producto de PCR a un tubo de reacción de enlace de ADN-bajo de 1,5 mL (ver tabla de materiales). Resuspend las perlas magnéticas (ver tabla de materiales) a fondo mediante vórtex y añada 50 μl de perlas a la reacción PCR. Mezclar bien. Incubar la muestra en un mezclador giratorio (ver tabla de materiales) durante 5 min a temperatura ambiente (a 15 rpm).
- Gire brevemente la muestra y coloque el tubo de microcentrífuga de 1,5 mL en un bastidor magnético (consulte la tabla de materiales) para pelar las cuentas magnéticas. Espere hasta que el sobrenadante se haya aclarado por completo antes de continuar con el siguiente paso.
- Aspirar el sobrenadante sin perturbar el pellet de perlas y desechar.
- Pipetear 200 μL de etanol al 70% en el tubo de reacción e incubar durante 30 s. aspirar el etanol sin perturbar el pellet y desecharlo. Repita para un total de dos lavados. Asegúrese de que no quede etanol. Puede ser necesario aspirar primero con una picada más grande (p. ej., 1000 μL), y luego retirar las gotas de etanol restantes con una picada más pequeña (p. ej., 10 μL).
- Secar al aire el pellet durante 1 min a temperatura ambiente.
- Extraer el tubo de reacción del bastidor magnético, Resuspender el pellet en 30 μL de agua sin nucleasa e incubar durante 2 min a temperatura ambiente.
- Vuelva a colocar el tubo de reacción en el bastidor magnético y espere hasta que las perlas estén completamente peladas.
- Retirar el sobrenadante sin perturbar el pellet y transferirlo a un nuevo tubo de reacción de 1,5 mL.
-
Adición de adaptadores de código de barras por PCR anidada
- Configure una PCR de touchdown de 50 μL con una polimerasa de ADN de alta fidelidad de inicio en caliente con tampón de reacción 5x y primer set 2 (ver tabla 1). Los imprimadores en este conjunto constan de una región específica de la secuencia de destino para permitir el enlace de los productos de PCR generados en el paso 3,2 (dentro de las secuencias del conjunto de imprimación 1), así como una secuencia de adaptador que se utiliza como objetivo en la reacción de PCR de código de barras subsiguiente (cf. sección 4). Si es posible, configure la reacción sobre hielo o en un bloque frío de 4 ° c. Utilizar 1 μL del producto de PCR purificado preparado en la sección 3,2 como plantilla.
- Incubar la mezcla de reacción en un termociclador utilizando un paso de desnaturalización inicial de 30 s a 98 ° c, seguido de 10 ciclos con desnaturalización a 98 ° c durante 10 s, recocido de imprimación para 20 s a 65 ° c, reduciendo la temperatura en 0,5 ° c por ciclo , y alargamiento durante 1 min a 72 ° c. Posteriormente, incubar la reacción de otros 30 ciclos durante 10 s a 98 ° c, 20 s a 71 ° c y 1 min a 72 ° c, seguida de un paso final de elongación de 5 min a 72 ° c.
- Limpie el producto de PCR utilizando cuentas magnéticas como se describe en la sección 3,2.
4. código de barras de NPC1 amplicons
- Para cada producto de PCR generado en la sección 3, configurar una reacción PCR de código de barras en un tubo de reacción de 0,2 ml utilizando 50 μl de Taq DNA polimerasa 2x Master Mix (ver tabla de materiales), 2 μl de uno de los imprimadores de código de barras de un kit de códigos de barra PCR (ver tabla de materiales y tabla 2), y 1 μl de producto PCR purificado desde el paso 3.3.2 como plantilla. Añadir 47 μL de agua libre de nucleasa para obtener un volumen final de 100 μL.
- Incubar la reacción en un termociclador a 95 ° c durante 3 min como una desnaturalización inicial. Posteriormente, ejecutar 15 ciclos para 15 s a 95 ° c, 15 s a 62 ° c y 1,5 min a 65 ° c. Como alargamiento final, Incube la reacción a 65 ° c durante 5 min.
- Purificar el producto de PCR como se describe en 3,2, pero utilice 100 μl de perlas magnéticas y eluyen en 30 μl de agua sin nucleasa.
- Si es posible, Cuantifique la muestra utilizando un espectrofotómetro UV.
5. preparación de la biblioteca
- Combine una cantidad igual de ADN con código de barras de cada muestra para un total de 1 μg de ADN en un volumen de 45 μL (si es necesario, añada agua sin nucleasa) en un tubo de reacción de 0,2 mL. Si no hay ningún espectrofotómetro UV disponible, utilice volúmenes iguales de cada muestra. Para el dA-tailing, añadir 7 μL de tampón de reacción de preparación final (ver tabla de materiales), 3 μl de mezcla enzimática de preparación final (ver tabla de materiales), y 5 μl de agua sin nucleasa. Mezcle suavemente moviendo el tubo.
- Incubar la reacción durante 5 min a 20 ° c, seguida de 5 min a 65 ° c en un termociclador.
- Purificar el producto de reacción como se describe en la sección 3,2, pero utilice 60 μL de perlas magnéticas y eluyen en 25 μl de agua sin nucleasa.
- Opcional: tomar 1 μL para cuantificar la concentración de la muestra utilizando un espectrofotómetro UV. La cantidad total debe ser superior a 700 ng.
- Combine 22,5 μL de ADN purificado a partir del paso 5,3 con 2,5 μL de adaptador 1D2 (ver tabla de materiales) y 25 μl de mezcla Master de ligasa Blunt/TA (ver tabla de materiales) en un nuevo tubo de reacción de enlace de adn-bajo de 1,5 ml, mezcle suavemente con un parpadeo y gire brevemente abajo. Incubar durante 10 min a temperatura ambiente.
- Purificar el producto de reacción como se describe en la sección 3,2, pero utilizar 20 μL de perlas magnéticas, aumentar el tiempo de incubación para la Unión de ADN a 10 min, realizar dos pasos de lavado con 1 mL de etanol cada uno, y eluir en 46 μL de agua sin nucleasa.
- Combinar 45 μL del producto de reacción del paso 5,6 con 5 μL de mezcla de adaptador de código de barras (ver tabla de materiales) y 50 μl de mezcla de Master de ligasa Blunt/TA en un tubo de reacción de enlace de ADN bajo. Mezclar suavemente deslizando e incubar durante 10 min a temperatura ambiente.
- Purificar el producto de reacción como se describe en la sección 3,2, pero utilizar 40 μL de perlas magnéticas, hacer dos pasos de lavado con 140 μL de tampón ABB (ver tabla de materiales) en lugar de etanol, resuspender las perlas por parpadeo y pellet en un rack magnético. Elute en 15 μL de tampón de elución (ver tabla de materiales). Aumentar los tiempos de incubación para la Unión inicial de ADN a las perlas, así como para el paso de elución a 10 min. Almacene el producto resultante sobre hielo o a 4 ° c hasta su uso.
6. control de calidad de la célula de flujo
- Realice una comprobación de calidad en la célula de flujo antes de utilizarla. Con este fin, conecte el dispositivo de secuenciación al equipo host y abra el software.
- Inserte una celda de flujo (consulte la tabla de materiales) en el dispositivo de secuenciación y elija el tipo de celda de flujo en el cuadro selector y confirme haciendo clic en disponible.
- Haga clic en verificar celda de flujo en la parte inferior de la pantalla y elija el tipo de celda de flujo correcto.
- Haga clic en iniciar prueba para iniciar la comprobación de calidad. Se requiere un mínimo de 800 nanoporos activos en total para que la célula de flujo sea utilizable.
7. cargando la celda de flujo e iniciando la ejecución de secuenciación
- Abra la tapa del puerto de cebado deslizándola en sentido horario. Fije una Piera P1000 a 200 μL e inserte la punta en el puerto de cebado. Ajuste la Piera a 230 μL manteniendo la punta en el puerto de cebado, para extraer 20-30 μL de tampón y remover cualquier burbuja de aire.
- En un nuevo tubo de reacción de enlace de ADN-bajo de 1,5 mL, prepare la mezcla de cebado combinando 576 μL de tampón RBF (ver tabla de materiales) con 624 μl de agua sin nucleasa.
- Pipetee cuidadosamente 800 μL de la mezcla de cebado preparada en el puerto de cebado y espere 5 min. Levante la tapa de la muestra del puerto y pipetee 200 μL adicionales de la mezcla de cebado preparada en el puerto de cebado.
- Pipetear 35 μL de tampón RBF en un nuevo y limpio tubo de reacción de bajo enlace de 1,5 mL de ADN. Mezcle bien las perlas LLB (consulte la tabla de materiales) pipeteando y añada 25,5 μl de las perlas al buffer Rbf. Añadir 2,5 μL de agua sin nucleasa y 12 μL de la biblioteca de ADN del paso 5,8 y mezclar mediante pipeteo.
- Añadir 75 μl de la mezcla de muestra de forma lenta y gota a gota a la célula de flujo a través del puerto de la muestra.
- Reemplace la cubierta del puerto de la muestra, cierre el puerto de cebado y cierre la tapa del dispositivo de secuenciación.
- En el software, confirme que la celda de flujo sigue estando disponible, abra un nuevo experimento y configure los parámetros de ejecución seleccionando el kit utilizado. Seleccione las llamadas base en vivo. Inicie la ejecución de secuenciación haciendo clic en iniciar experimento. Continúe la ejecución de la secuenciación hasta que se recopilen suficientes datos experimentales.
Resultados
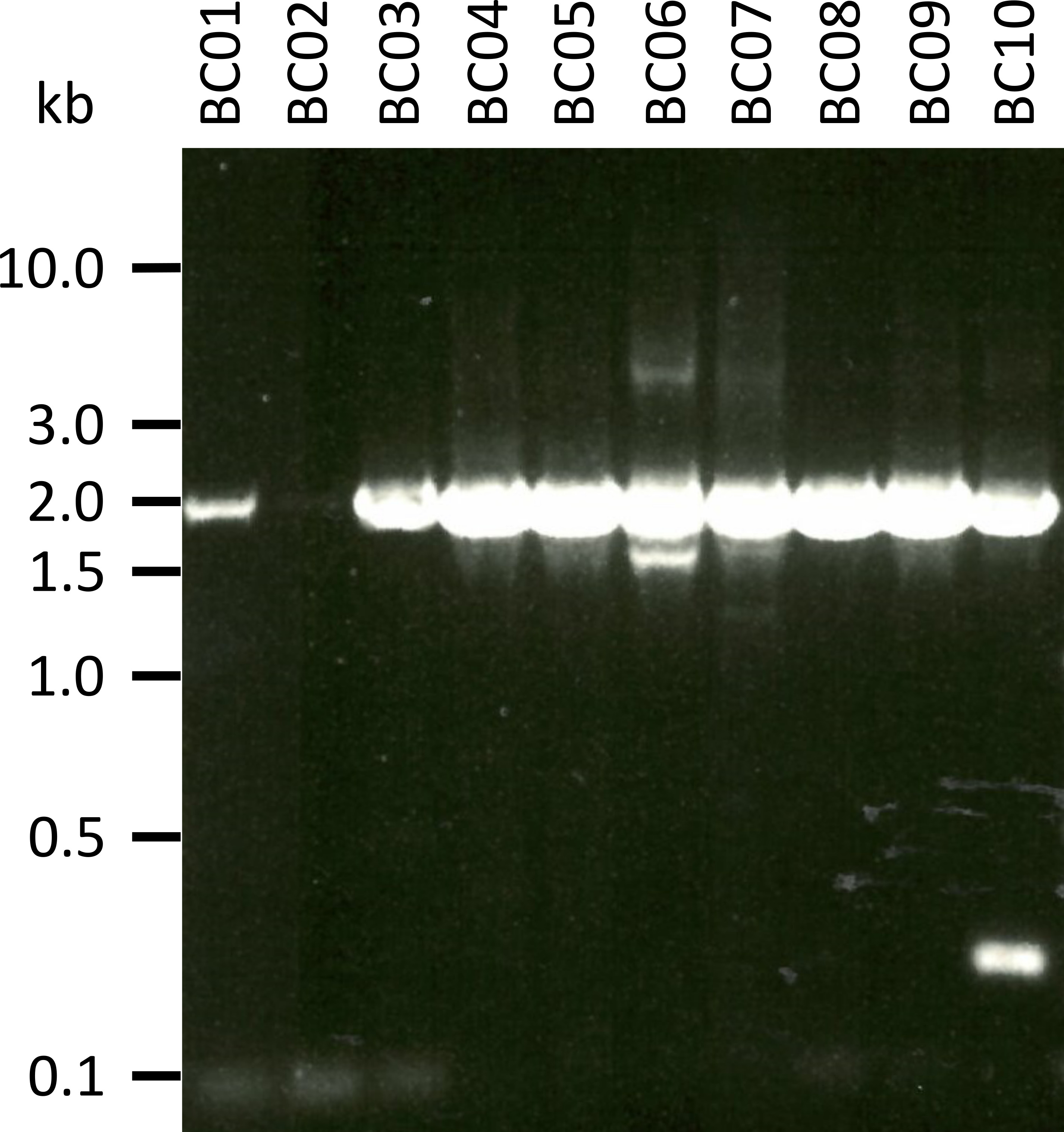
En un experimento representativo para probar el protocolo presentado, extraemos el ARN de 10 muestras de sangre diferentes de cinco especies animales (es decir, 2 individuos por especie (cabra, oveja, porcino, perro, ganado)) (tabla 3). El rendimiento de ARN y la calidad después de la extracción pueden variar ampliamente, en particular debido a las diferencias en el manejo y almacenamiento de muestras. En nuestro experimento representativo, observamos concentraciones de ARN entre 43 ng y 543 ng por μL (tabla 3). También, después de la amplificación por RT-PCR, análisis de gel de la NPC-1 PCR-Productos mostraron varios resultados (figura 2), con bandas notablemente más débiles para las muestras BC01 y BC02 (ambos cabra). Estas diferencias fueron probablemente causadas por diferencias en la calidad de la muestra, aunque no se pueden excluir las diferencias en la eficacia de PCR debido a las diferencias en la Unión de imprimación al gen NPC1 de diferentes especies. Sin embargo, estas diferencias en la eficiencia de rendimiento y/o amplificación no impactan marcadamente el resultado general de la secuenciación. Además, se produjo un producto adicional no específico de PCR en la muestra BC10 (bovinos). A diferencia de la secuenciación de Sanger, estos productos no específicos no influyen negativamente en los resultados de la secuenciación de nanoporos, ya que estas lecturas se descartan durante el mapeo de las lecturas obtenidas a una secuencia de referencia como parte del análisis de datos.
Antes de cada ejecución de secuenciación, se recomienda encarecidamente una comprobación de calidad de la célula de flujo que se va a utilizar, con un requisito mínimo de 800 poros totales. En nuestro experimento representativo, este cheque de calidad devolvió 1.102 poros disponibles para la secuenciación. Dado que los datos se proporcionan en tiempo real y se pueden analizar inmediatamente, la duración de una ejecución de secuenciación se puede ajustar para la aplicación individual (es decir, hasta que se produzcan suficientes datos de secuenciación para el análisis deseado). En nuestros experimentos, las ejecuciones de secuenciación normalmente se realizan durante la noche, y en el caso de nuestro experimento representativo hemos obtenido aproximadamente 1,4 millones lecturas durante una carrera de 14 h.
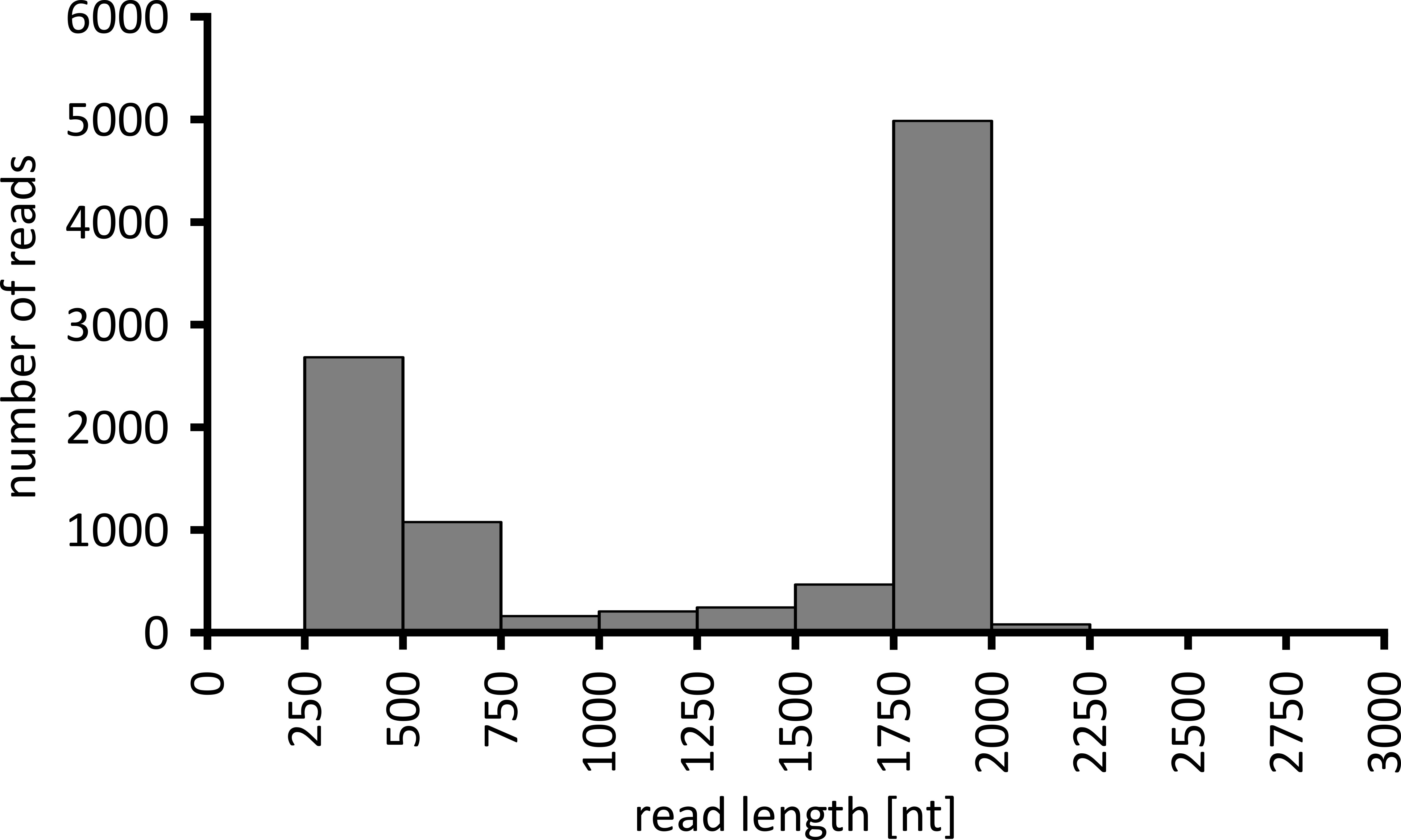
Dependiendo del tipo de análisis de datos a realizar, puede ser aconsejable procesar sólo un subconjunto de las lecturas obtenidas. En el caso de nuestro experimento representativo, se seleccionó un subconjunto de 10.000 lecturas para su posterior análisis. Con este fin, los archivos fastq generados durante la ejecución de secuenciación fueron procesados en un entorno Ubuntu 18,04 LTS, y demultiplexado usando FlexBar v 3.0.3 con parámetros optimizados para demultiplexación de datos de secuenciación de nanoporos (código de barras-longitud de cola 300 , código de barras-Error-Rate 0,2, código de barras-brecha-1)12. Después de la demultiplexación, el mapeo de lectura y la generación de consenso se pueden hacer usando una serie de herramientas diferentes, pero una discusión detallada del aspecto Bioinformático de la secuenciación de nanoporos va más allá del alcance de este manuscrito. Sin embargo, en el caso de nuestros resultados representativos, el mapeo de lectura a una secuencia de referencia se realizó usando Geneious 10.2.3. De las 10.000 lecturas analizadas, 5.457 mostraron una longitud entre 1.750 y 2.000 nucleótidos, que coincide con los tamaños esperados para los fragmentos de PCR amplificados como parte de nuestro flujo de trabajo (1.769 NT, figura 3). Se observó un pico adicional en la distribución de la longitud de las lecturas entre 250 y 500 nucleótidos, que pueden atribuirse a productos de PCR no específicos. La demultiplexación de lecturas permitió la asignación del 87,6% de las lecturas a uno de los 10 códigos de barras/muestras analizadas (figura 4). La proporción de lecturas demultiplexadas para cada código de barras osciló entre el 3,4% para el código de barras 1 al 16,9% para el código de barras 10; sin embargo, debido al gran número total de lecturas, esto todavía permitía un consenso significativo con una alta profundidad de lectura incluso para estos datasets de código de barras de menor abundancia. De hecho, el mapeo de las lecturas ordenadas a una secuencia de referencia de NPC1 dio como resultado entre 31,7% (código de barras 2) y 100% (código de barras 7 y 8) de lecturas de mapeo a la referencia, dando una profundidad de lectura de más de 90 lecturas en cualquier posición para cada muestra. Esto es más que suficiente para permitir una llamada base de consenso segura con una tasa de error insignificante.

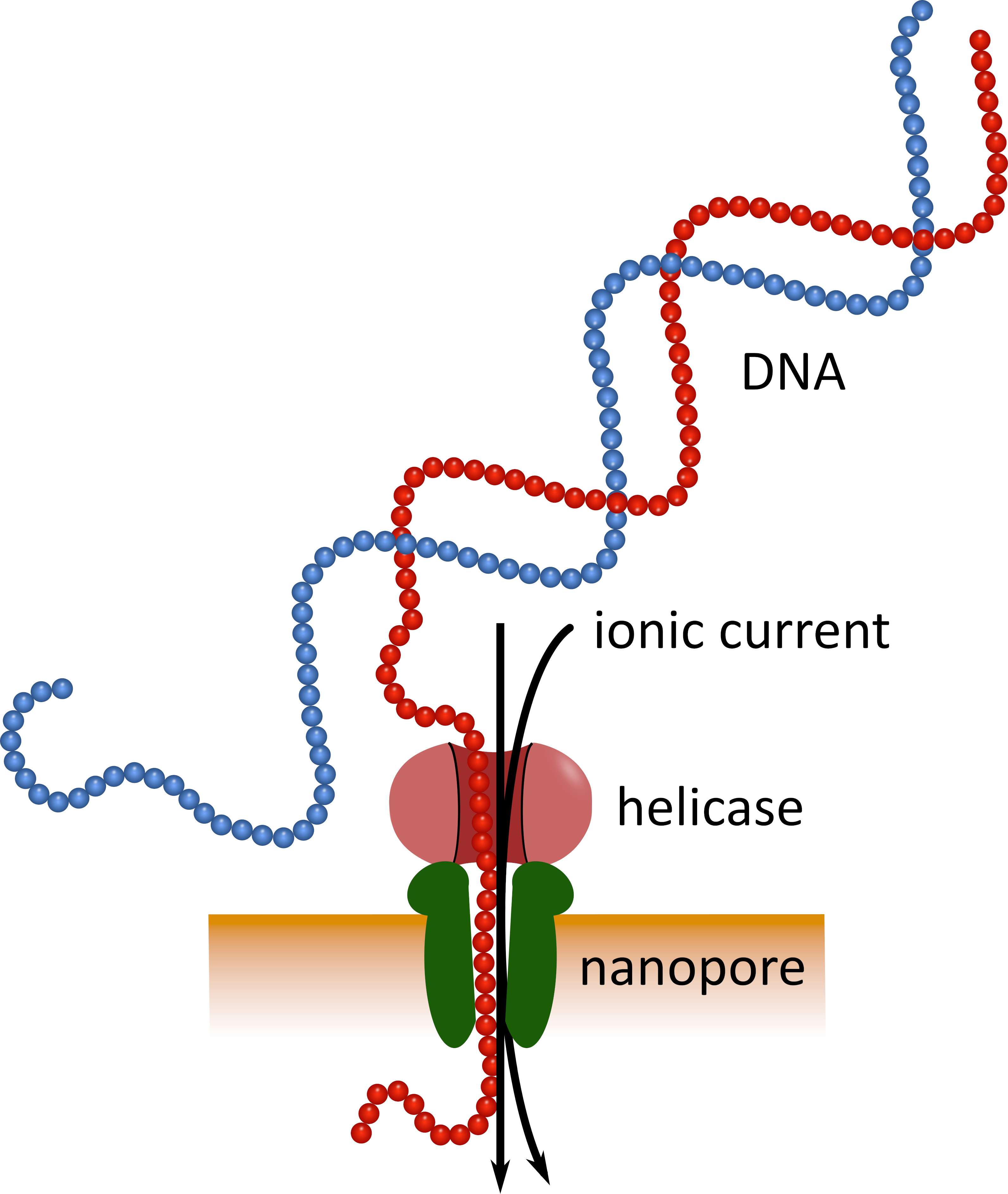
Figura 1: representación esquemática de la secuenciación de ADN utilizando tecnología de nanoporos. Una molécula de ADN de una sola cadena pasa a través de un nanoporo incrustado en una membrana resistente eléctricamente, con una helicasa que regula la velocidad de transición. Una corriente iónica pasa simultáneamente a través del poro y se mide continuamente. Las modulaciones de la corriente causadas por los nucleótidos presentes en el poro se detectan y se traducen de forma computacional a la secuencia nucleotídica de la hebra de ADN. Por favor, haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: amplificación de productos de PCR de Niemann-Pick C1 de mRNA. el mRNA fue aislado de cabra (BC01 y 02), ovejas (BC03 y 04), cerdos (BC05 y 06), perros (BC07 y 08), y bovinos (BC09 y 10). Los productos de PCR anidados fueron separados en un 0,8% gel de agarosa en 1x TAE buffer (preparado a partir de 50X TAE buffer: 242,28 g de Tris base, 57,1 mL de ácido acético glacial, 100 mL de 0,5 M EDTA, dH2O a 1 L, pH ajustado a 8,0) para 45 min a 100 V y manchado con SYBR Safe. Por favor, haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: distribución de la longitud de lectura de 10.000 lecturas del experimento representativo. Se indica el número de lecturas obtenidas con un intervalo de longitud de lectura determinado. Por favor, haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4: distribución de lecturas tras demultiplexación. Se muestra el número y el porcentaje de lecturas desmultiplexadas (grises) y mapeadas (negras) para cada código de barras. Por favor, haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Tabla 1: Resumen de los sets de imprimación utilizados. La amplificación inicial de las secuencias de destino se realizó con primer set 1. El primer set 2 fue entonces utilizado para la amplificación anidada y la adición del adaptador. Los adaptadores se indican en rojo. Por favor haga clic aquí para descargar este archivo.
Tabla 2: Resumen de las secuencias de códigos de barras. Se utilizaron códigos de barras individuales para identificar cada muestra secuenciada. Por favor haga clic aquí para descargar este archivo.
Tabla 3: concentraciones de ARN obtenidas tras la extracción de muestras de sangre secuenciadas en el experimento representativo. Se muestran las concentraciones de ARN de dos individuos de cada una de las cinco especies, y se indican las proporciones de las densidades ópticas a 260/280 nm y 260/230 Nm. Por favor haga clic aquí para descargar este archivo.
Discusión
En las últimas dos décadas, la secuenciación de muestras biológicas se ha convertido en un aspecto cada vez más importante de los estudios en una amplia gama de áreas temáticas. El desarrollo de sistemas de secuenciación de segunda generación basados en la secuenciación de una densa gama de características de ADN utilizando ciclos iterativos de manipulación enzimática y adquisición de datos basada en imágenes1 ha aumentado drásticamente el rendimiento en comparación con el técnica tradicional de secuenciación de Sanger, y permite el análisis de múltiples muestras, así como diversas especies de ácidos nucleicos en una muestra dada en paralelo4. Sin embargo, para la mayoría de los sistemas de segunda generación de uso común, sólo se producen lecturas cortas, y todas las plataformas dependen de equipos sensibles, voluminosos y caros3,4.
A diferencia de las plataformas de secuenciación de segunda generación, el dispositivo de secuenciación utilizado en este protocolo se basa en la tecnología de nanoporos. Aquí, una molécula de ácido nucleico de una sola cadena pasa a través de un nanoporo, lo que resulta en la modulación de una corriente iónica que también fluye a través del mismo nanoporo, y que se puede medir y volver a traducir para inferir la secuencia de la molécula de ácido nucleico. Este enfoque de secuenciación de tercera generación imparte una serie de ventajas sobre otros enfoques. Las principales ventajas que están directamente relacionadas con el principio de funcionamiento único de esta tecnología son la longitud de lectura extremadamente larga producida (se han reportado longitudes de lectura de hasta 8,8 x 105 nucleótidos6), la capacidad de secuenciar no sólo el ADN sino también ARN directamente, que se demostró recientemente para un genoma completo del virus de la gripe17, y la capacidad de analizar datos en tiempo real a medida que se generan, lo que permite la detección rápida metagenómica de patógenos en cuestión de minutos18. Las ventajas prácticas adicionales son el tamaño extremadamente pequeño del dispositivo de secuenciación de nanoporos, lo que permite su uso en cualquier laboratorio o en misiones de campo a ubicaciones remotas19,20y el bajo precio en comparación con otras secuencias Plataformas. En cuanto a los costes de ejecución, actualmente se requiere una nueva célula de flujo para cada ejecución de secuenciación, lo que da como resultado unos costes de aproximadamente $1.100 por ejecución para la célula de flujo y los reactivos de preparación de biblioteca. Estos costes pueden reducirse en algunos casos lavando y reutilizando la célula de flujo, o codificando y secuenciando varias muestras en una sola ejecución. También, un nuevo tipo de célula de flujo está siendo probado beta por un pequeño número de laboratorios, que requerirá el uso de un adaptador de célula de flujo (llamado un "flongle"), y debe reducir significativamente el precio de la célula de flujo y por lo tanto los costos de funcionamiento.
La mayor deficiencia de la secuenciación de nanoporos sigue siendo su precisión, con precisiones de lectura únicas en el rango de 83 a 86% que se notifican6,21,22, y la mayoría de las inexactitudes causadas por la inserción/deleciones ( InDels)5,21. Sin embargo, una alta profundidad de lectura puede compensar estas inexactitudes, y un estudio reciente sugerido basado en consideraciones teóricas que una profundidad de lectura de > 10 podría aumentar la precisión general a > 99,8%21. Sin embargo, se necesitarán mejoras adicionales en la precisión, especialmente si el análisis debe realizarse en un nivel de molécula única en lugar de en un nivel de secuencia de consenso. El uso de la tecnología 1D2 como se describe en este protocolo, que se basa en la adición de los adaptadores 1D2 y códigos de barras (cf. sección 5,5) que dan lugar a que ambas hebras de una sola molécula de ADN sean secuenciadas por el mismo nanoporo, aumenta la lectura precisión, ya que la información de ambas hebras de ADN puede utilizarse para la determinación de secuencia. Además, una estrategia de solución alternativa que se puede perseguir para combinar las ventajas de la secuenciación de nanoporos (especialmente larga longitud de lectura) con la mayor precisión de otras tecnologías de secuenciación es utilizar la información de secuenciación de nanoporos como un andamio, que es luego pulidas usando datos de secuenciación de otras plataformas6.
El factor más crítico para el éxito del protocolo presentado aquí es la calidad de la muestra, y en particular la cantidad y la calidad del ARN extraído. El almacenamiento adecuado y la extracción rápida del ARN ayudan a lograr un rendimiento de ARN adecuado. El uso de tubos de recolección de sangre apropiados permite el almacenamiento de muestras de sangre por hasta un mes, pero la coagulación de la sangre puede ser un problema, especialmente cuando las muestras se almacenan a temperaturas elevadas, que puede ser el caso en condiciones de campo. El segundo paso crítico es la amplificación de las secuencias de destino, y en particular en condiciones de campo las reacciones de PCR a menudo funcionan menos bien que en condiciones de laboratorio estándar7. Con este fin, el diseño y la optimización de imprimación cuidadosa es primordial para lograr una amplificación robusta. Además, los enfoques de PCR anidados y la PCR de touchdown, tal como se utilizan en este protocolo, pueden aumentar tanto la especificidad como la sensibilidad de la amplificación del gen objetivo4,7. De hecho, en nuestra experiencia en Liberia y Guinea con esta tecnología se requerían protocolos anidados en condiciones de campo con muestras de campo, incluso para los sets de imprimación que permitían la amplificación de objetivos a partir de muestras de laboratorio y en condiciones de laboratorio con una única ronda de PCR (7 y resultados inéditos).
En contraste con estos pasos más críticos, la preparación de la biblioteca y la ejecución de secuenciación en sí son procedimientos bastante robustos. Sin embargo, en condiciones de campo, las cuestiones prácticas, como la disponibilidad de ciertas piezas de equipo, pueden ser problemáticas. Por ejemplo, se necesita un espectrofotómetro UV para determinar las concentraciones de ADN antes de la preparación de la biblioteca de muestras con código de barras. Sin embargo, en caso de que este dispositivo no esté disponible en condiciones de campo, un volumen igual de cada muestra puede combinarse simplemente para formar los 45 μL requeridos para la preparación de la biblioteca, con diferencias en el material de entrada de la muestra, que generalmente se mitiga por el gran número de lecturas. Del mismo modo, la necesidad de conectividad a Internet para la ejecución de secuenciación puede ser un problema, aunque la llamada base ya no tenga que realizarse en línea, pero se puede realizar localmente; sin embargo, esta necesidad se puede eliminar en determinadas circunstancias por el fabricante, si es necesario.
En Resumen, el protocolo presentado permite una secuenciación relativamente de bajo costo en ubicaciones sin acceso a equipos de secuenciación tradicionales, incluso en ubicaciones remotas. Se puede adaptar fácilmente a cualquier ARN o ADN objetivo, lo que permite a los investigadores responder a numerosas preguntas biológicas.
Divulgaciones
TH participó en el programa de acceso anticipado Oxford Nanopore Technologies (ONT) MinION de 2014 a 2015, y recibió dispositivos MinION y células de flujo para un estudio anterior 7 realizado por los institutos nacionales de la salud, EE.UU., de forma gratuita o a costos reducidos . Fue invitado por ONT para presentar parte de ese trabajo en el London Calling 2015 reunión en Londres, Reino Unido, y ONT pagado para el transporte y alojamiento. Para el trabajo presentado en este manuscrito, no se obtuvieron beneficios (por ejemplo, hardware o reactivos a un costo reducido, reembolsos de viajes, etc.) de ONT. AM, KF y RS no tienen nada que revelar.
Agradecimientos
Los autores agradecen a Allison Groseth la lectura crítica del manuscrito. Este trabajo fue apoyado financieramente por el Ministerio Federal alemán de alimentación y agricultura (BMEL) basado en una decisión del Parlamento de la República Federal de Alemania a través de la Oficina Federal de agricultura y alimentación (BLE).
Materiales
| Name | Company | Catalog Number | Comments |
| 1D2 adapter, barcode adapter mix, ABB buffer, elution buffer, RBF buffer, LBB beads | Oxford Nanopore Technologies | SQK-LSK308 | 1D² Sequencing Kit |
| Blood collection tube with DNA/RNA stabilizing reagent | Zymo Research | R1150 | DNA/RNA Shield - Blood Collection Tube |
| Blunt/TA ligase master mix | New England Biolabs | M0367S | Blunt/TA Ligase Master Mix |
| DNA-low binding reaction tube | Eppendorf | 30108051 | DNA LoBind Tube |
| DNase buffer and DNase | ThermoFisher Scientific | 11766050 | SuperScript™ IV VILO™ Master Mix with ezDNase™ Enzyme |
| Flow cell | Oxford Nanopore Technologies | FLO-MIN105.24 | flow cell R9.4 |
| Hot start high fidelity DNA polymerase | New England Biolabs | M0493L | Q5 Hot Start High-Fidelity DNA Polymerase (500 U) |
| Magnetic beads | Beckman Coulter | A63881 | Agencourt AMPure XP beads |
| Magnetic rack | ThermoFisher Scientific | 12321D | DynaMag-2 Magnet |
| Nanopore sequencing device | Oxford Nanopore Technologies | - | MinION Mk 1B |
| PCR barcoding kit | Oxford Nanopore Technologies | EXP-PBC001 | PCR Barcoding Kit I (R9) |
| Reverse transcriptase master mix | ThermoFisher Scientific | 11766050 | SuperScript™ IV VILO™ Master Mix with ezDNase™ Enzyme |
| RNA purification spin column, DNA/RNA prep buffer, DNA/RNA wash buffer, DNase I, DNA digestion buffer | Zymo Research | R1151 | Quick-DNA/RNA Blood Tube Kit |
| Rotating mixer | ThermoFisher Scientific | 15920D | HulaMixer Sample Mixer |
| Taq DNA polymerase | New England Biolabs | M0287S | LongAmp Taq 2x Master Mix |
| Ultra II End-prep kit | New England Biolabs | E7546S | NEBNext Ultra II End-Repair/dA-tailing Modul |
| UV spectrophotometer | Implen | - | NanoPhotometer |
| Vacuum manifold | Zymo Research | S7000 | EZ-Vac Vacuum Manifold |
Referencias
- Shendure, J., Ji, H. Next-generation DNA sequencing. Nature Biotechnology. 26 (10), 1135-1145 (2008).
- Shendure, J., Lieberman Aiden, E. The expanding scope of DNA sequencing. Nature Biotechnology. 30 (11), 1084-1094 (2012).
- Liu, L., et al. Comparison of next-generation sequencing systems. Journal of Biomedicine and Biotechnology. 2012, 251364 (2012).
- Levy, S. E., Myers, R. M. Advancements in Next-Generation Sequencing. Annual Review of Genomics and Human Genetics. 17, 95-115 (2016).
- Lu, H., Giordano, F., Ning, Z. Oxford Nanopore MinION Sequencing and Genome Assembly. Genomics, Proteomics & Bioinformatics. 14 (5), 265-279 (2016).
- Jain, M., et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nature Biotechnology. 36 (4), 338-345 (2018).
- Hoenen, T., et al. Nanopore Sequencing as a Rapidly Deployable Ebola Outbreak Tool. Emerging Infectious Diseases. 22 (2), 331-334 (2016).
- Quick, J., et al. Real-time, portable genome sequencing for Ebola surveillance. Nature. 530 (7589), 228-232 (2016).
- Carette, J. E., et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature. 477 (7364), 340-343 (2011).
- Ndungo, E., et al. A Single Residue in Ebola Virus Receptor NPC1 Influences Cellular Host Range in Reptiles. mSphere. 1 (2), (2016).
- Martin, S., et al. A genome-wide siRNA screen identifies a druggable host pathway essential for the Ebola virus life cycle. Genome Medicine. 10 (1), 58 (2018).
- Roehr, J. T., Dieterich, C., Reinert, K. Flexbar 3.0 - SIMD and multicore parallelization. Bioinformatics. 33 (18), 2941-2942 (2017).
- Li, H., et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25 (16), 2078-2079 (2009).
- Kielbasa, S. M., Wan, R., Sato, K., Horton, P., Frith, M. C. Adaptive seeds tame genomic sequence comparison. Genome Research. 21 (3), 487-493 (2011).
- Don, R. H., Cox, P. T., Wainwright, B. J., Baker, K., Mattick, J. S. Touchdown' PCR to circumvent spurious priming during gene amplification. Nucleic Acids Research. 19 (14), 4008 (1991).
- Korbie, D. J., Mattick, J. S. Touchdown PCR for increased specificity and sensitivity in PCR amplification. Nature Protocols. 3 (9), 1452-1456 (2008).
- Keller, M. W., et al. Direct RNA Sequencing of the Coding Complete Influenza A Virus Genome. Scientific Reports. 8 (1), 14408 (2018).
- Greninger, A. L., et al. Rapid metagenomic identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis. Genome Medicine. 7, 99 (2015).
- Castro-Wallace, S. L., et al. Nanopore DNA Sequencing and Genome Assembly on the International Space Station. Scientific Reports. 7 (1), 18022 (2017).
- Goordial, J., et al. In Situ Field Sequencing and Life Detection in Remote (79 degrees 26'N) Canadian High Arctic Permafrost Ice Wedge Microbial Communities. Frontiers in Microbiology. 8, 2594 (2017).
- Runtuwene, L. R., et al. Nanopore sequencing of drug-resistance-associated genes in malaria parasites, Plasmodium falciparum. Scientific Reports. 8 (1), 8286 (2018).
- Rang, F. J., Kloosterman, W. P., de Ridder, J. From squiggle to basepair: computational approaches for improving nanopore sequencing read accuracy. Genome Biology. 19 (1), 90 (2018).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados