
Method Article
Sequenzierung von mRNA aus Vollblut mit Nanopore-Sequenzierung
In diesem Artikel
Zusammenfassung
Die Nanopore-Sequenzierung ist eine neuartige Technologie, die eine kostengünstige Sequenzierung an entfernten Orten und ressourcenarmen Umgebungen ermöglicht. Hier stellen wir ein Protokoll zur Sequenzierung von mRNAs aus Vollblut vor, das mit solchen Bedingungen kompatibel ist.
Zusammenfassung
Die Sequenzierung an entlegenen Orten und ressourcenarmen Umgebungen stellt einzigartige Herausforderungen dar. Die Nanosequenzierung kann unter solchen Bedingungen erfolgreich eingesetzt werden und wurde während der jüngsten Ebola-Virus-Epidemie nach Westafrika eingesetzt, was diese Möglichkeit unterstreicht. Neben den praktischen Vorteilen (kostengünstige, einfache Gerätetransporte und-nutzung) bietet diese Technologie auch grundlegende Vorteile gegenüber Sequenzierungsansätzen der zweiten Generation, insbesondere die sehr lange Leselänge, die Fähigkeit, direkt zu sequenzieren. RNA, und Echtzeitverfügbarkeit von Daten. Die Rohlesegenauigkeit ist geringer als bei anderen Sequenzierplattformen, was die Haupteinschränkung dieser Technologie darstellt; Dies kann jedoch durch die hohe Lesefe teilweise abgemildert werden. Hier stellen wir ein feldkompatibles Protokoll zur Sequenzierung der mRNA-Kodierung für Niemann-Pick C1 vor, das der zelluläre Rezeptor für ebolaviruses ist. Dieses Protokoll umfasst die Extraktion von RNA aus tierischen Blutproben, gefolgt von RT-PCR für die Zielanreicherung, Barcodierung, Bibliotheksvorbereitung und den Sequenzierungslauf selbst und kann leicht für den Einsatz mit anderen DNA oder RNA-Zielen angepasst werden.
Einleitung
Sequenzierung ist ein leistungsfähiges und wichtiges Werkzeug in der biologischen und biomedizinischen Forschung. Es ermöglicht die Analyse von Genomen, genetischen Variationen und RNA-Expressionsprofilen und spielt damit eine wichtige Rolle bei der Erforschung von menschlichen und tierischen Krankheiten gleichermaßen 1,2. Sanfere Sequenzierung, eine der ältesten Methoden zur DNA-Sequenzierung, wird bis heute routinemäßig eingesetzt und ist ein Eckpfeiler der Molekularbiologie. In den letzten 50 Jahren wurde diese Technologie verbessert, um Leselängen von mehr als 1.000 nt und eine Genauigkeit von bis zu 99,999% 1 zu erreichen. Die Sanger-Sequenzierung hat aber auch Einschränkungen. Die Sequenzierung eines größeren Samples oder die Analyse ganzer Genome mit dieser Methodeistzeitaufwendig und teuer 1,3. DNA-Sequenzierungsmethoden der zweiten Generation (nächste Generation) wie 454 Pyrosequenzierungsmethoden und Illumina-Technologie haben es uns ermöglicht, die Kosten und die Arbeitsbelastung, die für die Sequenzierung erforderlich sind, in den letzten zehn Jahren deutlich zu reduzieren, und haben zu einem enormen Anstieg der Anzahl der biologischen Sequenzinformationen verfügbar4. Dennoch sind einzelne Sequenzierungsläufe mit diesen Technologien der zweiten Generation teuer, und die Sequenzierung unter Feldbedingungen ist herausfordernd, da die notwendige Ausrüstung sperrig und zerbrechlich ist (ähnlich wie bei Sanger-Sequenzierungsgeräten), und muss oft zu Sie werden von speziell geschultem Personal kalibriert und gewartet. Auch für viele der Leserlängen der zweiten Generation sind die Leselängen eher begrenzt, was die nachgelagerte Bioinformatik-Analyse dieser Daten oft herausfordernd macht.
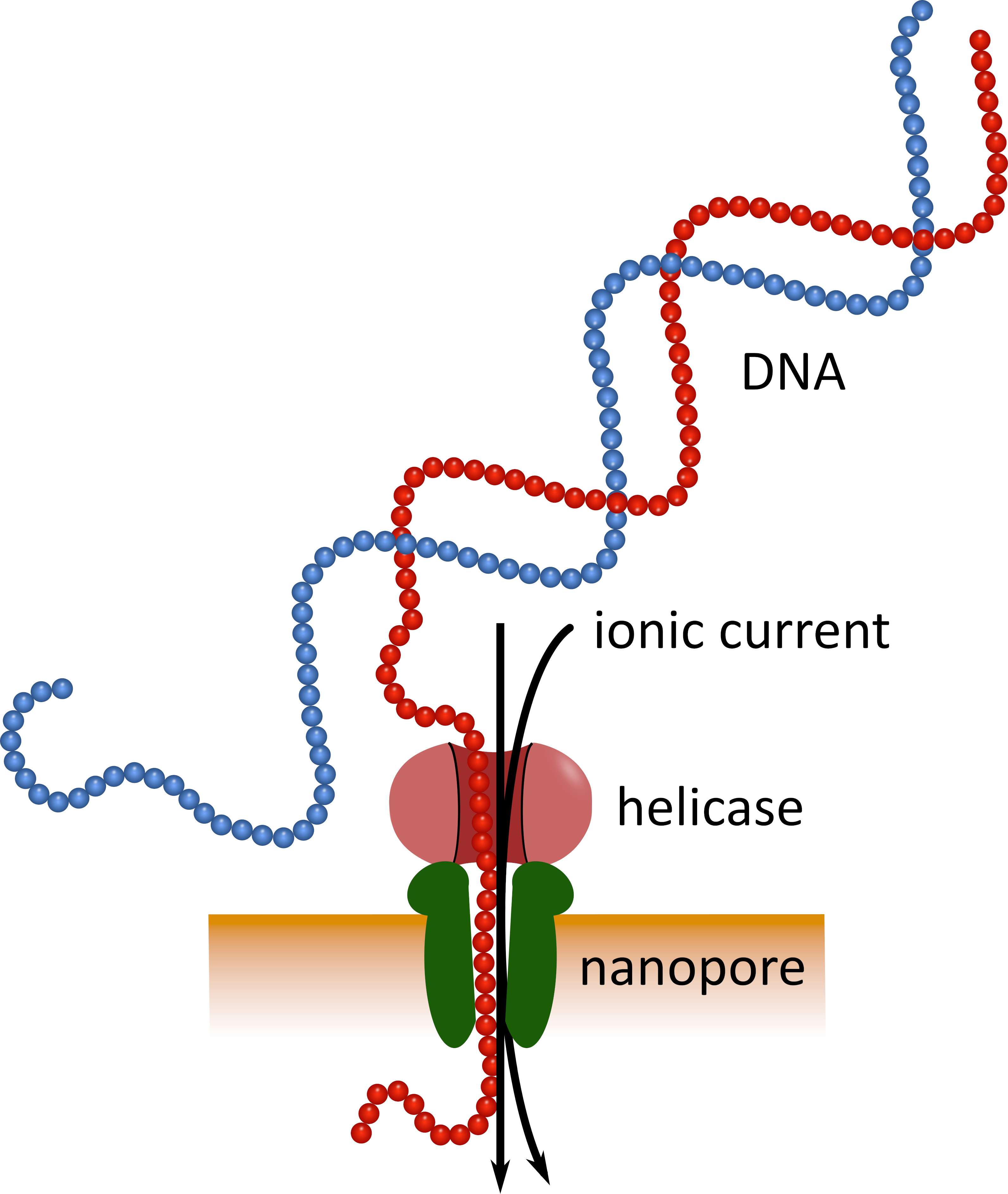
Die Sequenzierung der dritten Generation mit Hilfe von Nanopore-Sequenzierungsgeräten im Taschenformat (siehe Materialtabelle) kann als Alternative zu diesen etablierten Sequenzierplattformen dienen. Bei diesen Geräten durchläuft ein einsträngiges DNA-oder RNA-Molekül gleichzeitig ein Nanoporum mit einem ionischen Strom, der dann von einem Sensor gemessen wird (Abbildung1). Während der Strang den Nanopore durchquert, wird die Modulation des Stroms durch die in der Pore vorhandenen Nukleotide zu jeder Zeit erkannt und rechnerisch wieder in die Nukleotid-Sequenz 5 übersetzt. Aufgrund dieses Operationsprinzips ermöglicht die Nanopore-Sequenzierung sowohl die Generierung von sehr langen Lesezeichen (in der Nähe von 1 x 106 Nukleotiden 6) als auch die Analyse von SequenzierungsdateninEchtzeit. Die Barcodierung ist möglich, indem definierte Nukleotidsequenzen an den Nukleinsäuren in einer Probe befestigt werden, was die Analyse mehrerer Proben in einem einzigen Sequenzierungslauf ermöglicht und so den Probendurchsatz erhöht und die Probenkosten pro Probenkosten senkt. Aufgrund ihrer hohen Portabilität und ihrer einfachen Handhabung wurden Nanopore-Sequenziergeräte während der jüngsten Epidemie der Ebola-Viruserkrankungen in Westafrika erfolgreich vor Ort eingesetzt, was ihre Eignung für einen schnellen Einsatz in entlegenen Regionenunterstreicht . , 8.
Hier beschreiben wir ein detailliertes, feldkompatibles Protokoll zur Sequenzierung der mRNA-Kodierung für das Niemann-Pick C1 (NPC1)-Protein, das der obligatorische Einreiseempfänger für Filoviren wie ebolaviren ist und nachweislich die Anfälligkeit der Arten begrenzt. Diese Viren9,10. Das Protokoll umfasst die Extraktion ganzer RNA aus Blutproben, die spezifische Verstärkung von NPC1 mRNA durch RT-PCR, die Barcodierung von Proben, die Bibliotheksvorbereitung und die Sequenzierung mit einem Nanopore-Sequenzierungsgerät. Die Datenanalyse kann aufgrund von Platzbeschränkungen nicht diskutiert werden, obwohl in den repräsentativen Ergebnissen einige grundlegende Richtungen angegeben sind; Der interessierte Leser wird jedoch auf eine frühere Publikation11 für eine detailliertere Beschreibung des von uns verwendeten Arbeitsablaufs sowie auf Publikationen anderer12, 13,14 für detaillierte Informationen verwiesen. In Bezug auf die Analysewerkzeuge, die in diesem Workflow verwendet werden.
Protokoll
Die Proben wurden nach dem Protokoll Nr. 1 der Njala University Institutional Review Board (NUIRB) gesammelt. IRB0000886einhalb-FWA00018924.
1. RNA-Extraktion aus Blutproben
- Sammeln Sie 3 ml Vollblut von den Arten, die in eine Blutentnahme Röhre mit 6 mL DNA/RNA stabilisierenden Reagenz (siehe Tabelle der Materialien) vorgefüllt analysiert werden und mischen, indem Sie 5-mal. Die Blutprobe bis zu einem Monat bei 4 ° C aufbewahren.
- Den Inhalt des Blutentnahmerohrs in ein 50 mL-Sammelrohr geben, 120 μL Proteinase K hinzufügen und durch Wirbel für 5 s mischen.
- 9 ml Isopropanol in die Mischung und den Wirbel für 5 s geben.
- Legen Sie ein Reservoir auf eine RNA-Reinigungsspinnsäule (siehe Materialtabelle) und legen Sie die Montage auf einen Vakuumkrümmer (siehe Materialtabelle). Die Probe in den Stausee geben. Tragen Sie ein Vakuum auf, bis alle Flüssigkeit durch die Säule gelangt ist.
- Wenn kein Vakuumkrümmer zur Verfügung steht, kann das Blut in 700 μL-Portionen durch wiederholte Zentrifugation für 30 s bei 12.000 x g mit dem Durchfluss zwischen Zentrifugationsschritten durchzogen werden. Dazu sind aber etwa 26 Zentrifugationsschritte erforderlich.
- Legen Sie die RNA-Reinigungsspulsäule in ein Sammelrohr und fügen Sie 400 μL DNA/RNA-Prep-Puffer hinzu (siehe Materialtabelle). Zentrifuge bei 12.000 x g für 30 s und den Durchfluss abwerfen.
- 400 μL DNA/RNA-Waschpuffer (siehe Materialtabelle) in die Säule geben, Zentrifuge bei 12.000 x g für 30 s hinzufügen und den Durchfluss abwerfen.
- 5 μL DNase I (1 U/μL) (siehe Materialtabelle) mit 75 μL DNA-Verdauungspuffer (siehe Materialtabelle) mischen und das Gemisch auf die Säule legen. Bei Raumtemperatur 15 Minuten inkubieren.
- Fügen Sie 400 μL DNA/RNA-Prep-Puffer in die Spalte und Zentrifuge bei 12.000 x g für 30 s ein.
- Waschen Sie die Säule mit 700 μL von DNA/RNA-Waschpuffer und Zentrifuge bei 12.000 x g für 30 s.
- Wiederholen Sie den Schritt 1,9 mit 400 μL DNA/RNA-Waschpuffer und Zentrifuge bei 12.000 x g für 2 Minuten, um den gesamten Restwaschpuffer zu entfernen und die Säule zu trocknen. Achten Sie beim Entfernen der Säule aus dem Sammelrohr darauf, die Unterseite der Spalte nicht mit dem Puffer im Sammelrohr zu nassen.
-
Die Säule in ein neues 1,5 mL Mikrozentrifugenrohr geben und 70 μL kernfreies Wasser hinzufügen. Inkubieren Sie 1 min bei Raumtemperatur und Zentrifuge für 30 s bei 12.000 x g. Die RNA bei-80 ° C speichern, bis sie weiter verwendet wird (oder sofort verwendet wird).
- OPTIONAL: Um die RNA zu quantifizieren, nehmen Sie einen Aliquot und bestimmen Sie die Konzentration mit einem UV-Spektrophotometer (siehe Materialtabelle).
2. Umkehrung der Transcription von NPC1 mRNA in cDNA
- In einem 0,2 mL-Reaktionsrohr 8 μL Schablone RNA (1 pg bis 2,5 μg RNA) und 1 μL jeweils 10x DNase-Puffer und DNase-Enzym (siehe Materialtabelle) hinzufügen. Bei 37 ° C 2 min umatenfühlen. Anschließend zentrifuge die Reaktion kurz und auf Eis.
- Fügen Sie 4 μL des umgekehrten Transkriptase-Master-Mix (siehe Materialtabelle) und 6 μL kernfreies Wasser in das Reaktionsrohr und mischen Sie sanft. Die Reaktion in einem Thermocycler 10 min bei 25 ° C (für Primerglühen) umwandeln, gefolgt von 10 min bei 50 ° C (für die umgekehrte Transkription der RNA). Um das Enzym zu inaktivieren, inkubieren Sie 5 min bei 85 ° C.
- CDNA auf ein neues 1,5 mL Mikrozentrifugenrohr übertragen und bei-80 ° C bis zur weiteren Nutzung (oder sofort) lagern.
3. Verstärkung des NPC1 Open Reading Frame
-
Anfänglicher Verstärkungsschritt
- Richten Sie eine Touchdown PCR 15,16 ein, um die NPC1 cDNA mit Primer Set 1 zu verstärken (siehe Tabelle 1), mit einem heißen Start High Fidelity DNA Polymerase (siehe Tabelle der Materialien) mit dem entsprechenden Reaktionspuffer in einer 50 μL-Reaktion Volumen mit 1 μL Vorlage. Wenn möglich, die Reaktion auf Eis oder in einem 4 ° C kühlen Block aufstellen.
- Inkubieren Sie die Reaktion in einem Thermocycler mit einem ersten Denaturierungsschritt von 30 s bei 98 ° C, gefolgt von 10 Zyklen mit Denaturierung bei 98 ° C für 10 s, Primerglühen für 20 s bei 65 ° C, Senkung der Temperatur um 0,5 ° C pro Zyklus und Verlängerung für 1 min bei 72 ° C. Anschließend 20 Zyklen mit 10 s bei 98 ° C, 20 s bei 60 ° C und 1 min bei 72 ° C, gefolgt von einem letzten Dehnungsschritt von 5 min bei 72 ° C.
-
PCR-Reinigung mit Magnetperlen
- Überführung von 50 μL PCR-Produkt in ein 1,5 ml DNA-nieder-Bindungsreaktionsrohr (siehe Materialtabelle). Magnetperlen (siehe Materialtabelle) gründlich durch Wirbeln wiederbeleben und 50 μL-Perlen der PCR-Reaktion hinzufügen. Gut vermischen. Die Probe auf einem rotierenden Mixer (siehe Materialtabelle) 5 min bei Raumtemperatur (bei 15 U/min) einweichen.
- Kurz die Probe abdrehen und das 1,5 ml Mikrozentrifugenrohr auf ein Magnetgestell legen (siehe Materialtabelle), um die Magnetperlen zu pülen. Warten Sie, bis der Supernatant komplett geklärt ist, bevor es mit dem nächsten Schritt weitergeht.
- Aspirieren Sie den Supernatant, ohne die Perlenpellette zu stören und abwerfen.
- Pipette 200 μL von 70% Ethanol in das Reaktionsrohr und Inkubat für 30 s. Aspirieren Sie das Ethanol, ohne das Pellet zu stören und abwerfen. Wiederholen Sie für insgesamt zwei Waschungen. Achten Sie darauf, dass kein Ethanol übrig bleibt. Es kann notwendig sein, zuerst mit einer größeren Pipette (z.B. 1000 μL) zu saugen und dann alle verbleibenden Ethanoltröpfchen mit einer kleineren Pipette (z.B. 10 μL) zu entfernen.
- Luftrockengang des Pellets für 1 min bei Raumtemperatur.
- Entfernen Sie das Reaktionsrohr aus dem Magnetgestell, teilen Sie das Pellet in 30 μL kernfreiem Wasser wieder auf und inkubieren Sie bei Raumtemperatur 2 Minuten inkubieren.
- Legen Sie das Reaktionsrohr wieder auf das Magnetgestell und warten Sie, bis die Perlen vollständig gepellt sind.
- Entfernen Sie das Supernatant, ohne das Pellet zu stören und auf ein neues 1,5 ml Reaktionsrohr zu übertragen.
-
Zugabe von Barcode-Adaptern durch verschachtelte PCR
- Richten Sie eine 50 μL-Touchdown-PCR mit heißem Start High-Fidelity-DNA-Polymerase mit 5x Reaktionspuffer und Primer-Set 2 ein (siehe Tabelle1). Die Primer in diesem Satz bestehen aus einer zielsequenzspezifischen Region, die die Bindung von PCR-Produkten ermöglicht, die in Schritt 3.2 (innerhalb des Primer-Sets 1 Sequenzen) erzeugt werden, sowie einer Adaptersequenz, die als Ziel in der anschließenden Barcoding-PCR-Reaktion verwendet wird (vgl. Abschnitt 4). Wenn möglich, die Reaktion auf Eis oder in einem 4 ° C kühlen Block aufstellen. Verwenden Sie 1 μL des gereinigten PCR-Produktes, das in Abschnitt 3.2 als Vorlage hergestellt wird.
- Inkubieren Sie den Reaktionsmix in einem Thermocycler mit einem ersten Denaturierungsschritt von 30 s bei 98 ° C, gefolgt von 10 Zyklen mit Denaturierung bei 98 ° C für 10 s, Grundierglühen für 20 s bei 65 ° C, wodurch die Temperatur um 0,5 ° C pro Zyklus gesenkt wird. , und Verlängerung für 1 min bei 72 ° C. Anschließend die Reaktion für weitere 30 Takte für 10 s bei 98 ° C, 20 s bei 71 ° C und 1 min bei 72 ° C inkubieren, gefolgt von einem letzten Dehnungsschritt von 5 min bei 72 ° C.
- Reinigen Sie das PCR-Produkt mit Magnetperlen, wie in Abschnitt 3.2 beschrieben.
4. Barcodierung von NPC1 Amplicons
- Für jedes PCR-Produkt, das in Abschnitt 3 erzeugt wird, eine Barcoding PCR-Reaktion in einem 0,2 mL-Reaktionsrohr mit 50 μL Taq-DNA-Polymerase 2x Master-Mix (siehe Materialtabelle), 2 μL eines der Barcode-Primer aus einem PCR-Barcode-Kit (siehe Tabelle der und Tabelle 2), und 1 μL gereinigtes PCR-Produkt aus Schritt 3.3.2 als Vorlage. Um ein Endvolumen von 100 μL zu erhalten, können Sie 47 μL Nuklease-freies Wasser hinzufügen.
- Die Reaktion in einem Thermocycler bei 95 ° C 3 min als erste Denaturierung anfechten. Anschließend 15 Takte für 15 s bei 95 ° C, 15 s bei 62 ° C und 1,5 min bei 65 ° C. Als letzte Dehnung die Reaktion bei 65 ° C für 5 min inkubieren.
- Das PCR-Produkt wie unter 3.2 beschrieben reinigen, aber 100 μL Magnetperlen verwenden und in 30 μL kernfreiem Wasser elutschen.
- Wenn möglich, quantifizieren Sie die Probe mit einem UV-Spektrophotometer.
5. Bibliotheksvorbereitung
- Kombinieren Sie in einem 0,2 mL-Reaktionsrohr eine gleiche Menge barkodierter DNA aus jeder Probe für insgesamt 1 μg DNA in einem Volumen von 45 μL (bei Bedarf nuklebefreies Wasser hinzufügen). Wenn kein UV-Spektrophotometer zur Verfügung steht, verwenden Sie die gleichen Mengen jeder Probe. Für das DA-Einschneiden 7 μL End-Prep-Reaktionspuffer ( siehe Materialtabelle), 3 μL End-Prep-Enzym-Mix (siehe Materialtabelle) und 5 μL nukleasfreies Wasser hinzufügen. Mischen Sie sanft, indem Sie die Röhre flicken.
- Die Reaktion 5 min bei 20 ° C umgestalten, bei einem Thermocycler 5 min bei 65 ° C.
- Reinigen Sie das Reaktionsprodukt, wie in Abschnitt 3.2 beschrieben, verwenden Sie aber 60 μL Magnetperlen und eluteten in 25 μL nabelfreiem Wasser.
- OPTIONAL: Nehmen Sie 1 μL, um die Konzentration der Probe mit einem UV-Spektrophotometer zu quantifizieren. Der Gesamtbetrag soll mehr als 700 ng betragen.
- Kombinieren Sie 22,5 μL gereinigter DNA aus Schritt 5.3 mit 2,5 μL 1D2-Adapter (siehe Materialtabelle) und 25 μL mit Blunt/TA-Ligase-Master-Mix (siehe Materialtabelle) in einem neuen 1,5-ML DNA-Niederbindungsreaktionsrohr, mischen Sie sanft durch Flicken, und kurz drehen daunen. Bei Raumtemperatur 10 Minuten inkubieren.
- Das Reaktionsprodukt, wie in Abschnitt 3.2 beschrieben, reinigen, aber 20 μL Magnetperlen verwenden, die Inkubationszeit für die DNA-Bindung auf 10 min erhöhen, zwei Waschschritte mit je 1 ml Ethanol durchführen und in 46 μL natiesfreiem Wasser austrocknen.
- Kombinieren Sie 45 μL des Reaktionsprodukts aus Schritt 5.6 mit 5 μL Barcode-Adapter-Mix (siehe Materialtabelle) und 50 μL Blunt/TA-Ligase-Master-Mix in einem DNA-Niederbindungsreaktionsrohr. Mischen Sie sanft durch Flicken und Inkubieren für 10 Minuten bei Raumtemperatur.
- Das Reaktionsprodukt, wie in Abschnitt 3.2 beschrieben, reinigen, aber 40 μL Magnetperlen verwenden, zwei Waschschritte mit 140 μL ABB-Puffer (siehe Materialtabelle) anstelle von Ethanol durchführen, die Perlen durch Flicken und Pellet auf einem Magnetgestell wiederbeleben. Elit in 15 μL des Ellenationspuffers (siehe Materialtabelle). Erhöhen Sie die Inkubationszeiten für die anfängliche Bindung der DNA an die Perlen sowie für den Ellungsschritt auf 10 min. Bewahren Sie das resultierende Produkt bis zum Gebrauch auf Eis oder bei 4 ° C.
6. Qualitätsprüfung der Flow-Cell
- Führen Sie vor dem Einsatz eine Qualitätsprüfung der Strömungszelle durch. Verbinden Sie dazu das Sequenzierungsgerät mit dem Host-Computer und öffnen Sie die Software.
- Legen Sie eine Fließzelle (siehe Materialtabelle) in das Sequenzierungsgerät ein und wählen Sie den Strömungszellentyp aus dem Auswahlfeld aus und bestätigen Sie durch einen Klick zur Verfügung.
- Klicken Sie auf die Durchflusszelle am unteren Bildschirmrand und wählen Sie den richtigen Fließzellentyp.
- Klicken Sie auf den Start-Test , um die Qualitätsprüfung zu starten. Insgesamt sind mindestens 800 aktive Nanoporen erforderlich, damit die Fließzelle nutzbar ist.
7. Laden der Flow-Zelle und Start der Sequenzierung Lauf
- Öffnen Sie die grundierende Portwülle, indem Sie sie im Uhrzeigersinn verrutschen. Setzen Sie eine P1000-Pipette auf 200 μL und stecken Sie die Spitze in den Grundierhafen. Stellen Sie die Pipette auf 230 μL ein und halten Sie die Spitze im Grundierhafen, um 20-30 μL-Puffer zu zeichnen und Luftblasen zu entfernen.
- In einem neuen 1,5-ML DNA-Niederbindungsrohr bereiten Sie den Grundiermix vor, indem Sie 576 μL RBF-Puffer (siehe Materialtabelle) mit 624 μL natikfreies Wasser kombinieren.
- Die vorbereitete Grundierung wird vorsichtig mit 800 μL in den Grundierhafen gesteckt und wartet 5 Minuten.
- Pipette 35 μL des RBF-Puffers in ein neues, sauberes 1,5 ml DNA-niedrigbindendes Reaktionsrohr. LLB-Perlen (siehe Materialtabelle) gründlich durch Pipettieren mischen und 25,5 μL der Perlen in den RBF-Puffer einfügen. 2,5 μL nukleasfreies Wasser und 12 μL DNA-Bibliothek aus Schritt 5.8 hinzufügen und durch Pipetting mischen.
- Über den Probenport 75 μL des Probengemisches in einer langsamen Dropzeitri-Weise in die Fließzelle geben.
- Ersetzen Sie die Probenportabdeckung, schließen Sie den Grundierhafen und schließen Sie den Deckel des Sequenzierungsgerätes.
- Bestätigen Sie innerhalb der Software, dass die Fließzelle noch verfügbar ist, öffnen Sie ein neues Experiment und stellen Sie die Laufparameter ein, indem Sie den verwendeten Bausatz auswählen. Wählen Sie Live-Basen-Rufe. Starten Sie die Sequenzierung, die ausgeführt wird, indem Sie auf "Experimentbeginnen" klicken. Setzen Sie den Sequenzierungslauf fort, bis genügend experimentelle Daten gesammelt werden.
Ergebnisse
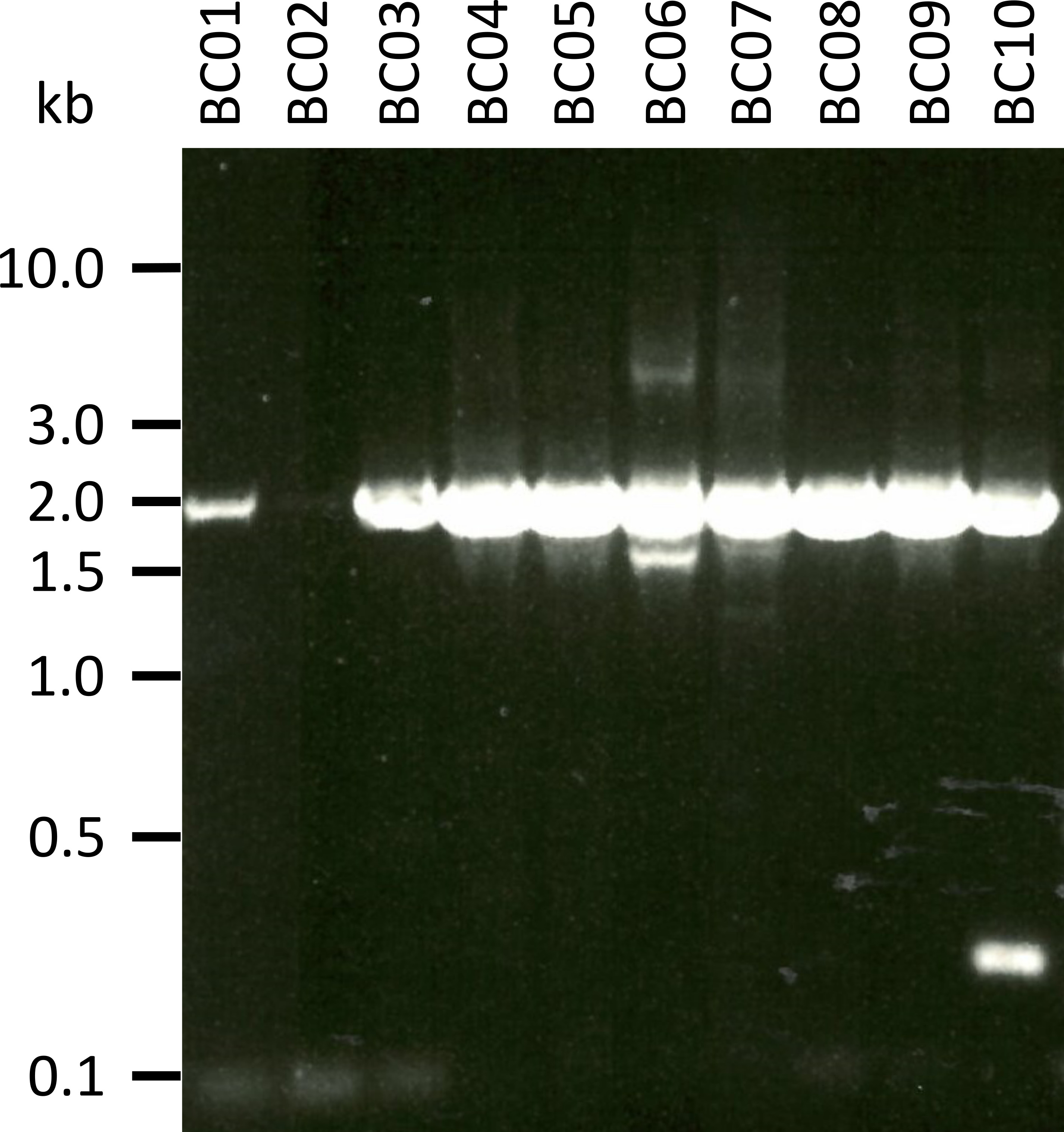
In einem repräsentativen Experiment zur Prüfung des vorgestellten Protokolls haben wir die RNA aus 10 verschiedenen Blutproben von fünf Tierarten extrahiert (d.h. 2 Individuen pro Art (Ziege, Schafe, Schweine, Hund, Vieh)) (Tabelle 3). RNA-Erträge und Qualität nach der Extraktion können sehr unterschiedlich sein, insbesondere aufgrund von Unterschieden in der Probenbehandlung und-lagerung. In unserem repräsentativen Experiment beobachteten wir RNA-Konzentrationen zwischen 43 ng und 543 ng pro μL (Tabelle 3). Auch nach der Verstärkung durch RT-PCR zeigte die Gel-Analyse der NPC-1-PCR-Produkte verschiedene Ergebnisse (Abbildung 2), mit deutlich schwächeren Bändern für die Proben BC01 und BC02 (beide Ziege). Diese Unterschiede wurden höchstwahrscheinlich durch Unterschiede in der Probenqualität verursacht, obwohl Unterschiede in der PCR-Wirksamkeit aufgrund von Unterschieden in der Primerbindung an das NPC1-Gen verschiedener Arten nicht ausgeschlossen werden können. Diese Unterschiede in der Ausbeute und der Effizienz der Verstärkung haben sich jedoch nicht signifikant auf das Gesamtergebnis der Sequenzierung ausgewirkt. Darüber hinaus kam es in der Probe BC10 (Rinder) zu einem zusätzlichen unspezifischen PCR-Produkt. Im Gegensatz zur Sanger-Sequenzierung beeinflussen solche unspezifischen Produkte die Ergebnisse der Nanopore-Sequenzierung nicht negativ, da diese Lesezeichen während der Kartierung der erhaltenen Lesezeichen in eine Referenzsequenz im Rahmen der Datenanalyse verworfen werden.
Vor jedem Sequenzierungslauf wird eine Qualitätsprüfung der zu verwendenden Fließzelle mit einem Mindestanforderung von 800 Poren dringend empfohlen. In unserem repräsentativen Experiment wurden bei dieser Qualitätsprüfung 1.102 Poren für die Sequenzierung zurückgegeben. Da die Daten in Echtzeit zur Verfügung gestellt werden und sofort ausgewertet werden können, kann die Länge eines Sequenzierungslaufs für die jeweilige Anwendung angepasst werden (d.h. bis für die gewünschte Analyse ausreichende Sequenzierungsdaten erstellt werden). In unseren Experimenten werden Sequenzierungsläufe in der Regel über Nacht durchgeführt, und im Falle unseres repräsentativen Experiments haben wir bei einem solchen 14-stünden-Run etwa 1,4 Millionen Lesezeiten erhalten.
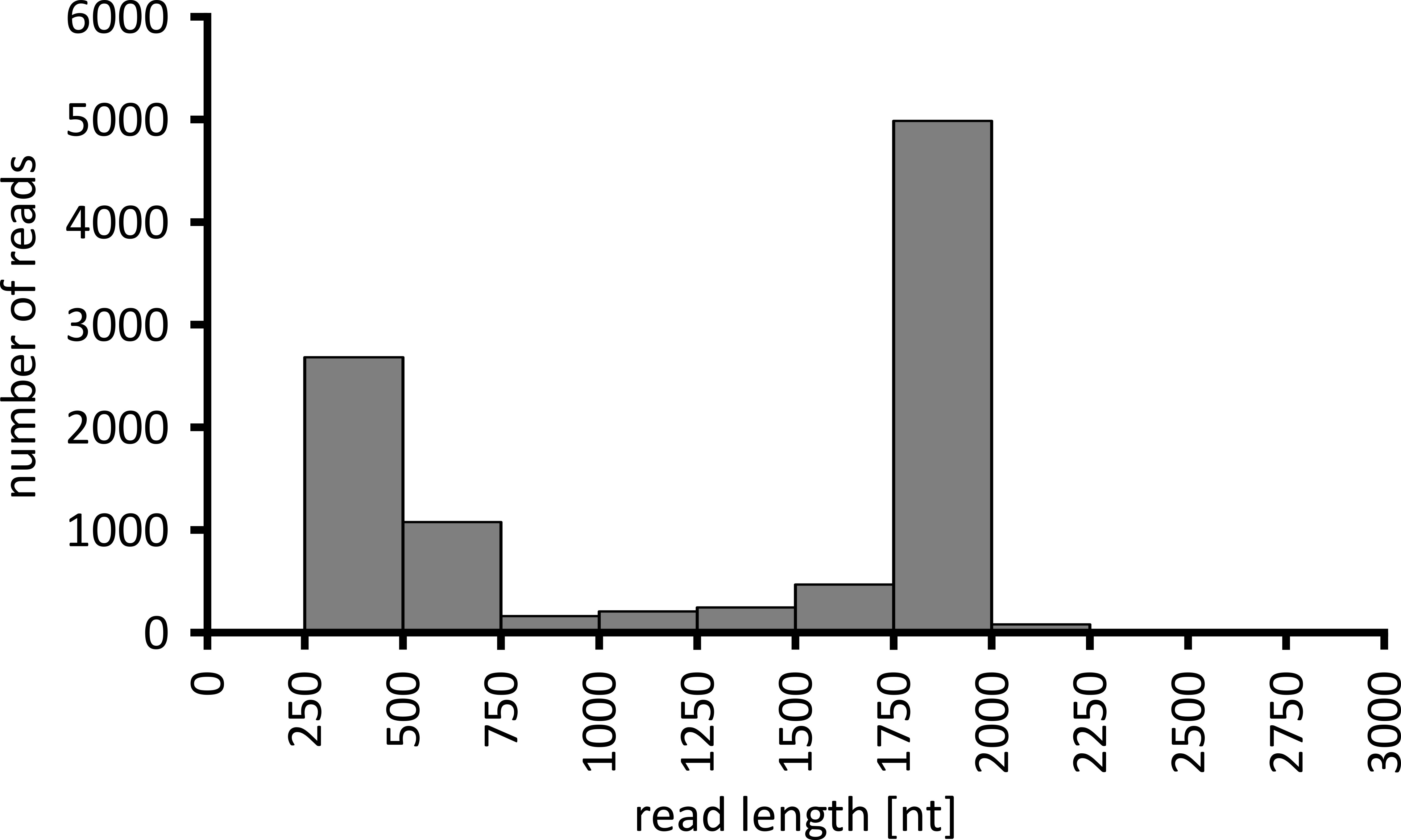
Je nach Art der zu durchführenden Datenanalyse kann es ratsam sein, nur eine Teilmenge der erhaltenen Lesevorgänge zu verarbeiten. Im Falle unseres repräsentativen Experiments wurde eine Teilmenge von 10.000 Lesezeichen für die weitere Analyse ausgewählt. Zu diesem Zweck wurden die während des Sequenzierungslaufs generierten Fastq-Dateien in einer Ubuntu 18.04 LTS-Umgebung weiterverarbeitet und mit Flexbar v3.0.3 mit Parametern dekultiplexed (Barcode-Schwanzlänge 300). , Barcode-Fehlerquote 0,2, Barcode-Gap-Strafe-1)12. Nach der Demultiplexing können dann mit verschiedenen Werkzeugen die Lesekartierung und Konsensgenerierung durchgeführt werden, aber eine ausführliche Diskussion über den bioinformatischen Aspekt der Nanopore-Sequenzierung geht über den Rahmen dieses Manuskripts hinaus. Bei unseren repräsentativen Ergebnissen wurde jedoch die Leseabbildung zu einer Referenzsequenz mit Geneious 10.2.3 durchgeführt. Von den 10.000 analysierten Lesungen zeigte 5.457 eine Länge zwischen 1.750 und 2.000 Nukleotiden, die den erwarteten Größen für die PCR-Fragmente entspricht, dieim Rahmen unseres Workflows verstärkt wurden (1.769 nt, Abbildung 3). Ein zusätzlicher Höhepunkt in der Längenverteilung der Lesewerte wurde zwischen 250 und 500 Nukleotiden beobachtet, die auf unspezifische PCR-Produkte zurückgeführt werden können. Die Entschlüsselung der Lesevorlesungen ermöglichte die Zuordnung von 87,6% der reads an eines der 10 untersuchten Barcodes/Proben (Abbildung4). Der Anteil der dekultiplexed reads für jeden Barcode reichte von 3,4% für Barcode 1 bis 16,9% für Barcode 10; Aufgrund der insgesamt großen Anzahl von Lesungen ermöglichte dies jedoch immer noch einen sinnvollen Konsens, der mit einer hohen Leseptiefe auch für diese Barcode-Datensätze mit niedrigerem Überfluss aufrief. In der Tat führte die Kartierung der sortierten Lesezeichen zu einer Referenzsequenz von NPC1 zwischen 31,7% (Barcode 2) und 100% (Barcode 7 und 8) der Lesekarten zu der Referenz, was eine Lesefe von mehr als 90 Lesezeichen an jeder beliebigen Position für jede Probe ergibt. Dies ist dann mehr als ausreichend, um einen selbstbewussten Konsens bei einer vernachlässigbaren Fehlerquote zu ermöglichen.

Abbildung 1: Schematische Darstellung der DNA-Sequenzierung mittels Nanopore-Technologie. Ein einsträngiges DNA-Molekül durchläuft ein Nanoporum, das in eine elektrisch resistente Membran eingebettet ist, wobei eine Helicase die Übergangsgeschwindigkeit reguliert. Ein ionischer Strom durchläuft gleichzeitig die Pore und wird kontinuierlich gemessen. Die Strömungsmodulationen, die durch die in der Pore vorhandenen Nukleotide verursacht werden, werden erkannt und rechnerisch wieder in die Nukleotidsequenz des DNA-Strandes übersetzt. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}

Abbildung 2: Verstärkung der PCR-Produkte von Niemann-Pick C1 von mRNA. mRNA wurde isoliert von Ziege (BC01 und 02), Schafen (BC03 und 04), Schweine (BC05 und 06), Hund (BC07 und 08) und Rinder (BC09 und 10). Nested PCR-Produkte wurden in einem 0,8%-Agarch-Gel im 1x TAE-Puffer (hergestellt aus 50x TAE-Puffer) getrennt: 242,28 g Tris-Basis, 57,1 mL Gletscherätersäure, 100 ml von 0,5 M EDTA, dH 2 O bis 1 L, pHauf8,0 eingestellt) für 45 min bei 100 V und mit Sybr sicher. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}

Abbildung 3: Langlange Verteilung von 10.000 Lesezeichen aus dem repräsentativen Experiment. Die Anzahl der gewonnenen Lesungen mit einem gegebenen Leselümintervall wird angegeben. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}

Abbildung 4: Verteilung der Lesevorgänge nach der Demultiplexing. Die Anzahl und der Prozentsatz der dekultiplexed (grau) und gemappten Lesezeichen (schwarz) für jeden Barcode werden angezeigt. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}
Tabelle 1: Übersicht der verwendeten Primer-Sets. Die erste Verstärkung der Zielsequenzen wurde mit Primer Set 1 durchgeführt. Das Primer Set 2 wurde dann für die verschachtelte Verstärkung und Adapterzusatzung verwendet. Adapter sind rot gekennzeichnet. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Tabelle 2: Übersicht über Barcode-Sequenzen. Es wurden einzelne Barcodes verwendet, um jede sequenzierte Probe zu identifizieren. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Tabelle 3: RNA-Konzentrationen, die nach der Extraktion aus Blutproben gewonnen wurden, die im repräsentativen Experiment sequenziert wurden. Die RNA-Konzentrationen von zwei Individuen aus je fünf Arten werden angezeigt, und die Verhältnisse der optischen Dichten bei 260/280 nm und 260/230 nm werden angezeigt. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Diskussion
In den letzten zwei Jahrzehnten ist die Sequenzierung biologischer Proben zu einem immer wichtigeren Aspekt der Studien in den verschiedensten Fachgebieten geworden. Die Entwicklung von Sequenzierungssystemen der zweiten Generation, die auf der Sequenzierung einer dichten Reihe von DNA-Features mit iterativen Zyklen der enzymatischen Manipulation und bildbasierten Datenerfassung1 basieren, hat den Durchsatz im Vergleich zu den Traditionelle Sanger-Sequenziertechnik, und ermöglicht die Analyse von mehreren Proben sowie verschiedenen Nukleinsäurearten in einer bestimmten Probein parallelen 4. Für die meisten der gängigen Systeme der zweiten Generation werden jedoch nur kurze Lesezeichen produziert, und alle Plattformen sind auf empfindliche, sperrige und teureGeräte 3,4angewiesen.
Im Gegensatz zu den Sequenzierplattformen der zweiten Generation basiert das in diesem Protokoll verwendete Sequenzierungsgerät auf der Nanopore-Technologie. Hier durchläuft ein einsträndisches Nukleinsäuremolekül ein Nanoporum, was zur Modulation eines ionischen Stroms führt, der ebenfalls durch denselben Nanopore fließt und gemessen und kopfüber übersetzt werden kann, um die Sequenz des Nukleinsäuremeküls abzuleiten. Dieser Sequenzierungsansatz der dritten Generation vergibt eine Reihe von Vorteilen gegenüber anderen Ansätzen. Die Hauptvorteile, die direkt mit dem einzigartigen Arbeitsprinzip dieser Technologie zusammenhängen, sind die extrem lange produzierte Leselänge (Leselängen von bis zu 8,8 x 105 Nukleotiden wurden angegeben 6), die Fähigkeit, nicht nur DNA, sondern auch DNA zu sequenzieren, sondern Auch RNA direkt, die kürzlich für ein komplettes Influenza-Virus-Genom17nachgewiesen wurde, und die Fähigkeit, Daten in Echtzeit zu analysieren, wie sie erzeugt werden, was eine schnelle Metagenomik-Erkennung von Krankheitserregern innerhalb von Minuten18 ermöglicht. Weitere praktische Vorteile sind die extrem geringe Größe des Nanopore-Sequenzierungsgerätes, das den Einsatz in jedem Labor oder bei Feldmissionen an entlegenen Orten19,20ermöglicht, und der niedrige Preis im Vergleich zu anderen Sequenzierungen Plattformen. Was die laufenden Kosten angeht, so ist derzeit für jeden Sequenzierungslauf eine neue Fließzelle erforderlich, was Kosten von etwa 1.100 Dollar pro Lauf für die Fließzelle und die Bibliotheksvorbereitung nach Reagenzien verursacht. Diese Kosten können in einigen Fällen durch das Waschen und Wiederverwenden der Strömungszelle oder durch Barcodierung und Sequenzierung mehrerer Proben in einem Durchlauf reduziert werden. Außerdem wird derzeit eine neuartige Art von Fließzelle von einer kleinen Anzahl von Labors getestet, die den Einsatz eines Strömungszelladapters ("Flongle" genannt) erfordern und den Fließzellpreis und damit die laufenden Kosten deutlich senken sollen.
Das größte Manko der Nanopore-Sequenzierung bleibt ihre Genauigkeit, wobei einzelne Lesgenauigkeiten im Bereich von 83 bis 86% zwischen6,21,22Jahren gemeldet werden und die meisten Ungenauigkeiten durch Einfügungen/Löschungen verursacht werden ( Indels)5,21. Eine hohe Leseptiefe kann diese Ungenauigkeiten jedoch kompensieren, und eine aktuelle Studie, die auf theoretischen Überlegungen beruht, dass eine Leseptiefe von & gt;10 die Gesamtgenauigkeit auf bis gt;99.8% 21 erhöhen könnte. Dennoch sind weitere Präzisionsverbesserungen erforderlich, insbesondere wenn die Analyse auf einer Ebene der einzelnen Moleküle und nicht auf einer Konsenssequenz durchgeführt werden soll. Die Verwendung der 1D2-Technologie , wie sie in diesem Protokoll beschrieben wird, die auf der Addition der 1D 2 und Barcode-Adapter (vgl. Abschnitt 5.5) basiert, die dazu führen, dass beide Stränge eines einzelnen DNA-Moleküls durch das gleiche Nanopore sequenziert werden, erhöht das Lesen Genauigkeit, da Informationen aus beiden DNA-Strängen zur Sequenzbestimmung verwendet werden können. Darüber hinaus ist eine Workaround-Strategie, die verfolgt werden kann, um die Vorteile der Nanopore-Sequenzierung (insbesondere lange Leselänge) mit der höheren Genauigkeit anderer Sequenzierungstechnologien zu kombinieren, die Verwendung von Nanopore-Sequenzierungsinformationen als Gerüst, das heißt Anschließend poliert mit Sequenzierungsdaten von anderen Plattformen6.
Der kritischste Faktor für den Erfolg des hier vorgestellten Protokolls ist die Probenqualität und insbesondere die Menge und Qualität der extrahierten RNA. Eine ordnungsgemäße Speicherung und schnelle Extraktion der RNA-Hilfe bei der Erreichung eines angemessenen RNA-Ausbeute. Der Einsatz geeigneter Blutentnahrohre ermöglicht die Lagerung von Blutproben für bis zu einem Monat, aber Blutgerinnung kann ein Problem sein, vor allem, wenn Proben bei erhöhten Temperaturen gelagert werden, was unter Feldbedingungen der Fall sein kann. Der zweite kritische Schritt ist die Verstärkung der Zielsequenzen, und insbesondere unter Feldbedingungen schneiden PCR-Reaktionen oft weniger gut ab als unternormalen Laborbedingungen 7. Dazu ist ein sorgfältiges Grundierungsdesign und-optimierung von entscheidender Bedeutung, um eine robuste Verstärkung zu erreichen. Darüber hinaus können verschachtelte PCR-Ansätze und Touchdown-PCR-Ansätze, wie sie in diesem Protokoll verwendet werden, sowohl die Spezifität als auch die Empfindlichkeit der Zielgenverstärkung4,7erhöhen. In der Tat, nach unserer Erfahrung in Liberia und Guinea mit dieser Technologie verschachtelte Protokolle wurden unter Feldbedingungen mit Feldproben auch für Primer-Sets, die Verstärkung von Zielen aus Laborproben und unter Laborbedingungen mit Eine einzige PCR-Runde (7 und unveröffentlichte Ergebnisse).
Im Gegensatz zu diesen kritischeren Schritten sind die Bibliotheksvorbereitung und der Sequenzierungslauf selbst eher robuste Verfahren. Unter feldbedingten Bedingungen können praktische Probleme wie die Verfügbarkeit bestimmter Geräte jedoch problematisch sein. So wird beispielsweise ein UV-Spektrophotometer benötigt, um die DNA-Konzentrationen vor der Bibliotheksvorbereitung von barkodierten Proben zu ermitteln. Sollte ein solches Gerät jedoch unter Feldbedingungen nicht verfügbar sein, kann ein gleichmäßiges Volumen jeder Probe einfach kombiniert werden, um die 45 μL zu bilden, die für die Bibliotheksvorbereitung benötigt werden, wobei die Unterschiede im Probeneingangsmaterial dann in der Regel durch die große Anzahl der liest. In ähnlicher Weise kann die Notwendigkeit einer Internetverbindung für den Sequenzierungslauf ein Problem sein, auch wenn die Grundanrufung nicht mehr online durchgeführt werden muss, sondern lokal durchgeführt werden kann; Diese Notwendigkeit kann der Hersteller jedoch unter bestimmten Umständen bei Bedarf beseitigen.
Zusammenfassend lässt sich sagen, dass das vorgestellte Protokoll eine relativ kostengünstige Sequenzierung an Orten ohne Zugriff auf herkömmliche Sequenzierungsgeräte ermöglicht, auch an entfernten Orten. Es lässt sich leicht an jede Zielgruppe RNA oder DNA anpassen, so dass die Forscher zahlreiche biologische Fragen beantworten können.
Offenlegungen
Von 2014 bis 2015 nahm TH am Early Access Programm von Oxford Nanopore Technologies (ONT) MinION teil und erhielt MinION-Geräte und Fließzellen für eine frühere Studie 7 , die von den National Institutes of Health, USA, durchgeführt wurde, kostenlos oder kostengünstig. . Er wurde von ONT eingeladen, einen Teil dieser Arbeit auf dem London Calling 2015 Meeting in London, Großbritannien, zu präsentieren, und ONT zahlte für Transport und Unterkunft. Für die in diesem Manuskript vorgestellten Arbeiten wurden bei ONT keine Vorteile (z.B. Hardware oder Reagenzien zu reduzierten Kosten, Reiseerstattungen etc.) erzielt. AM, KF und RS haben nichts zu offenbaren.
Danksagungen
Die Autoren danken Allison Groseth für die kritische Lektüre des Manuskripts. Diese Arbeit wurde vom Bundesministerium für Ernährung und Landwirtschaft (BMEL) auf der Grundlage eines Beschlusses des Bundestages durch das Bundesamt für Landwirtschaft und Ernährung (BLE) finanziell unterstützt.
Materialien
| Name | Company | Catalog Number | Comments |
| 1D2 adapter, barcode adapter mix, ABB buffer, elution buffer, RBF buffer, LBB beads | Oxford Nanopore Technologies | SQK-LSK308 | 1D² Sequencing Kit |
| Blood collection tube with DNA/RNA stabilizing reagent | Zymo Research | R1150 | DNA/RNA Shield - Blood Collection Tube |
| Blunt/TA ligase master mix | New England Biolabs | M0367S | Blunt/TA Ligase Master Mix |
| DNA-low binding reaction tube | Eppendorf | 30108051 | DNA LoBind Tube |
| DNase buffer and DNase | ThermoFisher Scientific | 11766050 | SuperScript™ IV VILO™ Master Mix with ezDNase™ Enzyme |
| Flow cell | Oxford Nanopore Technologies | FLO-MIN105.24 | flow cell R9.4 |
| Hot start high fidelity DNA polymerase | New England Biolabs | M0493L | Q5 Hot Start High-Fidelity DNA Polymerase (500 U) |
| Magnetic beads | Beckman Coulter | A63881 | Agencourt AMPure XP beads |
| Magnetic rack | ThermoFisher Scientific | 12321D | DynaMag-2 Magnet |
| Nanopore sequencing device | Oxford Nanopore Technologies | - | MinION Mk 1B |
| PCR barcoding kit | Oxford Nanopore Technologies | EXP-PBC001 | PCR Barcoding Kit I (R9) |
| Reverse transcriptase master mix | ThermoFisher Scientific | 11766050 | SuperScript™ IV VILO™ Master Mix with ezDNase™ Enzyme |
| RNA purification spin column, DNA/RNA prep buffer, DNA/RNA wash buffer, DNase I, DNA digestion buffer | Zymo Research | R1151 | Quick-DNA/RNA Blood Tube Kit |
| Rotating mixer | ThermoFisher Scientific | 15920D | HulaMixer Sample Mixer |
| Taq DNA polymerase | New England Biolabs | M0287S | LongAmp Taq 2x Master Mix |
| Ultra II End-prep kit | New England Biolabs | E7546S | NEBNext Ultra II End-Repair/dA-tailing Modul |
| UV spectrophotometer | Implen | - | NanoPhotometer |
| Vacuum manifold | Zymo Research | S7000 | EZ-Vac Vacuum Manifold |
Referenzen
- Shendure, J., Ji, H. Next-generation DNA sequencing. Nature Biotechnology. 26 (10), 1135-1145 (2008).
- Shendure, J., Lieberman Aiden, E. The expanding scope of DNA sequencing. Nature Biotechnology. 30 (11), 1084-1094 (2012).
- Liu, L., et al. Comparison of next-generation sequencing systems. Journal of Biomedicine and Biotechnology. 2012, 251364 (2012).
- Levy, S. E., Myers, R. M. Advancements in Next-Generation Sequencing. Annual Review of Genomics and Human Genetics. 17, 95-115 (2016).
- Lu, H., Giordano, F., Ning, Z. Oxford Nanopore MinION Sequencing and Genome Assembly. Genomics, Proteomics & Bioinformatics. 14 (5), 265-279 (2016).
- Jain, M., et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nature Biotechnology. 36 (4), 338-345 (2018).
- Hoenen, T., et al. Nanopore Sequencing as a Rapidly Deployable Ebola Outbreak Tool. Emerging Infectious Diseases. 22 (2), 331-334 (2016).
- Quick, J., et al. Real-time, portable genome sequencing for Ebola surveillance. Nature. 530 (7589), 228-232 (2016).
- Carette, J. E., et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature. 477 (7364), 340-343 (2011).
- Ndungo, E., et al. A Single Residue in Ebola Virus Receptor NPC1 Influences Cellular Host Range in Reptiles. mSphere. 1 (2), (2016).
- Martin, S., et al. A genome-wide siRNA screen identifies a druggable host pathway essential for the Ebola virus life cycle. Genome Medicine. 10 (1), 58 (2018).
- Roehr, J. T., Dieterich, C., Reinert, K. Flexbar 3.0 - SIMD and multicore parallelization. Bioinformatics. 33 (18), 2941-2942 (2017).
- Li, H., et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25 (16), 2078-2079 (2009).
- Kielbasa, S. M., Wan, R., Sato, K., Horton, P., Frith, M. C. Adaptive seeds tame genomic sequence comparison. Genome Research. 21 (3), 487-493 (2011).
- Don, R. H., Cox, P. T., Wainwright, B. J., Baker, K., Mattick, J. S. Touchdown' PCR to circumvent spurious priming during gene amplification. Nucleic Acids Research. 19 (14), 4008 (1991).
- Korbie, D. J., Mattick, J. S. Touchdown PCR for increased specificity and sensitivity in PCR amplification. Nature Protocols. 3 (9), 1452-1456 (2008).
- Keller, M. W., et al. Direct RNA Sequencing of the Coding Complete Influenza A Virus Genome. Scientific Reports. 8 (1), 14408 (2018).
- Greninger, A. L., et al. Rapid metagenomic identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis. Genome Medicine. 7, 99 (2015).
- Castro-Wallace, S. L., et al. Nanopore DNA Sequencing and Genome Assembly on the International Space Station. Scientific Reports. 7 (1), 18022 (2017).
- Goordial, J., et al. In Situ Field Sequencing and Life Detection in Remote (79 degrees 26'N) Canadian High Arctic Permafrost Ice Wedge Microbial Communities. Frontiers in Microbiology. 8, 2594 (2017).
- Runtuwene, L. R., et al. Nanopore sequencing of drug-resistance-associated genes in malaria parasites, Plasmodium falciparum. Scientific Reports. 8 (1), 8286 (2018).
- Rang, F. J., Kloosterman, W. P., de Ridder, J. From squiggle to basepair: computational approaches for improving nanopore sequencing read accuracy. Genome Biology. 19 (1), 90 (2018).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten