
Method Article
Séquençage de l’ARNm de sang total à l’aide du séquençage NANOPORE
Dans cet article
Résumé
Le séquençage nanopore est une nouvelle technologie qui permet un séquençage rentable dans les emplacements distants et les paramètres pauvres en ressources. Ici, nous présentons un protocole pour le séquençage des mRNA de sang entier qui est compatible avec de telles conditions.
Résumé
Le séquençage dans les emplacements distants et les paramètres pauvres en ressources présentent des défis uniques. Le séquençage des nanopores peut être utilisé avec succès dans de telles conditions et a été déployé en Afrique de l’ouest au cours de la récente épidémie de virus Ebola, soulignant cette possibilité. En plus de ses avantages pratiques (faible coût, facilité de transport et d’utilisation de l’équipement), cette technologie fournit également des avantages fondamentaux par rapport aux approches de séquençage de deuxième génération, en particulier la très longue longueur de lecture, la possibilité de séquencer directement L’ARN et la disponibilité en temps réel des données. La précision de lecture brute est inférieure à celle d’autres plateformes de séquençage, ce qui représente la principale limitation de cette technologie; Toutefois, cela peut être partiellement atténué par la profondeur de lecture élevée générée. Ici, nous présentons un protocole compatible avec le champ pour le séquençage de l’encodage mRNAs pour Niemann-Pick C1, qui est le récepteur cellulaire pour les ebolavirus. Ce protocole englobe l’extraction de l’ARN des échantillons de sang animal, suivi de RT-PCR pour l’enrichissement ciblé, le codage à barres, la préparation de la bibliothèque, et le séquençage lui-même, et peut être facilement adapté pour une utilisation avec d’autres cibles d’ADN ou d’ARN.
Introduction
Le séquençage est un outil puissant et important dans la recherche biologique et biomédicale. Il permet l’analyse des génomes, des variations génétiques et des profils d’expression d’ARN, et joue donc un rôle important dans l’investigation des maladies humaines et animales1,2. Le séquençage de Sanger, l’une des méthodes les plus anciennes disponibles pour le séquençage de l’ADN, est encore couramment utilisé à ce jour et a été une pierre angulaire de la biologie moléculaire. Au cours des 50 dernières années, cette technologie a été améliorée pour atteindre des longueurs de lecture de plus de 1 000 nt et une précision aussi élevée que 99,999%1. Cependant, le séquençage Sanger a également des limitations. Séquençage d’un plus grand ensemble d’échantillons ou l’analyse de génomes entiers avec cette méthode est longue et coûteuse1,3. Les méthodes de séquençage de l’ADN de deuxième génération (nouvelle génération) telles que le 454 pyroséquençage et la technologie Illumina nous ont permis de réduire sensiblement le coût et la charge de travail nécessaires au séquençage au cours de la dernière décennie, et ont entraîné une augmentation considérable de la quantité d’information sur la séquence biologique disponible4. Néanmoins, les séquences de séquençage individuelles utilisant ces technologies de deuxième génération sont coûteuses, et le séquençage dans des conditions de terrain est difficile, car l’équipement nécessaire est volumineux et fragile (similaire aux dispositifs de séquençage Sanger), et doit souvent être étalonnés et entretenus par du personnel spécialement formé. En outre, pour de nombreuses technologies de deuxième génération, les longueurs de lecture sont assez limitées, ce qui rend difficile l’analyse bioinformatique en aval de ces données.
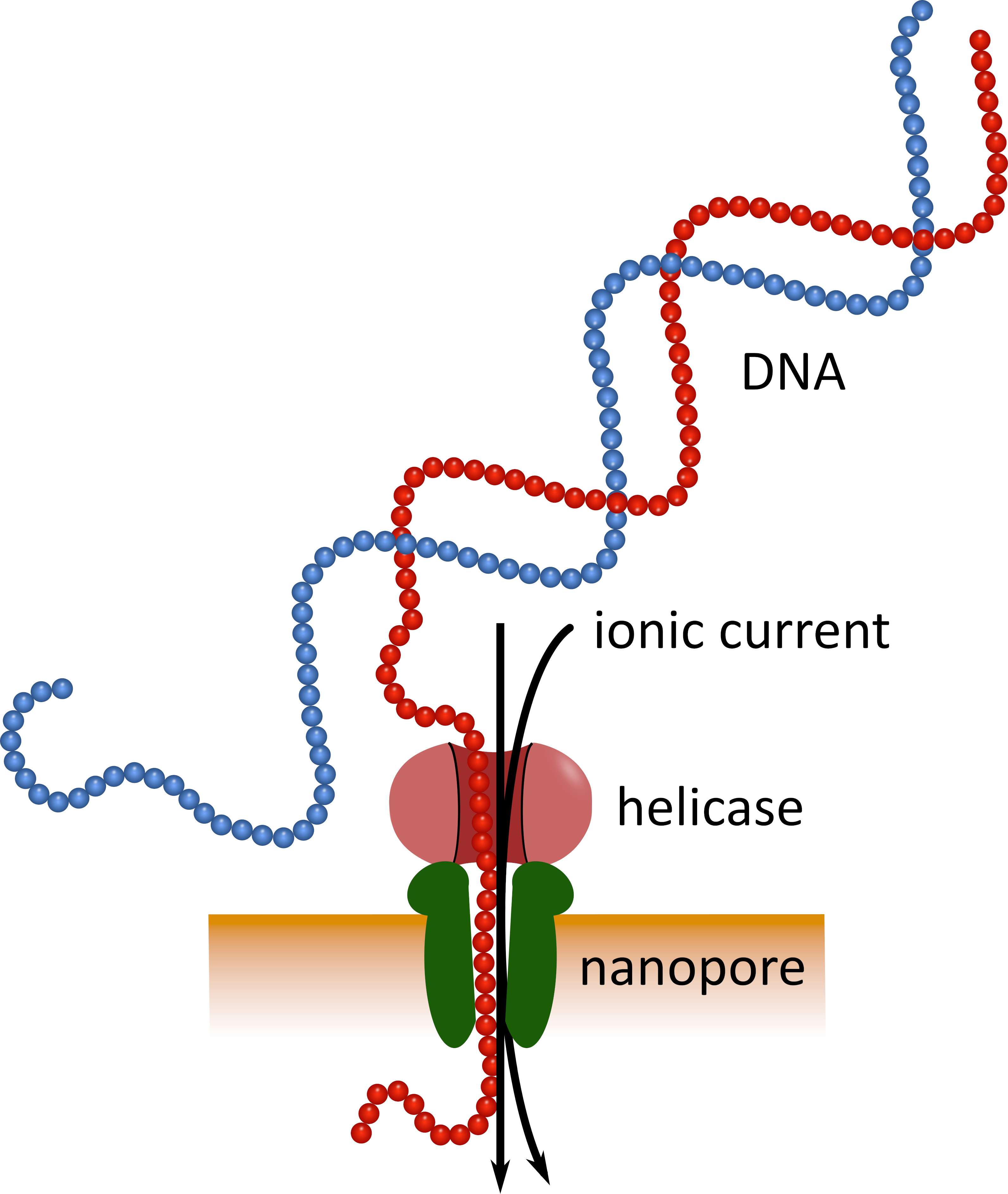
Le séquençage de troisième génération utilisant des dispositifs de séquençage de nanopores de poche (voir le tableau des matériaux) peut servir d’alternative à ces plateformes de séquençage établies. Dans ces dispositifs, une molécule d’ADN ou d’ARN monocaténaire traverse une nanopore simultanément avec un courant ionique qui est ensuite mesuré par un capteur (figure 1). Comme le brin traverse la NANOPORE, la modulation du courant par les nucléotides présents dans le pore à un moment donné est détectée, et computationellement en arrière-traduit dans la séquence de nucléotides5. En raison de ce principe opérationnel, le séquençage des nanopores permet à la fois la génération de lectures très longues (près de 1 x 106 nucléotides6) et l’analyse des données de séquençage en temps réel. Le codage à barres est possible en attachant des séquences de nucléotides définies aux acides nucléiques dans un échantillon, ce qui permet l’analyse de plusieurs échantillons en une seule séquence de séquençage, augmentant ainsi le débit des échantillons et réduisant les coûts par échantillon. En raison de leur grande portabilité et de leur facilité d’utilisation, les dispositifs de séquençage des nanopores ont été utilisés avec succès sur le terrain au cours de la récente épidémie de maladie à virus Ebola en Afrique de l’Ouest, soulignant leur aptitude à un déploiement rapide dans des régions éloignées7 , le 8.
Ici, nous décrivons un protocole détaillé compatible avec le champ pour le séquençage de l’encodage mRNA pour la protéine Niemann-Pick C1 (NPC1), qui est le récepteur d’entrée obligatoire pour les filovirus tels que les ebolavirus, et il a été montré pour limiter la susceptibilité des espèces à ces virus9,10. Le protocole englobe l’extraction de l’ARN entier à partir d’échantillons sanguins, l’amplification spécifique de l’ARNm de NPC1 par RT-PCR, le codage à barres des échantillons, la préparation de la bibliothèque et le séquençage avec un dispositif de séquençage des nanopores. L’analyse des données ne peut pas être discutée en raison de limitations de l’espace, bien que certaines directions générales soient fournies dans les résultats représentatifs; Cependant, le lecteur intéressé est renvoyé à une publication précédente11 pour une description plus détaillée du flux de travail que nous avons utilisé, ainsi que pour les publications d’autres12,13,14 pour des informations détaillées concernant les outils d’analyse utilisés dans ce Workflow.
Protocole
Des échantillons ont été recueillis à la suite du protocole no de la Commission d’examen institutionnel de l’Université Njala (NUIRB). IRB00008861/FWA00018924.
1. extraction d’ARN à partir d’échantillons sanguins
- Prélever 3 mL de sang total de l’espèce à analyser dans un tube de prélèvement sanguin prérempli avec 6 mL de réactif stabilisant ADN/ARN (voir tableau des matériaux) et mélanger en invertant 5 fois. Conserver l’échantillon sanguin jusqu’à un mois à 4 ° c.
- Transférer le contenu du tube de prélèvement sanguin dans un tube collecteur de 50 ml, ajouter 120 μl de protéinase K et mélanger par Vortex pendant 5 s. Incuber l’échantillon pendant 30 min à température ambiante.
- Ajouter 9 mL d’isopropanol au mélange et agiter pendant 5 s.
- Placez un réservoir sur une colonne de spin de purification de l’ARN (voir tableau des matériaux) et placez l’ensemble sur un collecteur de vide (voir tableau des matériaux). Ajouter le mélange d’échantillon dans le réservoir. Appliquez un aspirateur jusqu’à ce que tout le liquide ait traversé la colonne.
- Alternativement, si aucune tubulure de vide n’est disponible, le sang peut être transmis par la colonne dans 700 μL de portions par centrifugation répétée pendant 30 s à 12 000 x g avec l’écoulement à travers jeté entre les étapes de centrifugation. Toutefois, il faudra environ 26 étapes de centrifugation.
- Placer la colonne de spin de purification de l’ARN dans un tube collecteur et ajouter 400 μL de tampon de préparation d’ADN/ARN (voir tableau des matériaux). Centrifugez à 12 000 x g pendant 30 s et jetez l’écoulement.
- Ajouter 400 μL de tampon de lavage ADN/ARN (voir tableau des matériaux) dans la colonne, centrifuger à 12 000 x g pendant 30 s et jeter le flux.
- Mélanger 5 μL de DNase I (1 U/μL) (voir tableau des matériaux) avec 75 μl de tampon de digestion de l’ADN (voir tableau des matériaux) et ajouter le mélange sur la colonne. Incuber pendant 15 min à température ambiante.
- Ajouter 400 μL de tampon de préparation d’ADN/ARN à la colonne et centrifuger à 12 000 x g pendant 30 s. jetez le flux.
- Laver la colonne avec 700 μL de tampon de lavage ADN/ARN et centrifuger à 12 000 x g pendant 30 s. jetez le flux.
- Répéter l’étape 1,9 avec 400 μL de tampon de lavage ADN/ARN et centrifuger à 12 000 x g pendant 2 min pour enlever tous les tampons de lavage résiduels et sécher la colonne. Lors du retrait de la colonne du tube collecteur, veillez à ne pas mouiller le dessous de la colonne avec le tampon dans le tube collecteur.
-
Placer la colonne dans un nouveau tube de microcentrifugation de 1,5 mL et ajouter 70 μL d’eau exempte de nucléases. Incuber pendant 1 min à température ambiante et centrifuger pendant 30 s à 12 000 x g. Conserver l’ARN à-80 ° c jusqu’à utilisation ultérieure (ou utilisation immédiate).
- En option: Pour quantifier l’ARN, prendre une aliquote et déterminer la concentration à l’aide d’un spectrophotomètre UV (voir tableau des matériaux).
2. transcription inversée de l’ARNm NPC1 dans l’ADNc
- Dans un tube de réaction de 0,2 mL, ajouter 8 μL d’ARN de gabarit (1 PG à 2,5 μg d’ARN) et 1 μL chacun de tampon 10x DNase et d’enzyme DNase (voir tableau des matériaux). Incuber à 37 ° c pendant 2 min. Ensuite, Centrifugez brièvement la réaction et placez-la sur la glace.
- Ajouter 4 μL de mélange maître de transcriptase inverse (voir tableau des matériaux) et 6 μL d’eau exempte de nucléases dans le tube de réaction et mélanger doucement. Incuber la réaction dans un thermocycleur pendant 10 min à 25 ° c (pour le recuit d’amorce), suivie de 10 min à 50 ° c (pour la transcription inverse de l’ARN). Pour inactiver l’enzyme, incuber pendant 5 min à 85 ° c.
- Transférer l’ADNc à un nouveau tube de microcentrifugation de 1,5 mL et conserver à-80 ° c jusqu’à utilisation ultérieure (ou utilisation immédiate).
3. amplification du cadre de lecture ouvert NPC1
-
Étape d’amplification initiale
- Mettre en place une PCR de Touchdown15,16 pour amplifier le cDNA NPC1 avec le jeu d’amorces 1 (voir tableau 1), en utilisant une ADN polymérase haute fidélité à démarrage à chaud (voir tableau des matériaux) avec le tampon de réaction approprié dans une réaction de 50 μl volume avec un gabarit de 1 μL. Si possible, mettre en place la réaction sur la glace ou dans un bloc froid de 4 ° c.
- Incuber la réaction dans un thermocycleur avec une étape de dénaturation initiale de 30 s à 98 ° c, suivie de 10 cycles avec dénaturation à 98 ° c pendant 10 s, recuit d’amorce pour 20 s à 65 ° c, abaissement de la température de 0,5 ° c par cycle, et allongement pendant 1 min à 72 ° c. Ensuite, exécuter 20 cycles supplémentaires avec 10 s à 98 ° c, 20 s à 60 ° c, et 1 min à 72 ° c, suivi d’une étape finale d’allongement de 5 min à 72 ° c.
-
Purification par PCR à l’aide de billes magnétiques
- Transférer 50 μL de produit d’ACP dans un tube de réaction d’ADN-faible de 1,5 mL (voir tableau des matériaux). Resuspendre les billes magnétiques (voir tableau des matériaux) à fond en vortex et ajouter 50 μl de billes à la réaction PCR. Mélangez bien. Incuber l’échantillon sur un mélangeur rotatif (voir tableau des matériaux) pendant 5 min à température ambiante (à 15 tr/min).
- Tourner brièvement l’échantillon et placer le tube de microcentrifugation de 1,5 mL sur un rack magnétique (voir le tableau des matériaux) pour granulés les billes magnétiques. Attendez que le surnageant ait été complètement clarifié avant de poursuivre l’étape suivante.
- Aspirer le surnageant sans déranger le talon et le jeter.
- Pipetter 200 μL de 70% d’éthanol dans le tube de réaction et incuber pendant 30 s. aspirer l’éthanol sans déranger le culot et le jeter. Répéter pour un total de deux lavages. Assurez-vous qu’aucun éthanol n’est laissé. Il peut être nécessaire d’aspirer d’abord avec une plus grande pipette (p. ex., 1000 μL), puis d’enlever toute goutte d’éthanol restante avec une pipette plus petite (par exemple, 10 μL).
- Sécher à l’air le culot pendant 1 min à température ambiante.
- Retirez le tube de réaction du rack magnétique, Resuspendez le culot dans 30 μL d’eau exempte de nucléases et incubez pendant 2 min à température ambiante.
- Replacez le tube de réaction sur le rack magnétique et attendez que les billes soient complètement granulées.
- Enlevez le surnageant sans déranger le culot et transférez-le dans un nouveau tube de réaction de 1,5 mL.
-
Ajout d’adaptateurs de code-barres par PCR imbriquée
- Mettre en place une PCR de Touchdown de 50 μL avec l’ADN polymérase haute fidélité à démarrage à chaud avec tampon de réaction 5x et primer Set 2 (voir tableau 1). Les amorces de cet ensemble consistent en une région spécifique à la séquence cible pour permettre la liaison des produits PCR générés à l’étape 3,2 (à l’intérieur des séquences de l’ensemble d’amorce 1), ainsi qu’une séquence d’adaptateurs qui est utilisée comme cible dans la réaction PCR subséquente de codage à barres (cf. section 4). Si possible, mettre en place la réaction sur la glace ou dans un bloc froid de 4 ° c. Utiliser 1 μL du produit d’ACP purifié préparé dans la section 3,2 comme gabarit.
- Incuber le mélange réactionnel dans un thermocycleur à l’aide d’une étape de dénaturation initiale de 30 s à 98 ° c, suivie de 10 cycles avec dénaturation à 98 ° c pendant 10 s, recuit d’amorce pour 20 s à 65 ° c, abaissement de la température de 0,5 ° c par cycle et allongement pendant 1 min à 72 ° c. Ensuite, incuber la réaction pendant 30 cycles supplémentaires pendant 10 s à 98 ° c, 20 s à 71 ° c et 1 min à 72 ° c, suivie d’une étape finale d’allongement de 5 min à 72 ° c.
- Nettoyez le produit PCR à l’aide de billes magnétiques comme décrit dans la section 3,2.
4. codage à barres des amplicons NPC1
- Pour chaque produit de PCR généré à la section 3, mettre en place une réaction PCR à code à barres dans un tube de réaction de 0,2 mL à l’aide de 50 μL de mélange principal de Taq DNA polymérase 2x (voir tableau des matériaux), 2 μl de l’une des amorces de codes à barres d’un kit de codage à barres PCR (voir tableau des matériaux C1 > et tableau 2), et 1 μl de produit de PCR purifié à partir de l’étape 3.3.2 comme gabarit. Ajouter 47 μL d’eau libre de nucléase pour obtenir un volume final de 100 μL.
- Incuber la réaction dans un thermocycleur à 95 ° c pendant 3 min comme dénaturation initiale. Par la suite, exécuter 15 cycles pendant 15 s à 95 ° c, 15 s à 62 ° c et 1,5 min à 65 ° c. Pour une élongation finale, incuber la réaction à 65 ° c pendant 5 min.
- Purifier le produit PCR tel que décrit sous 3,2, mais utiliser 100 μL de billes magnétiques et éluer dans 30 μL d’eau exempte de nucléases.
- Si possible, quantifier l’échantillon à l’aide d’un spectrophotomètre UV.
5. préparation de la bibliothèque
- Combiner une quantité égale d’ADN à code à barres de chaque échantillon pour un total de 1 μg d’ADN dans un volume de 45 μL (si nécessaire, ajouter de l’eau exempte de nucléases) dans un tube de réaction de 0,2 mL. Si aucun spectrophotomètre UV n’est disponible, utilisez des volumes égaux de chaque échantillon. Pour le dosage du dA-tailing, ajouter 7 μL de tampon de réaction de préparation finale (voir tableau des matériaux), 3 μl de mélange enzymatique de préparation finale (voir le tableau des matériaux) et 5 μl d’eau exempte de nucléases. Mélanger doucement en faisant scintillement le tube.
- Incuber la réaction pendant 5 min à 20 ° c, suivie de 5 min à 65 ° c dans un thermocycleur.
- Purifier le produit de réaction comme décrit dans la section 3,2, mais utiliser 60 μL de billes magnétiques et éluer dans 25 μL d’eau exempte de nucléases.
- Facultatif: prendre 1 μl pour quantifier la concentration de l’échantillon à l’aide d’un SPECTROPHOTOMÈTRE UV. Le montant total devrait être supérieur à 700 ng.
- Combiner 22,5 μL d’ADN purifié à partir de l’étape 5,3 avec 2,5 μL d’adaptateur 1D2 (voir tableau des matériaux) et 25 μL de mélange principal Blunt/ta ligase (voir tableau des matériaux) dans un nouveau tube à réaction d’adn-faible de 1,5 ml, mélanger doucement en faisant un scintillement et tourner brièvement duvet. Incuber pendant 10 min à température ambiante.
- Purifier le produit de réaction comme décrit dans la section 3,2, mais utiliser 20 μL de billes magnétiques, augmenter le temps d’incubation pour la liaison de l’ADN à 10 min, effectuer deux étapes de lavage avec 1 mL d’éthanol chacun, et éluer dans 46 μL d’eau exempte de nucléases.
- Combiner 45 μL du produit de réaction de l’étape 5,6 avec 5 μL de mélange d’adaptateur de code-barres (voir tableau des matériaux) et 50 μl de mélange principal Blunt/ta ligase dans un tube de réaction à faible liaison à l’ADN. Mélanger doucement en feuillant et incuber pendant 10 min à température ambiante.
- Purifier le produit de réaction comme décrit dans la section 3,2, mais utiliser 40 μL de billes magnétiques, faire deux étapes de lavage avec 140 μL de tampon ABB (voir le tableau des matériaux) au lieu de l’éthanol, Resuspendre les billes en les feuillant et les granulés sur un rack magnétique. Éluer dans 15 μL de tampon d’élution (voir tableau des matériaux). Augmentez les temps d’incubation pour la liaison initiale de l’ADN aux billes, ainsi que pour l’étape d’élution à 10 min. conserver le produit obtenu sur la glace ou à 4 ° c jusqu’à l’utilisation.
6. contrôle de qualité de la cellule de débit
- Effectuer un contrôle de qualité sur la cellule de flux avant utilisation. À cette fin, connectez le périphérique de séquençage à l’ordinateur hôte et ouvrez le logiciel.
- Insérez une cellule de flux (voir tableau des matériaux) dans le dispositif de séquençage et choisissez le type de cellule de flux dans la boîte de sélection et confirmez en cliquant sur disponible.
- Cliquez sur Vérifier la cellule de flux en bas de l’écran et choisissez le type de cellule de flux correct.
- Cliquez sur Démarrer le test pour démarrer le contrôle de qualité. Un minimum de 800 nanopores actives au total est nécessaire pour que la cellule d’écoulement soit utilisable.
7. chargement de la cellule de flux et démarrage de l’exécution du séquençage
- Ouvrez le capot du port d’amorçage en le glissant dans le sens des aiguilles d’une montre. Placez une pipette P1000 à 200 μL et insérez la pointe dans le port d’amorçage. Ajuster la pipette à 230 μL tout en gardant l’embout dans le port d’amorçage, pour prélever 20-30 μL de tampon et enlever les bulles d’air.
- Dans un nouveau tube de réaction de 1,5 mL d’ADN à faible liaison, préparer le mélange d’amorçage en combinant 576 μL de tampon RBF (voir tableau des matériaux) avec 624 μL d’eau exempte de nucléases.
- Pipetter délicatement 800 μL du mélange d’amorçage préparé dans le port d’amorçage et attendre 5 min. Soulevez le couvercle de l’orifice d’échantillonnage et Pipettez un 200 μL additionnel du mélange d’amorçage préparé dans le port d’amorçage.
- Pipetter 35 μL de tampon RBF dans un nouveau tube de réaction à faible liaison de l’ADN 1,5 mL propre. Mélanger soigneusement les billes de LLB (voir tableau des matériaux) par pipetage et ajouter 25,5 μl de billes dans le tampon de la RBF. Ajouter 2,5 μL d’eau exempte de nucléases et 12 μL de bibliothèque d’ADN à partir de l’étape 5,8 et mélanger par pipetage.
- Ajouter 75 μL de mélange d’échantillon de façon lente à la cellule d’écoulement à l’aide du port d’échantillonnage.
- Remplacez le capot du port de l’échantillon, fermez le port d’amorçage et fermez le couvercle du dispositif de séquençage.
- Dans le logiciel, confirmez que la cellule de flux est toujours disponible, ouvrez une nouvelle expérience et configurez les paramètres d’exécution en sélectionnant le kit utilisé. Sélectionnez l’appel de base en direct. Démarrez l’exécution du séquençage en cliquant sur commencer l’expérience. Continuez l’exécution du séquençage jusqu’à ce que des données expérimentales suffisantes soient collectées.
Résultats
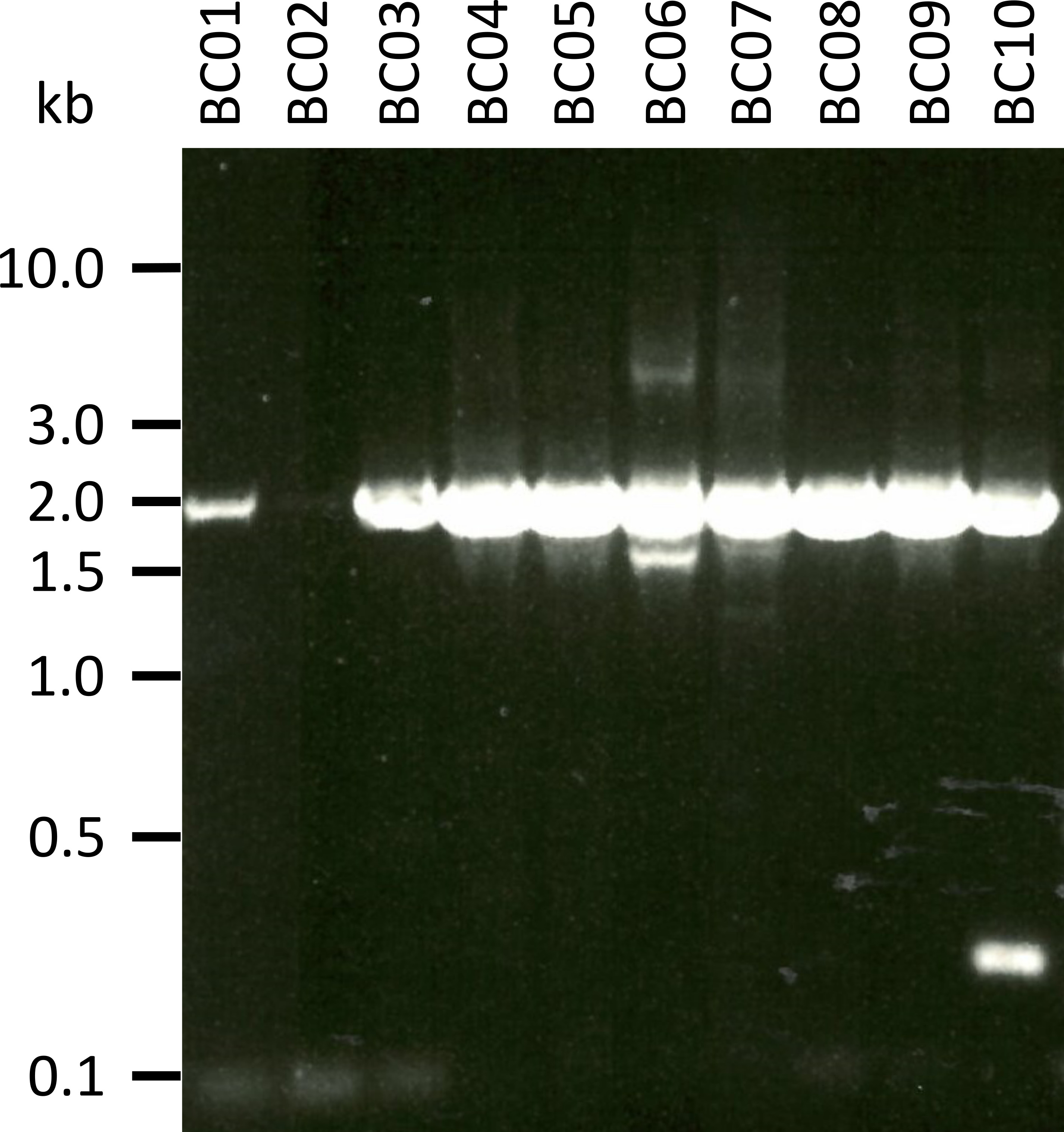
Dans une expérience représentative pour tester le protocole présenté, nous avons extrait l’ARN de 10 échantillons sanguins différents de cinq espèces animales (c.-à-d., 2 individus par espèce (chèvre, mouton, porc, chien, bétail)) (tableau 3). Les rendements d’ARN et la qualité après extraction peuvent varier considérablement, en particulier en raison des différences dans la manutention et le stockage des échantillons. Dans notre expérience représentative, nous avons observé des concentrations d’ARN comprises entre 43 ng et 543 ng par μL (tableau 3). En outre, après amplification par RT-PCR, l’analyse du gel des produits de PCR NPC-1 a montré divers résultats (figure 2), avec des bandes nettement plus faibles pour les échantillons BC01 et BC02 (les deux chèvres). Ces différences étaient probablement causées par des différences dans la qualité de l’échantillon, bien que les différences dans l’efficacité de la PCR en raison de différences dans la fixation des amorces au gène NPC1 de différentes espèces ne puissent pas être exclues. Cependant, ces différences de rendement et/ou d’efficacité d’amplification n’ont pas eu d’impact notable sur le résultat global du séquençage. De plus, un produit d’ACP supplémentaire non spécifique a été prélevé dans l’échantillon BC10 (bovins). Contrairement au séquençage de Sanger, ces produits non spécifiques n’influencent pas négativement les résultats du séquençage des nanopores, car ces lectures sont ignorées lors de la cartographie des lectures obtenues à une séquence de référence dans le cadre de l’analyse des données.
Avant chaque exécution de séquençage, un contrôle de qualité de la cellule d’écoulement à utiliser est fortement recommandé, avec une exigence minimale de 800 pores totaux. Dans notre expérience représentative, ce contrôle de qualité a retourné 1 102 pores disponibles pour le séquençage. Étant donné que les données sont fournies en temps réel et peuvent être analysées immédiatement, la longueur d’une exécution de séquençage peut être ajustée pour l’application individuelle (c.-à-d., jusqu’à ce que des données de séquençage suffisantes soient produites pour l’analyse souhaitée). Dans nos expériences, les séquences de séquençage sont généralement exécutées pendant la nuit, et dans le cas de notre expérience représentative, nous avons obtenu environ 1,4 million lectures pendant une telle course de 14 h.
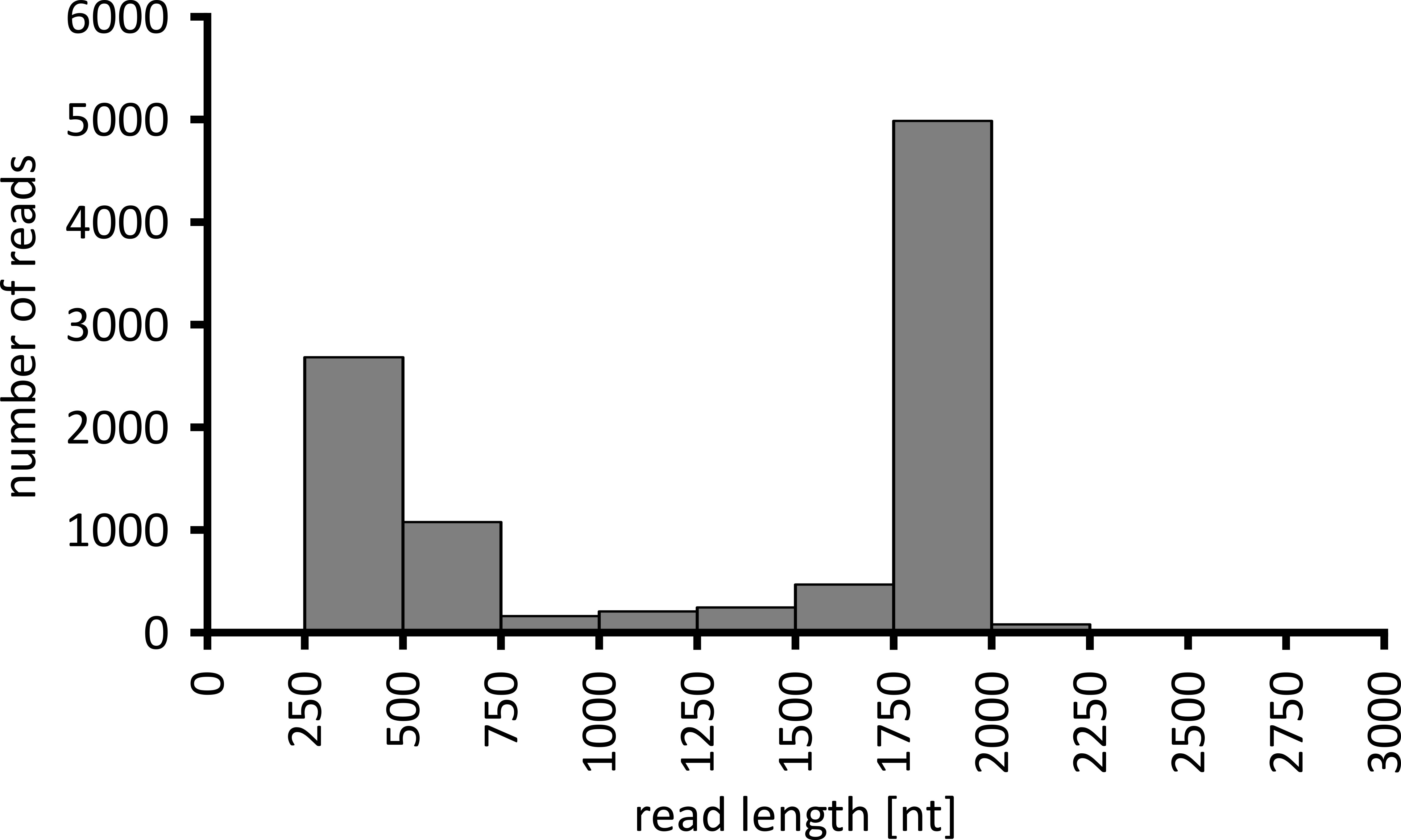
Selon le type d’analyse de données à effectuer, il peut être conseillé de traiter uniquement un sous-ensemble des lectures obtenues. Dans le cas de notre expérience représentative, un sous-ensemble de 10 000 lectures a été sélectionné pour une analyse plus approfondie. À cette fin, les fichiers FastQ générés au cours de la séquence de séquençage ont été traités ultérieurement dans un environnement Ubuntu 18,04 LTS, et démultiplexés à l’aide de FlexBar v 3.0.1 avec des paramètres optimisés pour le démultiplexage des données de séquençage des nanopores (code-barres-queue-longueur 300 , code-barres-erreur-rate 0,2, code-barres-Gap-pénalité-1)12. Après le démultiplexage, la cartographie de lecture et la génération de consensus peuvent alors être faites en utilisant un certain nombre d’outils différents, mais une discussion détaillée de l’aspect bioinformatique du séquençage des nanopores va au-delà de la portée de ce manuscrit. Cependant, dans le cas de nos résultats représentatifs, le mappage de lecture à une séquence de référence a été effectué à l’aide de Geneious. Sur les 10 000 lectures analysées, 5 457 a montré une longueur entre 1 750 et 2 000 nucléotides, qui correspond aux tailles attendues pour les fragments de PCR amplifiés dans le cadre de notre workflow (1 769 NT, figure 3). Un pic supplémentaire dans la distribution de la longueur des lectures a été observé entre 250 et 500 nucléotides, ce qui peut être attribué à des produits de PCR non spécifiques. Le démultiplexage des lectures a permis l’assignation de 87,6% des lectures à l’un des 10 codes à barres/échantillons analysés (figure 4). La proportion de lectures démultiplexées pour chaque code-barres variait de 3,4% pour le code-barres 1 à 16,9% pour le code à barres 10; Cependant, en raison du grand nombre total de lectures, cela a toujours permis un consensus significatif appelant avec une profondeur de lecture élevée, même pour ces jeux de codes à barres de faible abondance. En effet, la cartographie des lectures triées à une séquence de référence de NPC1 a entraîné entre 31,7% (code-barres 2) et 100% (code-barres 7 et 8) de lire le mappage à la référence, ce qui donne une profondeur de lecture de plus de 90 lectures à n’importe quelle position pour chaque échantillon. C’est alors plus que suffisant pour permettre un consensus confiant-appel de base avec un taux d’erreur négligeable.

Figure 1: représentation schématique du séquençage de l’ADN à l’aide de la technologie nanopore. Une molécule d’ADN monocaténaire traverse une nanopore incorporée dans une membrane électriquement résistante, avec une helicase régulant la vitesse de transition. Un courant ionique traverse simultanément le pore et est continuellement mesuré. Les modulations du courant causées par les nucléotides présents dans les pores sont détectées et traduites en arrière-plan dans la séquence nucléotidique du brin d’ADN. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 2: amplification des produits de PCR de Niemann-Pick C1 de l’ARNm. l’ARNm a été isolé à partir de chèvres (BC01 et 02), de moutons (BC03 et 04), de porcs (BC05 et 06), de chiens (BC07 et 08) et de bovins (BC09 et 10). Les produits de PCR imbriqués ont été séparés dans un gel d’d’agarose à 0,8% dans un tampon TAE (préparé à partir du tampon TAE 50x: 242,28 g de base Tris, 57,1 mL d’acide acétique glacial, 100 mL d’EDTA de 0,5 M,2O à 1 L de DH, pH ajusté à 8,0) pour 45 min à 100 V et teinté avec SYBR safe. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 3: distribution en lecture de longueur de 10 000 lectures de l’expérience représentative. Le nombre de lectures obtenues ayant un intervalle de longueur de lecture donné est indiqué. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 4: distribution des lectures après démultiplexage. Le nombre et le pourcentage de lectures démultiplexées (grises) et cartographiées (noires) pour chaque code-barres sont affichés. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}
Tableau 1: vue d’ensemble des ensembles d’amorces utilisés. L’amplification initiale des séquences cibles a été réalisée avec Primer Set 1. Primer Set 2 a ensuite été utilisé pour l’amplification imbriquée et l’addition d’adaptateur. Les adaptateurs sont indiqués en rouge. Veuillez cliquer ici pour télécharger ce fichier.
Tableau 2: vue d’ensemble des séquences de codes-barres. Des codes-barres individuels ont été utilisés pour identifier chaque échantillon séquencé. Veuillez cliquer ici pour télécharger ce fichier.
Tableau 3: concentrations d’ARN obtenues après extraction à partir d’échantillons sanguins séquencés dans l’expérience représentative. Les concentrations d’ARN de deux individus de chacune des cinq espèces sont montrées, et les ratios des densités optiques à 260/280 nm et 260/230 nm sont indiqués. Veuillez cliquer ici pour télécharger ce fichier.
Discussion
Au cours des deux dernières décennies, le séquençage des échantillons biologiques est devenu un aspect de plus en plus important des études dans un large éventail de sujets. Le développement de systèmes de séquençage de deuxième génération basés sur le séquençage d’un éventail dense de caractéristiques d’ADN utilisant des cycles itératifs de manipulation enzymatique et d’acquisition de données basée sur l’image1 a considérablement augmenté le débit par rapport à la technique de séquençage Sanger traditionnelle, et permet l’analyse de plusieurs échantillons ainsi que diverses espèces d’acide nucléique dans un échantillon donné en parallèle4. Cependant, pour la plupart des systèmes de deuxième génération couramment utilisés, seules les lectures courtes sont produites, et toutes les plateformes reposent sur des équipements sensibles, encombrants et coûteux3,4.
Contrairement aux plateformes de séquençage de deuxième génération, le dispositif de séquençage utilisé dans ce protocole est basé sur la technologie nanopore. Ici, une molécule d’acide nucléique monocaténaire traverse une NANOPORE, ce qui entraîne une modulation d’un courant ionique qui coule également à travers la même NANOPORE, et qui peut être mesurée et traduite en arrière pour déduire la séquence de la molécule d’acide nucléique. Cette approche de séquençage de troisième génération présente un certain nombre d’avantages par rapport à d’autres approches. Les principaux avantages qui sont directement liés au principe de fonctionnement unique de cette technologie sont la longueur de lecture extrêmement longue produite (longueurs de lecture jusqu’à 8,8 x 105 nucléotides ont été rapportés6), la capacité de séquencer non seulement l’ADN, mais aussi ARN directement, qui a récemment été démontrée pour un génome complet du virus de la grippe17, et la capacité d’analyser les données en temps réel à mesure qu’elles sont générées, ce qui permet la détection rapide des métagénomiques des agents pathogènes en quelques minutes18. Les avantages pratiques supplémentaires sont la taille extrêmement petite du dispositif de séquençage de NANOPORE, permettant son utilisation dans n’importe quel laboratoire ou sur des missions sur le terrain à des emplacements éloignés19,20, et le bas prix par rapport à d’autres séquençage Plates-formes. En ce qui concerne les coûts de fonctionnement, une nouvelle cellule de flux est actuellement requise pour chaque exécution de séquençage, ce qui entraîne des coûts d’environ $1 100 par course pour les réactifs de préparation de la cellule de débit et de la bibliothèque. Ces coûts peuvent être réduits dans certains cas par le lavage et la réutilisation de la cellule de flux, ou par barres et séquençage de plusieurs échantillons en une seule exécution. En outre, un nouveau type de cellule de flux est actuellement testé en version bêta par un petit nombre de laboratoires, ce qui nécessitera l’utilisation d’un adaptateur de cellule de débit (appelé «flongle»), et devrait réduire considérablement le prix des cellules de flux et donc les coûts de fonctionnement.
La lacune majeure du séquençage des nanopores reste sa précision, avec des précisions de lecture uniques dans la fourchette de 83 à 86% étant rapportées6,21,22, et la plupart des inexactitudes causées par l’insertion/suppression ( indels)5,21. Cependant, une profondeur de lecture élevée peut compenser ces inexactitudes, et une étude récente suggérée sur la base de considérations théoriques selon lesquelles une profondeur de lecture de > 10 pourrait augmenter la précision globale à > 99,8%21. Néanmoins, d’autres améliorations de l’exactitude seront nécessaires, en particulier si l’analyse doit être effectuée sur un seul niveau de molécule plutôt que sur un niveau de séquence consensuelle. L’utilisation de la technologie 1D2 telle que décrite dans ce protocole, qui est basée sur l’addition des adaptateurs 1D2 et Barcode (cf. section 5,5) qui entraînent la séquençage des deux brins d’une seule molécule d’ADN par la même NANOPORE, augmente la lecture précision puisque les informations des deux brins d’ADN peuvent être utilisées pour la détermination de la séquence. En outre, une stratégie de contournement qui peut être poursuivie afin de combiner les avantages du séquençage des nanopores (en particulier la longueur de lecture longue) avec la plus grande précision des autres technologies de séquençage est d’utiliser l’information de séquençage nanopore comme un échafaudage, qui est puis polie en utilisant les données de séquençage des autres plateformes6.
Le facteur le plus critique pour le succès du protocole présenté ici est la qualité de l’échantillon, et en particulier la quantité et la qualité de l’ARN extrait. Un stockage adéquat et une extraction rapide de l’ARN aident à obtenir un rendement d’ARN adéquat. L’utilisation de tubes de prélèvement de sang appropriés permet le stockage des échantillons de sang pendant un mois, mais la coagulation sanguine peut être un problème, en particulier lorsque des échantillons sont stockés à des températures élevées, ce qui peut être le cas dans des conditions de terrain. La deuxième étape critique est l’amplification des séquences cibles, et en particulier dans les conditions de terrain, les réactions de PCR se réalisent souvent moins bien que dans les conditions normales de laboratoire7. À cette fin, une conception et une optimisation soigneuses des amorces sont primordiales pour obtenir une amplification robuste. En outre, les approches PCR imbriquées et la PCR de Touchdown, telles qu’utilisées dans ce protocole, peuvent augmenter à la fois la spécificité et la sensibilité de l’amplification du gène cible4,7. En effet, dans notre expérience au Libéria et en Guinée avec cette technologie, des protocoles imbriqués étaient requis dans des conditions de terrain avec des échantillons de terrain, même pour les ensembles d’amorces qui permettaient l’amplification des cibles à partir d’échantillons de laboratoire et dans des conditions de laboratoire avec une seule série de PCR (7 et résultats non publiés).
Contrairement à ces étapes plus critiques, la préparation de la bibliothèque et l’exécution du séquençage sont des procédures plutôt robustes. Toutefois, dans des conditions de terrain, des questions pratiques telles que la disponibilité de certaines pièces d’équipement peuvent être problématiques. Par exemple, un spectrophotomètre UV est nécessaire pour déterminer les concentrations d’ADN avant la préparation de la bibliothèque d’échantillons à code à barres. Toutefois, si un tel dispositif n’est pas disponible dans des conditions de terrain, un volume égal de chaque échantillon peut simplement être combiné pour constituer le 45 μL requis pour la préparation de la bibliothèque, avec des différences dans le matériau d’entrée de l’échantillon, puis généralement atténué par la grande nombre de lectures. De même, le besoin de connectivité Internet pour la séquence de séquençage peut être un problème, même si l’appel de base ne doit plus être effectué en ligne, mais peut être effectué localement; Toutefois, cette nécessité peut être retirée dans certaines circonstances par le fabricant si nécessaire.
En résumé, le protocole présenté permet un séquençage relativement peu coûteux dans des endroits sans accès à l’équipement de séquençage traditionnel, y compris dans des endroits éloignés. Il peut facilement être adapté à n’importe quel ARN ou ADN cible, permettant ainsi aux chercheurs de répondre à de nombreuses questions biologiques.
Déclarations de divulgation
TH a participé au programme d’accès anticipé d’Oxford nanopore technologies (ONT) MinION de 2014 à 2015, et a reçu des dispositifs de MinION et des cellules d’écoulement pour une étude précédente 7 effectuée par les instituts nationaux de santé, États-Unis, gratuitement ou à des coûts réduits . Il a été invité par l’ONT à présenter une partie de ce travail lors de la réunion de Londres Calling 2015 à Londres, au Royaume-Uni, et ONT payé pour le transport et l’hébergement. Pour les travaux présentés dans ce manuscrit, aucun avantage (p. ex. matériel ou réactifs à coût réduit, remboursements de voyage, etc.) n’a été obtenu auprès d’ONT. AM, KF et RS n’ont rien à divulguer.
Remerciements
Les auteurs remercient Allison Groseth pour la lecture critique du manuscrit. Ce travail a été financé financièrement par le ministère fédéral allemand de l’alimentation et de l’agriculture (BMEL) sur la base d’une décision du Parlement de la République fédérale d’Allemagne par l’intermédiaire de l’Office fédéral de l’agriculture et de l’alimentation (BLE).
matériels
| Name | Company | Catalog Number | Comments |
| 1D2 adapter, barcode adapter mix, ABB buffer, elution buffer, RBF buffer, LBB beads | Oxford Nanopore Technologies | SQK-LSK308 | 1D² Sequencing Kit |
| Blood collection tube with DNA/RNA stabilizing reagent | Zymo Research | R1150 | DNA/RNA Shield - Blood Collection Tube |
| Blunt/TA ligase master mix | New England Biolabs | M0367S | Blunt/TA Ligase Master Mix |
| DNA-low binding reaction tube | Eppendorf | 30108051 | DNA LoBind Tube |
| DNase buffer and DNase | ThermoFisher Scientific | 11766050 | SuperScript™ IV VILO™ Master Mix with ezDNase™ Enzyme |
| Flow cell | Oxford Nanopore Technologies | FLO-MIN105.24 | flow cell R9.4 |
| Hot start high fidelity DNA polymerase | New England Biolabs | M0493L | Q5 Hot Start High-Fidelity DNA Polymerase (500 U) |
| Magnetic beads | Beckman Coulter | A63881 | Agencourt AMPure XP beads |
| Magnetic rack | ThermoFisher Scientific | 12321D | DynaMag-2 Magnet |
| Nanopore sequencing device | Oxford Nanopore Technologies | - | MinION Mk 1B |
| PCR barcoding kit | Oxford Nanopore Technologies | EXP-PBC001 | PCR Barcoding Kit I (R9) |
| Reverse transcriptase master mix | ThermoFisher Scientific | 11766050 | SuperScript™ IV VILO™ Master Mix with ezDNase™ Enzyme |
| RNA purification spin column, DNA/RNA prep buffer, DNA/RNA wash buffer, DNase I, DNA digestion buffer | Zymo Research | R1151 | Quick-DNA/RNA Blood Tube Kit |
| Rotating mixer | ThermoFisher Scientific | 15920D | HulaMixer Sample Mixer |
| Taq DNA polymerase | New England Biolabs | M0287S | LongAmp Taq 2x Master Mix |
| Ultra II End-prep kit | New England Biolabs | E7546S | NEBNext Ultra II End-Repair/dA-tailing Modul |
| UV spectrophotometer | Implen | - | NanoPhotometer |
| Vacuum manifold | Zymo Research | S7000 | EZ-Vac Vacuum Manifold |
Références
- Shendure, J., Ji, H. Next-generation DNA sequencing. Nature Biotechnology. 26 (10), 1135-1145 (2008).
- Shendure, J., Lieberman Aiden, E. The expanding scope of DNA sequencing. Nature Biotechnology. 30 (11), 1084-1094 (2012).
- Liu, L., et al. Comparison of next-generation sequencing systems. Journal of Biomedicine and Biotechnology. 2012, 251364 (2012).
- Levy, S. E., Myers, R. M. Advancements in Next-Generation Sequencing. Annual Review of Genomics and Human Genetics. 17, 95-115 (2016).
- Lu, H., Giordano, F., Ning, Z. Oxford Nanopore MinION Sequencing and Genome Assembly. Genomics, Proteomics & Bioinformatics. 14 (5), 265-279 (2016).
- Jain, M., et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nature Biotechnology. 36 (4), 338-345 (2018).
- Hoenen, T., et al. Nanopore Sequencing as a Rapidly Deployable Ebola Outbreak Tool. Emerging Infectious Diseases. 22 (2), 331-334 (2016).
- Quick, J., et al. Real-time, portable genome sequencing for Ebola surveillance. Nature. 530 (7589), 228-232 (2016).
- Carette, J. E., et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature. 477 (7364), 340-343 (2011).
- Ndungo, E., et al. A Single Residue in Ebola Virus Receptor NPC1 Influences Cellular Host Range in Reptiles. mSphere. 1 (2), (2016).
- Martin, S., et al. A genome-wide siRNA screen identifies a druggable host pathway essential for the Ebola virus life cycle. Genome Medicine. 10 (1), 58 (2018).
- Roehr, J. T., Dieterich, C., Reinert, K. Flexbar 3.0 - SIMD and multicore parallelization. Bioinformatics. 33 (18), 2941-2942 (2017).
- Li, H., et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25 (16), 2078-2079 (2009).
- Kielbasa, S. M., Wan, R., Sato, K., Horton, P., Frith, M. C. Adaptive seeds tame genomic sequence comparison. Genome Research. 21 (3), 487-493 (2011).
- Don, R. H., Cox, P. T., Wainwright, B. J., Baker, K., Mattick, J. S. Touchdown' PCR to circumvent spurious priming during gene amplification. Nucleic Acids Research. 19 (14), 4008 (1991).
- Korbie, D. J., Mattick, J. S. Touchdown PCR for increased specificity and sensitivity in PCR amplification. Nature Protocols. 3 (9), 1452-1456 (2008).
- Keller, M. W., et al. Direct RNA Sequencing of the Coding Complete Influenza A Virus Genome. Scientific Reports. 8 (1), 14408 (2018).
- Greninger, A. L., et al. Rapid metagenomic identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis. Genome Medicine. 7, 99 (2015).
- Castro-Wallace, S. L., et al. Nanopore DNA Sequencing and Genome Assembly on the International Space Station. Scientific Reports. 7 (1), 18022 (2017).
- Goordial, J., et al. In Situ Field Sequencing and Life Detection in Remote (79 degrees 26'N) Canadian High Arctic Permafrost Ice Wedge Microbial Communities. Frontiers in Microbiology. 8, 2594 (2017).
- Runtuwene, L. R., et al. Nanopore sequencing of drug-resistance-associated genes in malaria parasites, Plasmodium falciparum. Scientific Reports. 8 (1), 8286 (2018).
- Rang, F. J., Kloosterman, W. P., de Ridder, J. From squiggle to basepair: computational approaches for improving nanopore sequencing read accuracy. Genome Biology. 19 (1), 90 (2018).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.