
Method Article
Sequenziamento dell'mRNA da sangue intero utilizzando il sequenziamento di NanoPore
In questo articolo
Riepilogo
Il sequenziamento di nanopore è una nuova tecnologia che consente un sequenziamento conveniente in posizioni remote e impostazioni di scarsa risorse. Qui, presentiamo un protocollo per il sequenziamento di mRNA da sangue intero che è compatibile con tali condizioni.
Abstract
La sequenza in posizioni remote e le impostazioni di scarsa risorse presentano sfide uniche. Il sequenziamento di nanopore può essere utilizzato con successo in tali condizioni ed è stato distribuito in Africa occidentale durante la recente epidemia di virus Ebola, evidenziando questa possibilità. Oltre ai vantaggi pratici (basso costo, facilità di trasporto e utilizzo delle attrezzature), questa tecnologia offre anche vantaggi fondamentali rispetto agli approcci di sequenziamento di seconda generazione, in particolare la lunghezza di lettura molto lunga, la capacità di sequenziare direttamente L'RNA e la disponibilità in tempo reale dei dati. La precisione di lettura RAW è inferiore rispetto ad altre piattaforme di sequenziamento, che rappresenta la principale limitazione di questa tecnologia; Tuttavia, questo può essere parzialmente mitigato dall'alta profondità di lettura generata. Qui, presentiamo un protocollo compatibile sul campo per il sequenziamento della codifica mRNAs per Niemann-Pick C1, che è il recettore cellulare per ebolaviruses. Questo protocollo comprende l'estrazione di RNA da campioni di sangue animale, seguita da RT-PCR per l'arricchimento del bersaglio, barcoding, preparazione della libreria, e la sequenza di esecuzione stessa, e può essere facilmente adattato per l'uso con altri bersagli di DNA o RNA.
Introduzione
Il sequenziamento è uno strumento potente e importante nella ricerca biologica e biomedica. Esso consente l'analisi di Genomes, variazioni genetiche, e profili di espressione di RNA, e quindi svolge un ruolo importante nell'indagine delle malattie umane e animali allo stesso modo1,2. Il sequenziamento con metodo Sanger, uno dei metodi più antichi disponibili per il sequenziamento del DNA, è ancora abitualmente utilizzato fino ad oggi ed è stato un angolo-pietra della biologia molecolare. Negli ultimi 50 anni, questa tecnologia è stata migliorata per ottenere una lunghezza di lettura superiore a 1.000 NT e una precisione superiore a 99,999%1. Tuttavia, il sequenziamento di Sanger ha anche dei limiti. Sequenziare un insieme più ampio di campioni o l'analisi di interi geniti con questo metodo richiede molto tempo e costoso1,3. I metodi di sequenziamento del DNA di seconda generazione (di nuova generazione), come il pirosequenziamento 454 e la tecnologia illumina, ci hanno permesso di ridurre significativamente i costi e il carico di lavoro richiesti per il sequenziamento nell'ultimo decennio, e hanno portato ad un enorme aumento del quantità di informazioni sulla sequenza biologica disponibili4. Tuttavia, le singole sequenze di sequenziamento che utilizzano queste tecnologie di seconda generazione sono costose e il sequenziamento in condizioni di campo è impegnativo, in quanto l'equipaggiamento necessario è ingombrante e fragile (simile ai dispositivi di sequenziamento Sanger) e spesso deve essere calibrato e servito da personale appositamente addestrato. Inoltre, per molte delle tecnologie di seconda generazione, le lunghezze di lettura sono piuttosto limitate, il che spesso rende difficile l'analisi bioinformatica a valle di questi dati.
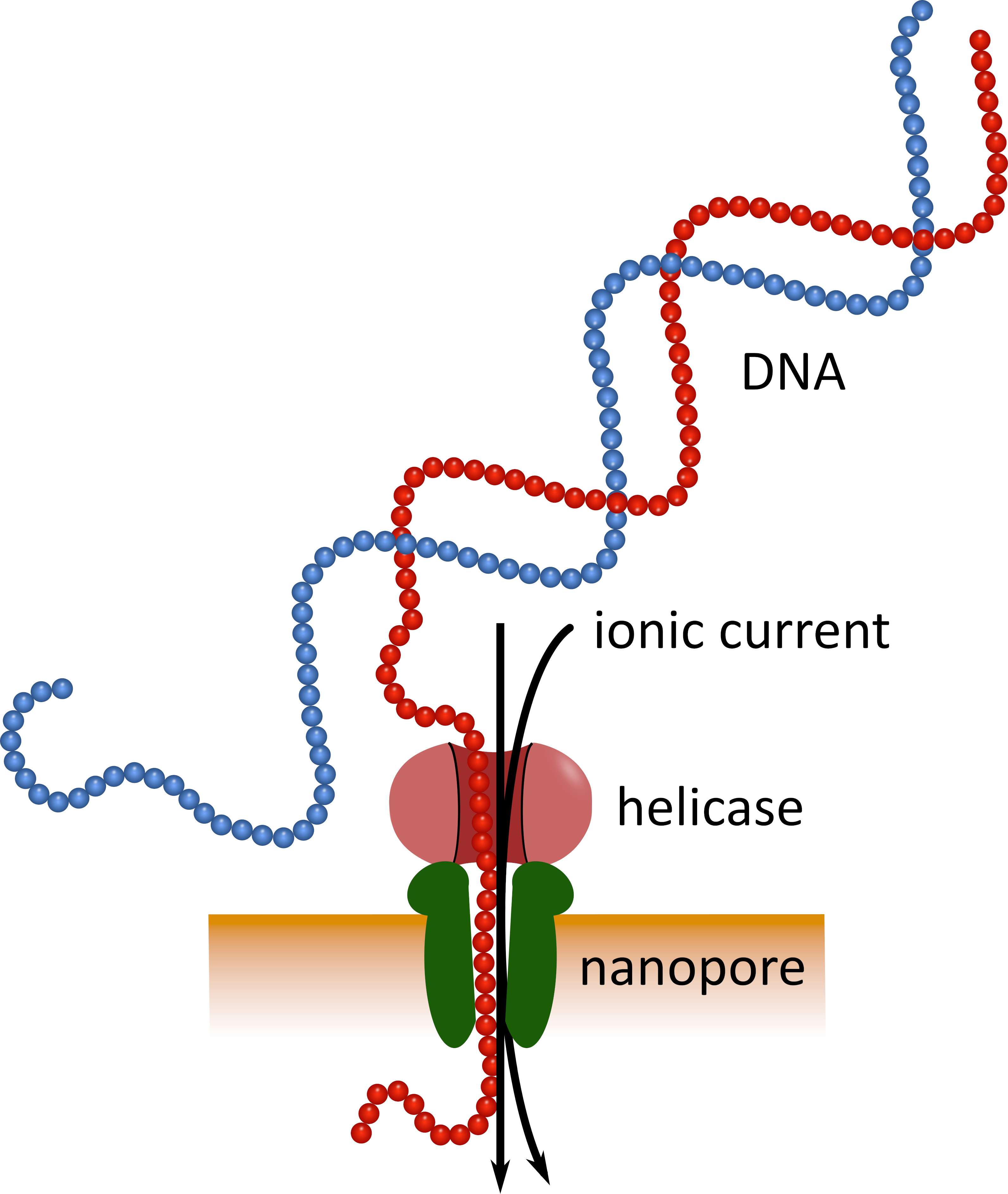
Il sequenziamento di terza generazione che utilizza dispositivi di sequenziamento di nanoparticelle di dimensioni tascabili (Vedi tabella dei materiali) può fungere da alternativa a queste piattaforme di sequenziamento consolidate. In questi dispositivi una molecola di DNA o RNA a filamento singolo passa attraverso un nanoporo simultaneamente con una corrente ionica che viene poi misurata da un sensore (Figura 1). Come il filo attraversa il nanopore, la modulazione della corrente dai nucleotidi presenti nel poro in un dato momento viene rilevata, e computazionalmente indietro-tradotto nella sequenza nucleotide5. A causa di questo principio operativo, il sequenziamento del nanoporo consente sia la generazione di letture molto lunghe (vicino a 1 x 106 nucleotidi6) e l'analisi dei dati di sequenziamento in tempo reale. Il barcoding è possibile collegando sequenze nucleotidiche definite agli acidi nucleici in un campione, che consente l'analisi di più campioni in una singola sequenza di sequenziamento, aumentando così la velocità effettiva del campione e abbassando i costi per campione. Grazie alla loro elevata portabilità e facilità d'uso, i dispositivi di sequenziamento delle nanoparticelle sono stati utilizzati con successo nel campo durante la recente epidemia di malattia da virus Ebola in Africa occidentale, evidenziando la loro idoneità per un rapido dispiegamento in regioni remote7 , 8. il
Qui, descriviamo un protocollo dettagliato compatibile con il campo per il sequenziamento della codifica mRNA per la proteina Niemann-Pick C1 (NPC1), che è il recettore di ingresso obbligato per filovirus come ebolaviruses, e ha dimostrato di limitare la suscettibilità delle specie a questi virus9,10. Il protocollo comprende l'estrazione dell'intero RNA da campioni di sangue, l'amplificazione specifica di NPC1 mRNA da RT-PCR, la barcoding dei campioni, la preparazione della libreria e il sequenziamento con un dispositivo di sequenziamento di nanoporo. L'analisi dei dati non può essere discussa a causa di limitazioni di spazio, anche se alcune direzioni di base sono fornite nei risultati rappresentativi; Tuttavia, il lettore interessato è riferito a una precedente pubblicazione11 per una descrizione più dettagliata del flusso di lavoro che abbiamo utilizzato, nonché alle pubblicazioni di altri12,13,14 per informazioni dettagliate per quanto riguarda gli strumenti di analisi utilizzati in questo flusso di lavoro.
Protocollo
I campioni sono stati raccolti seguendo il protocollo NUIRB (Njala University Institutional Review Board). IRB00008861/FWA00018924.
1. estrazione di RNA da campioni di sangue
- Raccogliere 3 mL di sangue intero dalla specie da analizzare in un tubo di raccolta del sangue precompilato con 6 mL di reagente stabilizzante DNA/RNA (vedere tabella dei materiali) e mescolare invertendo 5 volte. Conservare il campione di sangue per un massimo di un mese a 4 ° c.
- Trasferire il contenuto del tubo di raccolta del sangue in un tubo di raccolta da 50 mL, aggiungere 120 μL di proteinasi K e mescolare per 5 s. Incubare il campione per 30 minuti a temperatura ambiente.
- Aggiungere 9 mL di isopropanolo alla miscela e al vortice per 5 s.
- Collocare un serbatoio su una colonna di centrifuga di purificazione dell'RNA (vedere tabella dei materiali) e posizionare l'assieme su un collettore sottovuoto (vedere tabella dei materiali). Aggiungere la miscela campione nel serbatoio. Applicare un vuoto fino a quando tutto il liquido è passato attraverso la colonna.
- In alternativa, se non è disponibile un collettore sottovuoto, il sangue può essere passato attraverso la colonna in 700 μL di porzioni mediante centrifugazione ripetuta per 30 s a 12.000 x g con il flusso passante scartato tra le fasi di centrifugazione. Tuttavia, questo richiederà circa 26 fasi di centrifugazione.
- Posizionare la colonna di centrifuga di purificazione dell'RNA in un tubo di raccolta e aggiungere 400 μL di tampone di preparazione DNA/RNA (vedere tabella dei materiali). Centrifugare a 12.000 x g per 30 s e scartare il flusso passante.
- Aggiungere 400 μL di tampone di lavaggio DNA/RNA (vedere tabella dei materiali) alla colonna, centrifugare a 12.000 x g per 30 s e scartare il flusso passante.
- Miscelare 5 μL di DNase I (1 U/μL) (vedere tabella dei materiali) con 75 μl di tampone di digestione del DNA (vedere tabella dei materiali) e aggiungere la miscela sulla colonna. Incubare per 15 minuti a temperatura ambiente.
- Aggiungere 400 μL di tampone di preparazione DNA/RNA alla colonna e centrifugare a 12.000 x g per 30 s. eliminare il flusso passante.
- Lavare la colonna con 700 μL di tampone di lavaggio DNA/RNA e centrifugare a 12.000 x g per 30 s. eliminare il flusso passante.
- Ripetere il passo 1,9 con 400 μL di tampone di lavaggio DNA/RNA e centrifugare a 12.000 x g per 2 min per rimuovere tutto il tampone di lavaggio residuo e asciugare la colonna. Quando si rimuove la colonna dal tubo di raccolta, assicurarsi di non bagnare la parte inferiore della colonna con il tampone nel tubo di raccolta.
-
Collocare la colonna in una nuova provetta da microcentrifuga da 1,5 mL e aggiungere 70 μL di acqua priva di nucleasi. Incubare per 1 min a temperatura ambiente e centrifugare per 30 s a 12.000 x g. Conservare l'RNA a-80 ° c fino a ulteriore uso (o usare immediatamente).
- Facoltativo: Per quantificare l'RNA, prendere un'aliquota e determinare la concentrazione utilizzando uno spettrofotometro UV (vedere tabella dei materiali).
2. trascrizione inversa di NPC1 mRNA in cDNA
- In un tubo di reazione da 0,2 mL, aggiungere 8 μL di RNA di template (da 1 PG a 2,5 μg di RNA) e 1 μL ciascuno di buffer 10x DNase e enzima dnasi (vedere tabella dei materiali). Incubare a 37 ° c per 2 min. Successivamente, centrifugare brevemente la reazione e posizionarla sul ghiaccio.
- Aggiungere 4 μL di miscela Master della trascrittasi inversa (vedere tabella dei materiali) e 6 μl di acqua priva di nucleasi al tubo di reazione e mescolare delicatamente. Incubare la reazione in un termociclatore per 10 minuti a 25 ° c (per la ricottura del primer), seguita da 10 minuti a 50 ° c (per la trascrizione inversa dell'RNA). Per inattivare l'enzima, incubare per 5 min a 85 ° c.
- Trasferire cDNA in una nuova provetta da microcentrifuga da 1,5 mL e conservare a-80 ° c fino a ulteriore uso (o usare immediatamente).
3. amplificazione della cornice di lettura NPC1 Open
-
Fase di amplificazione iniziale
- Impostare un touchdown PCR15,16 per amplificare il NPC1 cDNA con primer set 1 (vedere tabella 1), utilizzando un hot start ad alta fedeltà DNA polimerasi (Vedi tabella dei materiali) con il tampone di reazione appropriato in una reazione di 50 μl volume con 1 μL di modello. Se possibile, impostare la reazione sul ghiaccio o in un blocco freddo di 4 ° c.
- Incubare la reazione in un termociclatore con una fase iniziale di denaturazione di 30 s a 98 ° c, seguita da 10 cicli con denaturazione a 98 ° c per 10 s, ricottura di fondo per 20 s a 65 ° c, abbassamento della temperatura di 0,5 ° c per ciclo e allungamento per 1 min a 72 ° c. Successivamente, eseguire un ulteriore 20 cicli con 10 s a 98 ° c, 20 s a 60 ° c, e 1 min a 72 ° c, seguita da una fase di allungamento finale di 5 min a 72 ° c.
-
Purificazione PCR con perle magnetiche
- Trasferire 50 μL di prodotto PCR in un tubo di reazione a legame di DNA-basso di 1,5 mL (vedere tabella dei materiali). Risospendere le perle magnetiche (vedere tabella dei materiali) accuratamente con vortex e aggiungere 50 μl di perle alla reazione PCR. Mescolare bene. Incubare il campione su un mixer rotante (vedere tabella dei materiali) per 5 minuti a temperatura ambiente (a 15 rpm).
- Ruotare brevemente il campione e posizionare il tubo microcentrifuga da 1,5 mL su un rack magnetico (vedere tabella dei materiali) per pellet le perle magnetiche. Attendere fino a quando il supernatante è stato completamente chiarito prima di continuare con il passo successivo.
- Aspirare il surnatante senza disturbare il pellet di perline e scartare.
- Pipettare 200 μL di etanolo 70% nel tubo di reazione e incubare per 30 s. aspirare l'etanolo senza disturbare il pellet e scartare. Ripetere per un totale di due Lavini. Assicurarsi che non venga lasciato etanolo. Può essere necessario aspirare prima con una pipetta più grande (ad es. 1000 μL) e quindi rimuovere le goccioline di etanolo rimanenti con una pipetta più piccola (ad es. 10 μL).
- Asciugare ad aria il pellet per 1 min a temperatura ambiente.
- Rimuovere il tubo di reazione dal rack magnetico, risospendere il pellet in 30 μL di acqua priva di nucleasi e incubare per 2 minuti a temperatura ambiente.
- Posizionare il tubo di reazione sul rack magnetico e attendere fino a quando le perle sono completamente pellettati.
- Rimuovere il supernatante senza disturbare il pellet e trasferirlo in un nuovo tubo di reazione da 1,5 mL.
-
Aggiunta di adattatori per codici a barre mediante PCR nidificata
- Impostare una PCR da touchdown da 50 μL con avvio a caldo di DNA polimerasi ad alta fedeltà con tampone di reazione 5x e primer set 2 (vedere tabella 1). I primer in questo set consistono in una regione specifica per la sequenza di destinazione per consentire il binding dei prodotti PCR generati nel passaggio 3,2 (all'interno del set di primer 1 sequenze), nonché una sequenza di adattatori utilizzata come bersaglio nella successiva reazione di PCR a barcoding (cfr. paragrafo 4). Se possibile, impostare la reazione sul ghiaccio o in un blocco freddo di 4 ° c. Utilizzare 1 μL del prodotto di PCR purificato preparato nella sezione 3,2 come dima.
- Incubare la miscela di reazione in un termociclatore utilizzando una fase iniziale di denaturazione di 30 s a 98 ° c, seguita da 10 cicli con denaturazione a 98 ° c per 10 s, ricottura di fondo per 20 s a 65 ° c, abbassamento della temperatura di 0,5 ° c per ciclo e allungamento per 1 min a 72 ° c. Successivamente, incubare la reazione per altri 30 cicli per 10 s a 98 ° c, 20 s a 71 ° c e 1 min a 72 ° c, seguita da una fase di allungamento finale di 5 min a 72 ° c.
- Pulire il prodotto PCR utilizzando perline magnetiche come descritto nel paragrafo 3,2.
4. barcoding di NPC1 amplicons
- Per ogni prodotto PCR generato nella sezione 3, impostare una reazione PCR a barcoding in un tubo di reazione da 0,2 mL utilizzando 50 μL di miscela Master Taq DNA polimerasi 2x (vedere tabella dei materiali), 2 μl di uno dei primer per codici a barre da un kit di codifica a barcoding PCR (vedere tabella dei materiali e tabella 2) e 1 μl di prodotto PCR purificato dalla fase 3.3.2 come modello. Aggiungere 47 μL di acqua libera nucleasi per ottenere un volume finale di 100 μL.
- Incubare la reazione in un termociclatore a 95 ° c per 3 minuti come denaturazione iniziale. Successivamente, eseguire 15 cicli per 15 s a 95 ° c, 15 s a 62 ° c e 1,5 min a 65 ° c. Come allungamento finale, incubare la reazione a 65 ° c per 5 min.
- Purificare il prodotto PCR come descritto sotto 3,2, ma utilizzare 100 μL di perle magnetiche e eluto in 30 μL di acqua priva di nucleasi.
- Se possibile, quantificare il campione utilizzando uno spettrofotometro UV.
5. preparazione della biblioteca
- Combinare una quantità uguale di DNA con codice a barre da ciascun campione per un totale di 1 μg di DNA in un volume di 45 μL (se necessario, aggiungere acqua priva di nucleasi) in un tubo di reazione da 0,2 mL. Se non è disponibile alcuno spettrofotometro UV, utilizzare volumi uguali di ciascun campione. Per dA-tailing, aggiungere 7 μL di tampone di reazione alla preparazione finale (vedere tabella dei materiali), 3 μL di miscela enzimatica end-Prep (vedere tabella dei materiali) e 5 μl di acqua priva di nucleasi. Mescolare delicatamente agitando il tubo.
- Incubare la reazione per 5 min a 20 ° c, seguita da 5 min a 65 ° c in un termociclatore.
- Purificare il prodotto di reazione come descritto nel paragrafo 3,2, ma utilizzare 60 μL di perle magnetiche e eluto in 25 μL di acqua priva di nucleasi.
- Facoltativo: assumere 1 μl per quantificare la concentrazione del campione utilizzando uno spettrofotometro UV. L'importo totale deve essere superiore a 700 ng.
- Unire 22,5 μL di DNA purificato dal passo 5,3 con 2,5 μL di adattatore 1D2 (vedere tabella dei materiali) e 25 μl di miscela Master Blunt/ta ligasi (vedere tabella dei materiali) in un nuovo tubo di reazione a basso legame di dna-bassa da 1,5 ml, mescolare delicatamente mediante la sfogliatura e ruotare brevemente giù. Incubare per 10 minuti a temperatura ambiente.
- Purificare il prodotto di reazione come descritto nella sezione 3,2, ma utilizzare 20 μL di perle magnetiche, aumentare il tempo di incubazione per il legame del DNA a 10 min, eseguire due passaggi di lavaggio con 1 mL di etanolo ciascuno e eluto in 46 μL di acqua priva di nucleasi.
- Combinare 45 μL del prodotto di reazione dal punto 5,6 con 5 μL di miscela di adattatori per codici a barre (vedere tabella dei materiali) e 50 μl di miscela Master Blunt/ta ligasi in un tubo di reazione a basso legame con DNA. Mescolare delicatamente sfogliando e incubare per 10 minuti a temperatura ambiente.
- Purificare il prodotto di reazione come descritto nella sezione 3,2, ma utilizzare 40 μL di perle magnetiche, fare due passaggi di lavaggio con 140 μL di tampone ABB (vedere tabella dei materiali) invece di etanolo, risospendere le perle da sfogliando e pellet su un rack magnetico. Elute in 15 μL di tampone di eluizione (vedere tabella dei materiali). Aumentare i tempi di incubazione per il legame iniziale del DNA alle perle e per la fase di eluizione a 10 min. conservare il prodotto risultante su ghiaccio o a 4 ° c fino all'uso.
6. controllo di qualità della cella di flusso
- Eseguire un controllo di qualità sulla cella di flusso prima dell'uso. A tal fine, collegare il dispositivo di sequenziazione al computer host e aprire il software.
- Inserire una cella di flusso (vedere tabella dei materiali) nel dispositivo di sequenziazione e scegliere il tipo di cella di flusso dalla casella di selezione e confermare facendo clic su disponibile.
- Fare clic su Controlla cella di flusso nella parte inferiore dello schermo e scegliere il tipo di cella di flusso corretto.
- Fare clic su Avvia test per avviare il controllo qualità. Per poter usare la cella di flusso è necessario un minimo di 800 nanoporti attivi in totale.
7. caricamento della cella di flusso e avvio della sequenza di esecuzione
- Aprire il coperchio della porta di adescamento facendolo scorrere in senso orario. Impostare una pipetta P1000 su 200 μL e inserire la punta nella porta di adescamento. Regolare la pipetta a 230 μL mantenendo la punta nella porta di adescamento, per aspirare 20-30 μL di tampone e rimuovere eventuali bolle d'aria.
- In un nuovo tubo di reazione a legame di DNA-basso da 1,5 mL, preparare il mix di adescamento combinando 576 μL di tampone RBF (vedere tabella dei materiali) con 624 μl di acqua priva di nucleasi.
- Pipettare con cautela 800 μL del mix di adescamento preparato nella porta di adescamento e attendere 5 minuti sollevare il coperchio della porta del campione e Pipettare un ulteriore 200 μL del mix di adescamento preparato nella porta di adescamento.
- Pipettare 35 μL di tampone RBF in un nuovo tubo di reazione a basso legame con DNA 1,5 mL pulito. Mescolare accuratamente le perle LLB (vedere tabella dei materiali) pipettando e aggiungere 25,5 μl di perline al tampone RBF. Aggiungere 2,5 μL di acqua priva di nucleasi e 12 μL di libreria del DNA dal punto 5,8 e miscelare mediante pipettaggio.
- Aggiungere 75 μL di miscela di campione in modo lento e graduale alla cella di flusso tramite la porta del campione.
- Sostituire il coperchio della porta di campionamento, chiudere la porta di adescamento e chiudere il coperchio del dispositivo di sequenziazione.
- All'interno del software, verificare che la cella di flusso sia ancora disponibile, aprire un nuovo esperimento e impostare i parametri di esecuzione selezionando il kit utilizzato. Selezionare Live base-Calling. Avviare l'esecuzione della sequenza facendo clic su inizia esperimento. Continuare la sequenza di esecuzione fino a quando non vengono raccolti dati sperimentali sufficienti.
Risultati
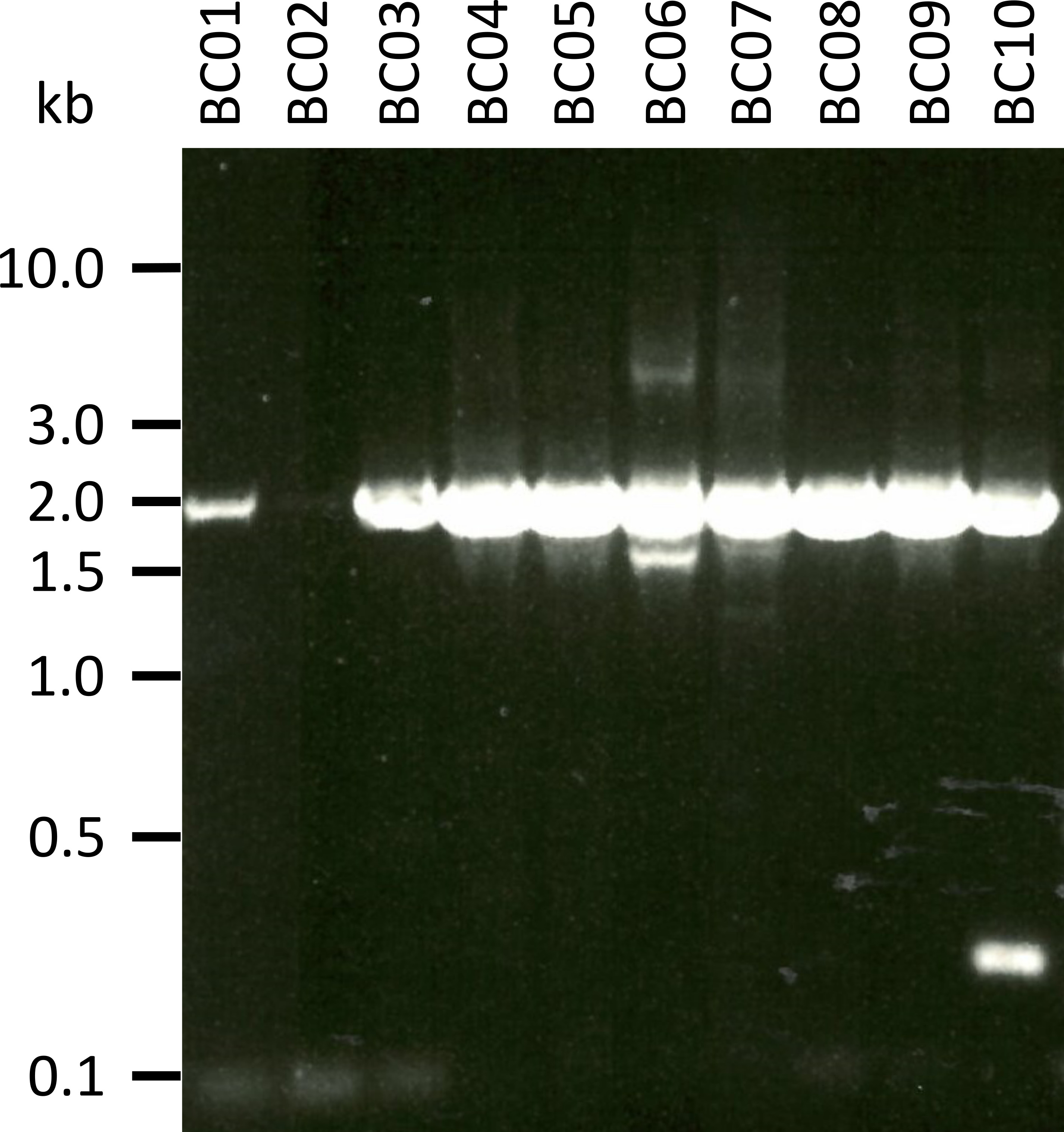
In un esperimento rappresentativo per testare il protocollo presentato abbiamo estratto l'RNA da 10 diversi campioni di sangue di cinque specie animali (cioè 2 individui per specie (capra, pecore, suini, cani, bovini)) (tabella 3). I rendimenti dell'RNA e la qualità dopo l'estrazione possono variare notevolmente, in particolare a causa delle differenze nella manipolazione e nello stoccaggio dei campioni. Nel nostro esperimento rappresentativo, abbiamo osservato concentrazioni di RNA tra 43 ng e 543 ng per μL (tabella 3). Inoltre, dopo l'amplificazione di RT-PCR, l'analisi del gel dei prodotti PCR NPC-1 ha mostrato vari esiti (Figura 2), con bande nettamente più deboli per i campioni BC01 e bc02 (entrambi caprini). Queste differenze sono state probabilmente causate da differenze nella qualità del campione, sebbene non sia possibile escludere differenze nell'efficacia della PCR dovute alle differenze nel legame del primer con il gene NPC1 di diverse specie. Tuttavia, queste differenze nella resa e/o nell'efficienza di amplificazione non hanno influito significativamente sull'esito complessivo del sequenziamento. Inoltre, si è verificato un ulteriore prodotto PCR non specifico nel campione BC10 (bovini). Contrariamente al sequenziamento di Sanger, tali prodotti non specifici non influenzano negativamente i risultati del sequenziamento delle nanoparticelle, poiché queste letture vengono scartate durante la mappatura delle letture ottenute in una sequenza di riferimento come parte dell'analisi dei dati.
Prima di ogni sequenza di sequenziamento, è fortemente raccomandato un controllo di qualità della cella di flusso da utilizzare, con un requisito minimo di 800 pori totali. Nel nostro esperimento rappresentativo, questo controllo di qualità ha restituito 1.102 pori disponibili per il sequenziamento. Poiché i dati sono forniti in tempo reale e possono essere analizzati immediatamente, la lunghezza di una sequenza di sequenziamento può essere regolata per la singola applicazione (ad esempio, fino a quando non vengono prodotti sufficienti dati di sequenziamento per l'analisi desiderata). Nei nostri esperimenti, le sequenze di sequenziamento sono tipicamente eseguite durante la notte, e nel caso del nostro esperimento rappresentativo abbiamo ottenuto circa 1,4 milioni letture durante una corsa di 14 ore.
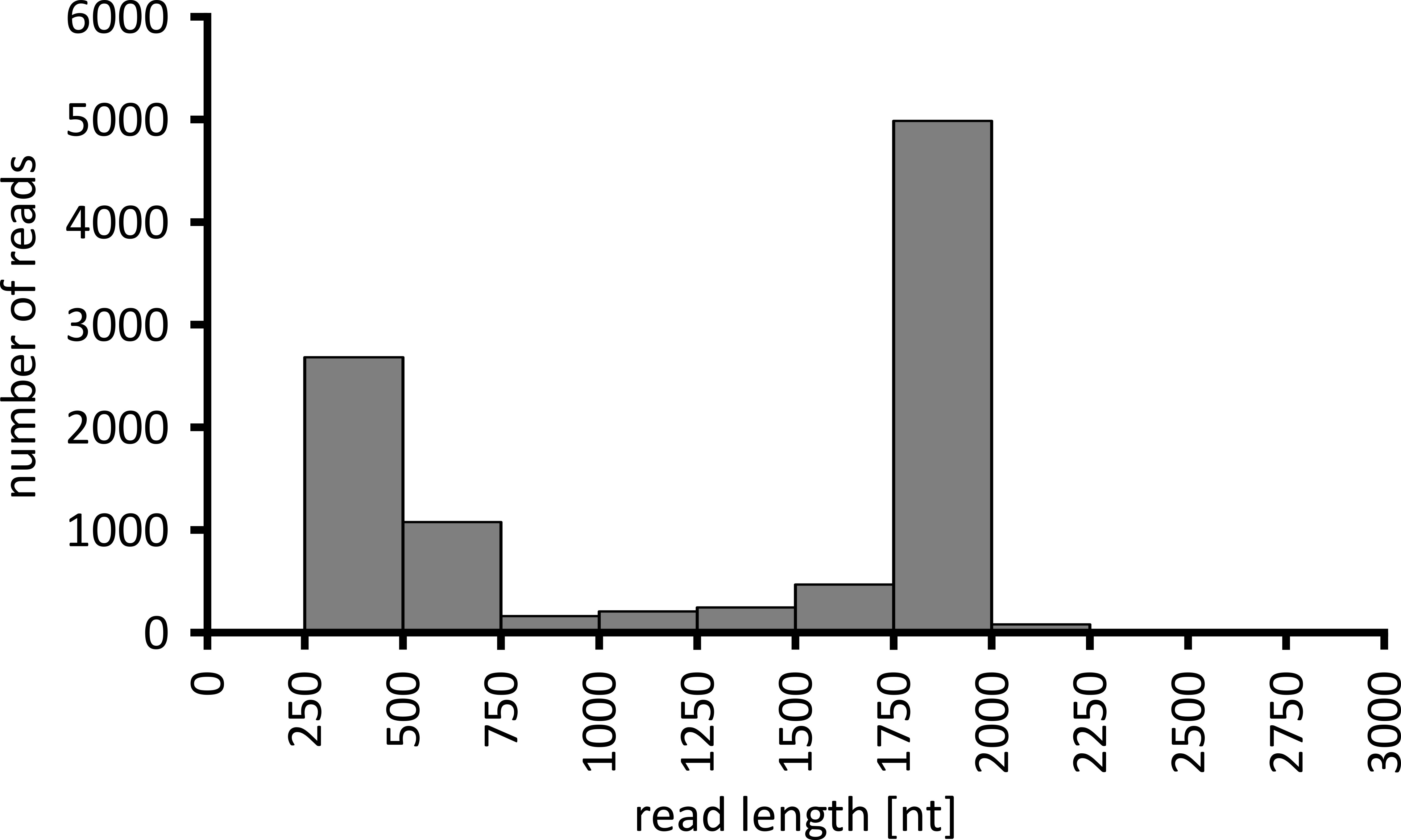
A seconda del tipo di analisi dei dati da eseguire, può essere consigliabile elaborare solo un sottoinsieme delle letture ottenute. Nel caso del nostro esperimento rappresentativo, è stato selezionato un sottoinsieme di 10.000 letture per ulteriori analisi. A tal fine, i file FASTQ generati durante la sequenza di sequenziamento sono stati ulteriormente elaborati in un ambiente LTS Ubuntu 18,04 e demultiplate utilizzando Flexbar v 3.0.3 con parametri ottimizzati per demultiplexing dei dati di sequenziamento nanoporo (Barcode-Tail-lunghezza 300 , codice a barre-error-rate 0,2, codice a barre-Gap-penalità-1)12. Dopo la demultiplexing, la mappatura di lettura e la generazione di consensi possono essere eseguite utilizzando una serie di strumenti diversi, ma una discussione dettagliata dell'aspetto bioinformatico del sequenziamento del nanoporo va oltre l'ambito di questo manoscritto. Tuttavia, nel caso dei nostri risultati rappresentativi, la mappatura di lettura a una sequenza di riferimento è stata eseguita utilizzando Geneious 10.2.3. Di 10.000 letture analizzate, 5.457 ha mostrato una lunghezza tra 1.750 e 2.000 nucleotidi, che corrisponde alle dimensioni attese per i frammenti PCR amplificati come parte del nostro flusso di lavoro (1.769 NT, Figura 3). È stato osservato un ulteriore picco nella distribuzione della lunghezza delle letture tra 250 e 500 nucleotidi, che possono essere attribuiti a prodotti PCR non specifici. La demultiplexing delle letture ha consentito l'assegnazione di 87,6% delle letture a uno dei 10 codici a barre/campioni analizzati (Figura 4). La proporzione di letture demultiplexate per ogni codice a barre varia da 3,4% per codice a barre da 1 a 16,9% per codice a barre 10; Tuttavia, a causa del gran numero complessivo di letture questo ha ancora permesso un consenso significativo chiamando con una profondità di lettura elevata anche per questi DataSet di codici a barre di abbondanza inferiore. Infatti, la mappatura delle letture ordinate a una sequenza di riferimento di NPC1 ha comportato tra il 31,7% (codice a barre 2) e il 100% (codice a barre 7 e 8) di lettura del mapping al riferimento, dando una profondità di lettura superiore a 90 letture in qualsiasi posizione per ogni campione. Questo è poi più che sufficiente per consentire fiducioso consenso base-chiamando con un tasso di errore trascurabile.

Figura 1: rappresentazione schematica del sequenziamento del DNA utilizzando la tecnologia nanoporo. Una molecola di DNA a filamento singolo passa attraverso un nanoporo incorporato in una membrana resistente elettricamente, con una elicasi che regola la velocità di transizione. Una corrente ionica passa simultaneamente attraverso il poro e viene misurata continuamente. Le modulazioni della corrente causate dai nucleotidi presenti nel poro vengono rilevate e ritradotte computazionalmente nella sequenza nucleotide del filamento del DNA. Si prega di cliccare qui per visualizzare una versione più grande di questa cifra.
{kind=link}

Figura 2: amplificazione dei prodotti di PCR di Niemann-Pick C1 da mRNA. mRNA è stato isolato da capra (BC01), pecore (BC03 e 04), suini (BC05 e 06), cane (BC07 e 08), e bovini (BC09 e 10). I prodotti PCR nidificati sono stati separati in un gel di agarosio 0,8% in tampone TAE 1x (preparato con tampone TAE 50x: 242,28 g di base tris, 57,1 mL di acido acetico glaciale, 100 mL di 0,5 M EDTA, dH2O a 1 L, pH regolato a 8,0) per 45 min a 100 V e macchiato con SYBR safe. Si prega di cliccare qui per visualizzare una versione più grande di questa cifra.
{kind=link}

Figura 3: distribuzione della lunghezza di lettura di 10.000 letture dall'esperimento rappresentativo. Viene indicato il numero di letture ottenute con un determinato intervallo di lunghezza di lettura. Si prega di cliccare qui per visualizzare una versione più grande di questa cifra.
{kind=link}

Figura 4: distribuzione delle letture dopo il demultiplexing. Vengono mostrati il numero e la percentuale di letture demultiplate (grigie) e mappate (nere) per ogni codice a barre. Si prega di cliccare qui per visualizzare una versione più grande di questa cifra.
{kind=link}
Tabella 1: Panoramica dei set di primer utilizzati. L'amplificazione iniziale delle sequenze di destinazione è stata eseguita con primer set 1. Primer set 2 è stato quindi utilizzato per l'amplificazione nidificata e l'aggiunta di adattatori. Gli adattatori sono indicati in rosso. Per favore clicca qui per scaricare questo file.
Tabella 2: Panoramica delle sequenze di codici a barre. I singoli codici a barre sono stati utilizzati per identificare ciascun campione sequenziato. Per favore clicca qui per scaricare questo file.
Tabella 3: concentrazioni di RNA ottenute in seguito all'estrazione da campioni di sangue sequenziati nell'esperimento rappresentativo. Vengono mostrate le concentrazioni di RNA di due individui da ciascuna delle cinque specie e vengono indicati i rapporti delle densità ottiche a 260/280 nm e 260/230 Nm. Per favore clicca qui per scaricare questo file.
Discussione
Negli ultimi due decenni, il sequenziamento dei campioni biologici è diventato un aspetto sempre più importante degli studi in una vasta gamma di aree tematiche. Lo sviluppo di sistemi di sequenziamento di seconda generazione basati sul sequenziamento di una fitta gamma di caratteristiche del DNA che utilizzano cicli iterativi di manipolazione enzimatica e acquisizione dati basata su immagini1 ha aumentato drasticamente il throughput rispetto al tecnica di sequenziamento Sanger tradizionale e consente l'analisi di campioni multipli e di varie specie di acido nucleico in un dato campione in parallelo4. Tuttavia, per la maggior parte dei sistemi di seconda generazione comunemente usati, vengono prodotte solo letture brevi e tutte le piattaforme si basano su apparecchiature sensibili, ingombranti e costose3,4.
A differenza delle piattaforme di sequenziamento di seconda generazione, il dispositivo di sequenziamento utilizzato in questo protocollo si basa sulla tecnologia nanoporo. Qui una molecola di acido nucleico a filamento singolo passa attraverso un nanopore, con conseguente modulazione di una corrente ionica che scorre anche attraverso lo stesso nanopore, e che può essere misurata e ritradotta per dedurre la sequenza della molecola di acido nucleico. Questo approccio di sequenziamento di terza generazione conferisce una serie di vantaggi rispetto ad altri approcci. I principali vantaggi che sono direttamente correlati al principio di funzionamento unico di questa tecnologia sono la lunghezza di lettura estremamente lunga prodotta (lunghezze di lettura fino a 8,8 x 105 nucleotidi sono stati segnalati6), la capacità di sequenziare non solo il DNA, ma anche RNA direttamente, che è stato recentemente dimostrato per un genoma completo del virus dell'influenza17, e la capacità di analizzare i dati in tempo reale come vengono generati, che consente la rapida rilevazione metagenomica di agenti patogeni in minuti18. Ulteriori vantaggi pratici sono le dimensioni estremamente ridotte del dispositivo di sequenziamento di nanoporo, che ne consente l'utilizzo in qualsiasi laboratorio o in missioni sul campo in sedi remote19,20e il prezzo basso rispetto ad altri sequenziamento Piattaforme. In termini di costi di gestione, attualmente è necessaria una nuova cella di flusso per ogni esecuzione di sequenziamento, che comporta costi di circa $1.100 per corsa per la cella di flusso e i reagenti di preparazione della libreria. Questi costi possono essere ridotti in alcuni casi mediante il lavaggio e il riuso della cella di flusso, oppure mediante barcoding e sequenziamento di più campioni in una singola corsa. Inoltre, un nuovo tipo di cella di flusso è attualmente in fase di beta-testato da un piccolo numero di laboratori, che richiederà l'uso di un adattatore di cella di flusso (chiamato "flongle"), e dovrebbe ridurre significativamente il prezzo della cella di flusso e quindi i costi di funzionamento.
La maggiore carenza di sequenziamento del nanoporo rimane la sua accuratezza, con singole precisioni di lettura nell'intervallo da 83 a 86% segnalati6,21,22e la maggior parte delle imprecisioni causate da inserimento/eliminazione ( indels)5,21. Tuttavia, un'elevata profondità di lettura può compensare queste imprecisioni e un recente studio suggerito sulla base di considerazioni teoriche che una profondità di lettura di > 10 potrebbe aumentare la precisione complessiva a > 99,8%21. Tuttavia, saranno necessari ulteriori miglioramenti nella precisione, in particolare se l'analisi deve essere eseguita su un livello di singola molecola piuttosto che su un livello di sequenza di consenso. L'uso della tecnologia 1D2 come descritto in questo protocollo, che si basa sull'aggiunta degli adattatori 1D2 e Barcode (cfr. sezione 5,5) che determinano la sequenzione di entrambe le ciocche di una singola molecola di DNA, aumenta la lettura precisione poiché le informazioni provenienti da entrambi i filamenti di DNA possono essere utilizzate per la determinazione della sequenza. Inoltre, una strategia alternativa che può essere perseguito al fine di combinare i vantaggi del sequenziamento di nanoporo (particolarmente lunga lunghezza di lettura) con la maggiore accuratezza di altre tecnologie di sequenziamento è quello di utilizzare le informazioni di sequenziamento di nanoporo come un scaffold, che è poi lucidato utilizzando i dati di sequenziamento da altre piattaforme6.
Il fattore più critico per il successo del protocollo qui presentato è la qualità del campione, e in particolare la quantità e la qualità dell'RNA estratto. La corretta conservazione e l'estrazione tempestiva dell'RNA contribuiscono a ottenere un adeguato rendimento di RNA. L'uso di adeguati tubi di raccolta del sangue consente lo stoccaggio di campioni di sangue per un massimo di un mese, ma la coagulazione del sangue può essere un problema, in particolare quando i campioni vengono conservati a temperature elevate, che può essere il caso in condizioni di campo. Il secondo passo critico è l'amplificazione delle sequenze bersaglio, e in particolare in condizioni di campo le reazioni di PCR spesso eseguono meno bene rispetto alle condizioni di laboratorio standard7. A tal fine, un'attenta progettazione e ottimizzazione del primer è fondamentale per ottenere un'amplificazione robusta. Inoltre, gli approcci di PCR nidificati e la PCR da touchdown, utilizzati in questo protocollo, possono aumentare sia la specificità che la sensibilità dell'amplificazione del gene bersaglio4,7. Infatti, nella nostra esperienza in Liberia e in Guinea con questa tecnologia sono stati richiesti protocolli nidificati in condizioni di campo con campioni di campo anche per set di primer che hanno consentito l'amplificazione di bersagli da campioni di laboratorio e in condizioni di laboratorio con un singolo round di PCR (7 e risultati non pubblicati).
A differenza di questi passaggi più critici, la preparazione della libreria e la sequenza di esecuzione sono procedure piuttosto robuste. Tuttavia, in condizioni di campo, questioni pratiche come la disponibilità di alcuni pezzi di attrezzature possono essere problematici. Ad esempio, è necessario uno spettrofotometro UV per determinare le concentrazioni di DNA prima della preparazione della libreria di campioni con codice a barre. Tuttavia, se un tale dispositivo non fosse disponibile in condizioni di campo, un volume uguale di ciascun campione può essere semplicemente combinato per creare i 45 μL necessari per la preparazione della libreria, con differenze nel materiale di input del campione, che viene di solito mitigato dalla grande numero di letture. Analogamente, la necessità di connettività Internet per l'esecuzione del sequenziamento può essere un problema, anche se la chiamata di base non deve più essere eseguita online, ma può essere eseguita localmente; Tuttavia, questa necessità può essere rimossa in determinate circostanze dal fabbricante, se necessario.
In sintesi, il protocollo presentato consente un sequenziamento relativamente basso costo in posizioni senza accesso alle apparecchiature di sequenziamento tradizionali, anche in luoghi remoti. Può essere facilmente adattato a qualsiasi RNA o DNA target, permettendo così ai ricercatori di rispondere a numerose domande biologiche.
Divulgazioni
TH ha partecipato al programma di accesso anticipato di Oxford nanopore Technologies (ONT) MinION da 2014 a 2015 e ha ricevuto dispositivi MinION e celle di flusso per un precedente studio 7 eseguito dagli istituti nazionali di salute, USA, a titolo gratuito o a costi ridotti . È stato invitato da ONT a presentare parte di quel lavoro all'incontro di Londra Calling 2015 a Londra, Regno Unito, e ONT pagato per il trasporto e l'alloggio. Per il lavoro presentato in questo manoscritto non sono stati ottenuti benefici (ad es. hardware o reagenti a costi ridotti, rimborsi di viaggio, ecc.) da parte di ONT. AM, KF e RS non hanno nulla da rivelare.
Riconoscimenti
Gli autori ringraziano Allison Groseth per la lettura critica del manoscritto. Questo lavoro è stato sostenuto finanziariamente dal Ministero federale tedesco dell'alimentazione e dell'agricoltura (BMEL) sulla base di una decisione del Parlamento della Repubblica federale di Germania attraverso l'Ufficio federale dell'agricoltura e dell'alimentazione (BLE).
Materiali
| Name | Company | Catalog Number | Comments |
| 1D2 adapter, barcode adapter mix, ABB buffer, elution buffer, RBF buffer, LBB beads | Oxford Nanopore Technologies | SQK-LSK308 | 1D² Sequencing Kit |
| Blood collection tube with DNA/RNA stabilizing reagent | Zymo Research | R1150 | DNA/RNA Shield - Blood Collection Tube |
| Blunt/TA ligase master mix | New England Biolabs | M0367S | Blunt/TA Ligase Master Mix |
| DNA-low binding reaction tube | Eppendorf | 30108051 | DNA LoBind Tube |
| DNase buffer and DNase | ThermoFisher Scientific | 11766050 | SuperScript™ IV VILO™ Master Mix with ezDNase™ Enzyme |
| Flow cell | Oxford Nanopore Technologies | FLO-MIN105.24 | flow cell R9.4 |
| Hot start high fidelity DNA polymerase | New England Biolabs | M0493L | Q5 Hot Start High-Fidelity DNA Polymerase (500 U) |
| Magnetic beads | Beckman Coulter | A63881 | Agencourt AMPure XP beads |
| Magnetic rack | ThermoFisher Scientific | 12321D | DynaMag-2 Magnet |
| Nanopore sequencing device | Oxford Nanopore Technologies | - | MinION Mk 1B |
| PCR barcoding kit | Oxford Nanopore Technologies | EXP-PBC001 | PCR Barcoding Kit I (R9) |
| Reverse transcriptase master mix | ThermoFisher Scientific | 11766050 | SuperScript™ IV VILO™ Master Mix with ezDNase™ Enzyme |
| RNA purification spin column, DNA/RNA prep buffer, DNA/RNA wash buffer, DNase I, DNA digestion buffer | Zymo Research | R1151 | Quick-DNA/RNA Blood Tube Kit |
| Rotating mixer | ThermoFisher Scientific | 15920D | HulaMixer Sample Mixer |
| Taq DNA polymerase | New England Biolabs | M0287S | LongAmp Taq 2x Master Mix |
| Ultra II End-prep kit | New England Biolabs | E7546S | NEBNext Ultra II End-Repair/dA-tailing Modul |
| UV spectrophotometer | Implen | - | NanoPhotometer |
| Vacuum manifold | Zymo Research | S7000 | EZ-Vac Vacuum Manifold |
Riferimenti
- Shendure, J., Ji, H. Next-generation DNA sequencing. Nature Biotechnology. 26 (10), 1135-1145 (2008).
- Shendure, J., Lieberman Aiden, E. The expanding scope of DNA sequencing. Nature Biotechnology. 30 (11), 1084-1094 (2012).
- Liu, L., et al. Comparison of next-generation sequencing systems. Journal of Biomedicine and Biotechnology. 2012, 251364 (2012).
- Levy, S. E., Myers, R. M. Advancements in Next-Generation Sequencing. Annual Review of Genomics and Human Genetics. 17, 95-115 (2016).
- Lu, H., Giordano, F., Ning, Z. Oxford Nanopore MinION Sequencing and Genome Assembly. Genomics, Proteomics & Bioinformatics. 14 (5), 265-279 (2016).
- Jain, M., et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nature Biotechnology. 36 (4), 338-345 (2018).
- Hoenen, T., et al. Nanopore Sequencing as a Rapidly Deployable Ebola Outbreak Tool. Emerging Infectious Diseases. 22 (2), 331-334 (2016).
- Quick, J., et al. Real-time, portable genome sequencing for Ebola surveillance. Nature. 530 (7589), 228-232 (2016).
- Carette, J. E., et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature. 477 (7364), 340-343 (2011).
- Ndungo, E., et al. A Single Residue in Ebola Virus Receptor NPC1 Influences Cellular Host Range in Reptiles. mSphere. 1 (2), (2016).
- Martin, S., et al. A genome-wide siRNA screen identifies a druggable host pathway essential for the Ebola virus life cycle. Genome Medicine. 10 (1), 58 (2018).
- Roehr, J. T., Dieterich, C., Reinert, K. Flexbar 3.0 - SIMD and multicore parallelization. Bioinformatics. 33 (18), 2941-2942 (2017).
- Li, H., et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25 (16), 2078-2079 (2009).
- Kielbasa, S. M., Wan, R., Sato, K., Horton, P., Frith, M. C. Adaptive seeds tame genomic sequence comparison. Genome Research. 21 (3), 487-493 (2011).
- Don, R. H., Cox, P. T., Wainwright, B. J., Baker, K., Mattick, J. S. Touchdown' PCR to circumvent spurious priming during gene amplification. Nucleic Acids Research. 19 (14), 4008 (1991).
- Korbie, D. J., Mattick, J. S. Touchdown PCR for increased specificity and sensitivity in PCR amplification. Nature Protocols. 3 (9), 1452-1456 (2008).
- Keller, M. W., et al. Direct RNA Sequencing of the Coding Complete Influenza A Virus Genome. Scientific Reports. 8 (1), 14408 (2018).
- Greninger, A. L., et al. Rapid metagenomic identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis. Genome Medicine. 7, 99 (2015).
- Castro-Wallace, S. L., et al. Nanopore DNA Sequencing and Genome Assembly on the International Space Station. Scientific Reports. 7 (1), 18022 (2017).
- Goordial, J., et al. In Situ Field Sequencing and Life Detection in Remote (79 degrees 26'N) Canadian High Arctic Permafrost Ice Wedge Microbial Communities. Frontiers in Microbiology. 8, 2594 (2017).
- Runtuwene, L. R., et al. Nanopore sequencing of drug-resistance-associated genes in malaria parasites, Plasmodium falciparum. Scientific Reports. 8 (1), 8286 (2018).
- Rang, F. J., Kloosterman, W. P., de Ridder, J. From squiggle to basepair: computational approaches for improving nanopore sequencing read accuracy. Genome Biology. 19 (1), 90 (2018).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati