
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Visualización simultánea de la dinámica de microtúbulos reticulados y únicos in vitro mediante microscopía TIRF
* Estos autores han contribuido por igual
En este artículo
Resumen
Aquí, se presenta un ensayo de reconstitución in vitro basado en microscopía TIRF para cuantificar y comparar simultáneamente la dinámica de dos poblaciones de microtúbulos. Se describe un método para ver simultáneamente la actividad colectiva de múltiples proteínas asociadas a microtúbulos en haces de microtúbulos reticulados y microtúbulos individuales.
Resumen
Los microtúbulos son polímeros de heterodímeros de αβ-tubulina que se organizan en estructuras distintas en las células. Las arquitecturas y redes basadas en microtúbulos a menudo contienen subconjuntos de matrices de microtúbulos que difieren en sus propiedades dinámicas. Por ejemplo, en las células en división, los haces estables de microtúbulos reticulados coexisten muy cerca de los microtúbulos dinámicos no reticulados. Los estudios de reconstitución in vitro basados en microscopía TIRF permiten la visualización simultánea de la dinámica de estas diferentes matrices de microtúbulos. En este ensayo, se ensambla una cámara de imágenes con microtúbulos inmovilizados en la superficie, que están presentes como filamentos individuales u organizados en haces reticulados. La introducción de tubulina, nucleótidos y reguladores de proteínas permite la visualización directa de proteínas asociadas y de las propiedades dinámicas de microtúbulos simples y reticulados. Además, los cambios que ocurren a medida que los microtúbulos individuales dinámicos se organizan en haces se pueden monitorear en tiempo real. El método descrito aquí permite una evaluación sistemática de la actividad y localización de proteínas individuales, así como los efectos sinérgicos de los reguladores de proteínas en dos subconjuntos de microtúbulos diferentes en condiciones experimentales idénticas, proporcionando así información mecanicista que es inaccesible por otros métodos.
Introducción
Los microtúbulos son biopolímeros que forman andamios estructurales esenciales para múltiples procesos celulares, que van desde el transporte intracelular y el posicionamiento de orgánulos hasta la división celular y el alargamiento. Para ejecutar estas diversas funciones, los microtúbulos individuales se organizan en matrices del tamaño de micras, como husos mitóticos, axonemas ciliares, haces neuronales, matrices interfase y matrices corticales de plantas. Un motivo arquitectónico omnipresente que se encuentra en estas estructuras es un haz de microtúbulos reticulados a lo largo de sus longitudes1. Una característica intrigante de varias estructuras basadas en microtúbulos es la coexistencia de microtúbulos agrupados y microtúbulos individuales no reticulados en estrecha proximidad espacial. Estas subpoblaciones de microtúbulos pueden mostrar una dinámica de polimerización marcadamente diferente entre sí, según sea necesario para su correcto funcionamiento2,3,4,5. Por ejemplo, dentro del huso mitótico, haces reticulados estables y microtúbulos individuales dinámicos están presentes dentro de una región a escala de micras en el centro celular6. Estudiar cómo se especifican las propiedades dinámicas de las poblaciones coexistentes de microtúbulos es, por lo tanto, fundamental para comprender el ensamblaje y la función de las estructuras basadas en microtúbulos.
Los microtúbulos son polímeros dinámicos que circulan entre las fases de polimerización y despolimerización, cambiando entre las dos fases en eventos conocidos como catástrofe y rescate7. La dinámica de los microtúbulos celulares está regulada por una miríada de proteínas asociadas a microtúbulos (MAP) que modulan las tasas de polimerización y despolimerización de microtúbulos y las frecuencias de catástrofes y eventos de rescate. Es difícil investigar la actividad de los MAP en matrices espacialmente proximales en células, debido a las limitaciones de la resolución espacial en microscopía de luz, especialmente en regiones de alta densidad de microtúbulos. Además, la presencia de múltiples MAP en la misma región celular dificulta las interpretaciones de los estudios biológicos celulares. Los ensayos de reconstitución in vitro, realizados junto con la microscopía de fluorescencia de reflexión interna total (TIRF), evitan los desafíos de examinar los mecanismos por los cuales subconjuntos específicos de MAP regulan la dinámica de las matrices de microtúbulos celulares proximales. Aquí, la dinámica de los microtúbulos ensamblados in vitro se examina en presencia de uno o más MAP recombinantes en condiciones controladas8,9,10. Sin embargo, los ensayos de reconstitución convencionales se realizan típicamente en microtúbulos individuales o en un tipo de matriz, lo que impide la visualización de poblaciones coexistentes.
Aquí, presentamos ensayos de reconstitución in vitro que permiten la visualización simultánea de dos poblaciones de microtúbulos en las mismas condiciones de solución11. Describimos un método para ver simultáneamente la actividad colectiva de múltiples MAP en microtúbulos individuales y en haces de microtúbulos reticulados por la proteína PRC1 asociada al huso mitótico. La proteína PRC1 se une preferentemente a la superposición entre microtúbulos antiparalelos, reticulándolos9. Brevemente, este protocolo consta de los siguientes pasos: (i) preparación de soluciones madre y reactivos, (ii) limpieza y tratamiento superficial de los cubrebocas utilizados para crear la cámara de imágenes para experimentos de microscopía, (iii) preparación de "semillas" de microtúbulos estables a partir de las cuales se inicia la polimerización durante el experimento, (iv) especificación de los ajustes del microscopio TIRF para visualizar la dinámica de los microtúbulos, (v) inmovilización de semillas de microtúbulos y generación de haces de microtúbulos reticulados en la cámara de imágenes, y (vi) visualización de la dinámica de microtúbulos en la cámara de imágenes a través de microscopía TIRF, tras la adición de tubulina soluble, MAP y nucleótidos. Estos ensayos permiten la evaluación cualitativa y el examen cuantitativo de la localización de MAP y su efecto en la dinámica de dos poblaciones de microtúbulos. Además, facilitan la evaluación de los efectos sinérgicos de múltiples MAP en estas poblaciones de microtúbulos, a través de una amplia gama de condiciones experimentales.
Protocolo
1. Preparar reactivos
- Prepare tampones y reactivos como se describe en la Tabla 1 y la Tabla 2. Durante el experimento, mantenga todas las soluciones en hielo, a menos que se indique lo contrario.
| Solución | Componentes | Duración de almacenamiento recomendada | Notas | ||
| 5X BRB80 | 400 mM K-PIPES, 5 mM MgCl2, 5 mM EGTA, pH 6.8 con KOH, filtro esterilizar | hasta 2 años | Conservar a 4 °C | ||
| 1X BRB80 | 80 mM K-PIPES, 1 mM MgCl2, 1 mM EGTA, pH 6.8 | hasta 2 años | Conservar a 4 °C | ||
| BRB80-TDT | 1X BRB80, 1 mM TDT | hasta 2 días | |||
| Búfer de ensayo | 80 mM K-PIPES, 3 mM MgCl2, 1 mM EGTA, pH 6.8, 5% sacarosa (OR 1X BRB80, 5% sacarosa, 2 mM MgCl2) | hasta 1 año | Conservar a 4 °C | ||
| Búfer maestro (MB) | Búfer de ensayo, 5mM TCEP | 1 semana | Prepárese el día del experimento; Separado en dos tubos: MB-warm a temperatura ambiente y MB-cold on ice; incluir 1 mM de TDT si se utilizan colorantes fluorescentes | ||
| Master Buffer con metilcelulosa (MBMC) | 1X BRB80, 0,8% metilcelulosa, 5 mM TCEP, 5 mM MgCl2 | 1 semana | Prepárese el día del experimento; incluir 1 mM de TDT si se utilizan colorantes fluorescentes | ||
| Tampón de dilución de proteínas (DB) | MB, 1 mg/ml de albúmina sérica bovina (BSA), 1 μM de ATP | 1 día, sobre hielo | Prepárese el día del experimento; incluir 1 mM de TDT si se utilizan colorantes fluorescentes | ||
| Mezcla de eliminación de oxígeno (OSM) | MB, 389 μg/ml de catalasa, 4,44 mg/ml de glucosa oxidasa, 15,9 mM de 2-mercaptoetanol (BME) | 1 día, sobre hielo | Prepárese el día del experimento | ||
| Final de eliminación de oxígeno (OSF) | MB, 350 μg/ml de catalasa, 4 mg/ml de glucosa oxidasa, 14,3 mM de BME, 15 mg/ml de glucosa | usar en 30 min | Preparar inmediatamente antes de su uso añadiendo 1 μL de glucosa a 9 μL de OSM | ||
Tabla 1: Lista de búferes utilizados en este protocolo y sus componentes. Consulte la columna "Duración de almacenamiento recomendada" para obtener orientación sobre la antelación con la que se puede preparar cada búfer.
| Reactivo | Concentración de almacenamiento | Disolvente de almacenamiento | Temperatura de almacenamiento | Concentración de trabajo | Concentración final | Duración de almacenamiento recomendada | Notas | |||||||||
| Neutravidina (NA) | 5 mg/ml | 1X BRB80 | -80°C | 0,2 mg/ml | 0,2 mg/ml | hasta 1 año | Se utiliza para inmovilizar microtúbulos a través de un enlace biotina-neutravidina-biotina; almacenar en pequeñas alícuotas | |||||||||
| Kappa-caseína (KC) | 5 mg/ml | 1X BRB80 | -80°C | 0,5 mg/ml | 0,5 mg/ml | hasta 2 años | Se utiliza para bloquear la superficie de la cámara de imágenes; Almacenar en pequeñas alícuotas; El día del experimento, deje a un lado un pequeño volumen a temperatura ambiente | |||||||||
| Albúmina sérica bovina (BSA) | 50 mg/ml | 1X BRB80 | -20°C | 1 mg/ml (en DB) | N/A | hasta 2 años | almacenar en pequeñas alícuotas | |||||||||
| Catalasa | 3,5 mg/ml | 1X BRB80 | -80°C | 350 μg/ml (en OSF) | 35 μg/ml | hasta 2 años | componente de la mezcla de eliminación de oxígeno; almacenar en pequeñas alícuotas | |||||||||
| Glucosa oxidasa | 40 mg/ml | 1X BRB80 | -80°C | 4 mg/ml (en la FSO) | 0,4 mg/ml | hasta 2 años | componente de la mezcla de eliminación de oxígeno; almacenar en pequeñas alícuotas | |||||||||
| Tubulina | Liofilizado | N/A | 4°C | 10 mg/ml | 2,12 mg/ml (en mezcla de tubulina) | hasta 1 año | Una vez que la tubulina esté en solución, manténgala fría para evitar la polimerización. | |||||||||
| Trifosfato de adenosina (ATP) | 100 metros | agua ultrapura | -20°C | 10 mM | 1 mM | 6 meses | Prepare la solución en agua esterilizada por filtro, ajuste el pH a ~ 7.0 y congele en pequeñas alícuotas. | |||||||||
| Trifosfato de guanosina (GTP) | 100 metros | agua ultrapura | -20°C | 10 mM | 1.29 mM (en mezcla de tubulina) | 6 meses | Prepare la solución en agua esterilizada por filtro, ajuste el pH a ~ 7.0 y congele en pequeñas alícuotas. | |||||||||
| Guanosina-5'-[(α,β)-metileno] trifosfato (GMPCPP) | 10 mM | agua ultrapura | -20°C | 10 μM | 0,5 μM | 6 meses | ||||||||||
| Ditiotreitol (TDT) | 1 M | agua estéril | -20°C | 1 mM | N/A | hasta 2 años | ||||||||||
| Tris(2-carboxietilo) fosfina (TCEP) | 0,5 M | agua esterilizada por filtro | Temperatura ambiente | 5 mM | N/A | hasta 2 años | ||||||||||
| Metilcelulosa | 1% | agua estéril | Temperatura ambiente | 0.8% (en MBMC) | 0.21% (en mezcla de tubulina) | hasta 1 año | Disuelva la metilcelulosa agregándola lentamente al agua casi hirviendo. Dejar enfriar mientras se agita continuamente. | |||||||||
| Beta-mercaptoetanol (BME) | 143 metros | agua estéril | Temperatura ambiente | 14,3 mM (en OSF) | 1,43 mM | hasta 5 años | 143 mM es una dilución 1:100 de stock BME | |||||||||
| Glucosa | 150 mg/ml | 1X BRB80 | -80°C | 15 mg/ml (en OSF) | 1,5 mg/ml | hasta 2 años | Agregar a OSM inmediatamente antes de usarlo | |||||||||
| (±)-6-Hydroxy-2,5,7,8-tetramethyl chromane-2-carboxylic acid (Trolox) | 10mM | 1X BRB80 | -80°C | 10 mM | 1 mM | hasta 1 año | No se disuelve por completo. Agregue un poco de NaOH, revuelva durante ~ 4 horas y esterilice el filtro antes de usarlo | |||||||||
| mPEG-Succinimidil Valerato, MW 5.000 | polvo | N/A | -20°C | 333 mg/ml (en 0,1 M de bicarbonato de sodio) | 324 mg/ml (en 0,1 M de bicarbonato de sodio) | 6 meses | Prepare ~ 34 mg de alícuotas, marcando cada tubo con un peso exacto de polvo. Pase el gas nitrógeno sobre el sólido, selle los tubos con parafilm y guárdelo a -20 ° C en un recipiente con desecante. | |||||||||
| Biotina-PEG-SVA, MW 5.000 | polvo | N/A | -20°C | 111 mg/ml (en 0,1 M de bicarbonato de sodio) | 3,24 mg/ml (en 0,1 M de bicarbonato de sodio) | 6 meses | Prepare ~ 3 mg de alícuotas, marcando cada tubo con un peso exacto de polvo. Pase el gas nitrógeno sobre el sólido, selle los tubos con parafilm y guárdelo a -20 ° C en un recipiente con desecante. | |||||||||
Tabla 2: Lista de reactivos utilizados en este protocolo. Se incluyen las condiciones y concentraciones de almacenamiento recomendadas, las concentraciones de trabajo de las soluciones madre utilizadas durante el experimento y la concentración final en la cámara de imágenes. Se dan notas adicionales en la columna de extrema derecha.
2. Preparar diapositivas biotina-PEG
NOTA: Prepare cámaras de imágenes lo más cerca posible del inicio de un experimento y no con más de 2 semanas de anticipación.
- Cubiertas limpias
- Coloque un número igual de cubiertas de 24 x 60 mm y 18 x 18 mm #1.5 (Figura 1F) en frascos de tinción deslizante y bastidores de lavado deslizantes, respectivamente (Figura 1A, B). Coloque las rejillas de lavado deslizantes que contienen hojas de cubierta de 18 x 18 mm en un vaso de precipitados de 100 ml.
- Enjuague todas las hojas de cubierta 5-6 veces en agua ultrapura (resistividad de 18,2 MΩ-cm) y retire el exceso de líquido después de cada enjuague con una punta de pipeta unida a un tubo de vacío (Figura 1C).
- Llene los vasos de precipitados y los frascos de tinción deslizante que contienen los cubrehojas con agua ultrapura, selle con parafilm y sonice durante 10 minutos.
- Llene dos vasos de precipitados de 150 ml con etanol a prueba de 200. Usando pinzas, sumerja cada funda en un vaso de precipitados lleno de etanol y luego en el otro.
- Con pinzas, transfiera las cubiertas a la rejilla de secado deslizante (Figura 1D), séquelas bajo una corriente de gas nitrógeno e incube a 37 ° C hasta que estén completamente secas (~ 15 min).
- Coloque las fundas secas en una sola capa dentro del limpiador de plasma. Forme un sello de vacío y luego establezca el nivel de radiofrecuencia (RF) del limpiador de plasma en ~ 8 MHz.
- Una vez que se genera el plasma, deje las cubiertas en el limpiador de plasma durante 5 minutos. Apague el limpiador de plasma y suelte la aspiradora lentamente.
- Una vez que se libere el sello de vacío, voltee los cubrecolchas y repita la limpieza con plasma durante 5 minutos para el otro lado de los cubrecolchas.
- Alternativa a la limpieza por plasma: En lugar de los pasos 2.1.2-2.1.3, sonicar las fundas en una solución tibia de detergente al 2% (en agua ultrapura) durante 10 min. Luego, lave bien las fundas con agua ultrapura y sonice en agua ultrapura 2-3 veces (10 minutos cada una). A continuación, lavar en etanol y secar como en los pasos 2.1.4-2.1.5. Omita los pasos 2.1.6-2.1.8.
- Tratamiento con biotina-PEG
- Inmediatamente antes de su uso, disolver 400 μL de 3-Aminopropiltrietoxisilano en 40 ml de acetona. Con pinzas, mueva las cubiertas limpias con plasma a la rejilla de lavado deslizante y a los frascos que manchan las diapositivas. Sumergir los recubiertos en solución de 3-aminopropiltrietoxisilano e incubar durante 5 min12,13.
- Lave todas las fundas 5-6 veces con agua ultrapura.
- Transfiera los cubrehojas a la rejilla de secado deslizante, séquelos bajo una corriente de gas nitrógeno e incube a 37 ° C hasta que estén completamente secos (~ 20 min).
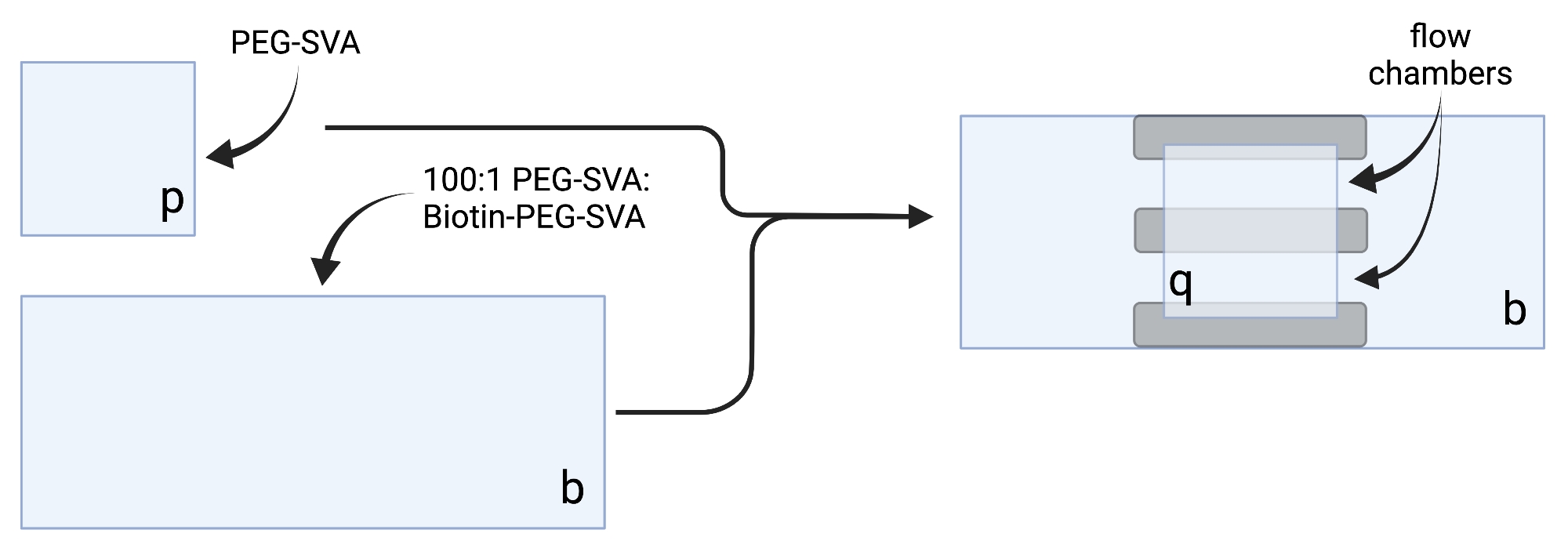
- Coloque los cubrehojas secas en toallitas de tareas delicadas y etiquete cada funda en una esquina, por ejemplo, 'p' en cada funda de 18 x 18 mm y 'b' en cada funda de 24 x 60 mm (ver Figura 2).
- El día del experimento, prepare una solución fresca de bicarbonato de sodio de 0,1 M disolviendo 0,84 mg de NaHCO3 en 10 ml de agua ultrapura.
- Lleve las alícuotas de mPEG-Succinimidyl Valerate (PEG-SVA) y Biotin-PEG-SVA a temperatura ambiente, inmediatamente antes de su uso. Véanse las notas sobre la preparación de alícuotas de polietilenglicol (PEG) en la Tabla 2.
NOTA: Trabaje rápidamente porque la vida media de hidrólisis de la fracción de valerato de succinimidil (SVA) es de ~ 30 min. - Añadir 102 μL de 0,1 M de NaHCO3 a 34 mg de PEG-SVA, hilar en una microcentrífuga de sobremesa a 2.656 x g durante 20 s, y luego mezclar mediante pipeteo hacia arriba y hacia abajo. Disolver 3 mg de Biotina-PEG-SVA en 27 μL de 0,1 M de NaHCO3 mediante pipeteo hacia arriba y hacia abajo. Ajustar los volúmenes de dilución de acuerdo con el peso exacto de PEG observado en los tubos (ver Tabla 2).
- Preparar 100:1 p/p de mezcla PEG:biotina-PEG combinando 75 μL de solución de PEG-SVA y 2,25 μL de solución de Biotina-PEG-SVA para 20 fundas, 100 μL y 3 μL para 30 fundas, o 125 μL y 3,75 μL para 40 fundas.
- Construya una cámara de hidratación colocando toallas de papel húmedas debajo del estante de punta en el fondo de una caja de punta vacía de 10 μL (Figura 1E). Esto evitará la evaporación de las soluciones PEG.
- Pipeta 6 μL de mezcla 100:1 PEG-SVA:biotina-PEG en el centro de una funda de 24 x 60 mm en el lado etiquetado. Coloque otro trozo de 24 x 60 mm en la parte superior del primer cobertor de modo que el par forme una forma de X, con los lados etiquetados como 'b' uno frente al otro. Coloque el par en una rejilla de punta vacía en la cámara de hidratación y repita para los cubrehojas restantes de 24 x 60 mm.
- Pipete 6 μL de PEG-SVA en el centro de una cubierta de 18 x 18 mm en el lado etiquetado. Coloque otro trozo de cubierta de 18 x 18 mm en la parte superior del primer cobertor, con los lados etiquetados como 'p' uno frente al otro. Coloque el par en una rejilla de punta vacía en la cámara de hidratación y repita para los cubrehojas restantes de 18 x 18 mm.
- Cierre la cámara de hidratación e incube durante 3 h o durante la noche.
- Separe los pares de fundas y enjuague en agua ultrapura.
- Seque los cubrecolchas con una corriente de nitrógeno y colóquelos en una incubadora de 37 ° C para que se sequen completamente.
- Para construir la cámara de imágenes, pegue tres tiras de cinta adhesiva de doble cara en una cubierta de 24 x 60 mm en el lado etiquetado como 'b'. Al otro lado de las tiras de cinta, coloque un cubrebocas de 18 x 18 mm con su lado etiquetado como 'p' frente a la cubierta más grande. Esto forma dos cámaras de flujo para experimentos de microscopía, con superficies tratadas una frente a la otra (Figura 2 y Figura 1G).

Figura 1: Equipo para el tratamiento de hojas de cubierta y preparación de cámaras de imagen. (A) frascos de tinción deslizante para fundas de 24 x 60 mm, (B) bastidores de lavado deslizantes para latas de 18 x 18 mm, (C) configuración al vacío, (D) estante de secado deslizante, (E) cámara de hidratación, (F) fundas, (G) cámara de imagen, (H) soporte deslizante. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Esquema para la preparación de cámaras de imagen utilizando cinta de doble cara (gris) y cubiertas tratadas con PEG/Biotina-PEG. Creado con BioRender.com. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
3. Polimerizar microtúbulos
- Preparar semillas GMPCPP
NOTA: Prepare las semillas GMPCPP en una cámara frigorífica, manteniendo todos los reactivos, puntas y tubos a 4 ° C. Las semillas GMPCPP se pueden preparar con anticipación y almacenar a -80 ° C durante un máximo de 1 año. Coloque el rotor de la ultracentrífuga de ángulo fijo, que contiene tubos de centrífuga de 1 ml, en la ultracentrífuga y ajuste la temperatura a 4 ° C.- Resuspend tubulina liofilizada (Tabla 2) a ~10 mg/ml en 1X BRB80, inmediatamente antes de su uso.
- Mezcle los componentes de las semillas GMPCPP como se describe en la Tabla 3.
NOTA: Mantenga todos los componentes de la tubulina en hielo tanto como sea posible para minimizar la polimerización de la tubulina soluble. - Clarificar la mezcla en un rotor de ultracentrífuga de ángulo fijo a 352.700 x g durante 5 min a 4 °C.
- Separe el sobrenadante en alícuotas de 5 μL, congélelos en nitrógeno líquido y guárdelos a -80 °C.
- Polimerizar semillas el día del experimento
- Caliente 1-2 ml de BRB80-TDT (Tabla 1) a 37 °C.
- Coloque 5 μL de alícuota de semillas GMPCPP (-80 °C) del paso 3.1.4 sobre hielo y disuelva inmediatamente en 20 μL de BRB80-TDT caliente. Girar a 2.000 x g durante 5 s a temperatura ambiente y tocar para mezclar.
NOTA: El volumen de dilución inicial puede variar entre 13 μL y 21 μL y se determina empíricamente para cada lote de semillas. Si las semillas no se polimerizan, solucione los problemas complementando el tampón de dilución inicial (paso 3.2.2) con 0,5 μM de GMPCPP. - Proteger de la luz e incubar a 37 °C durante 30-45 min.
NOTA: La longitud de los microtúbulos depende de la duración de la incubación. Para microtúbulos cortos, el tiempo de incubación puede ser tan corto como 15 min. Para microtúbulos largos, el tiempo de incubación puede ser de hasta 2 h. Los microtúbulos biotinilados tienden a requerir tiempos de incubación más largos que los microtúbulos no biotinilados. - Coloque el rotor de la ultracentrífuga de ángulo fijo, que contiene tubos de centrífuga de 500 μL, en la ultracentrífuga y precalentado a 30 °C.
- Después de la incubación, agregue 50 μL de BRB80-TDT caliente (paso 3.2.1) a las semillas GMPCPP polimerizadas y transfiera la mezcla a un tubo de centrífuga de 500 μL. Lave el tubo vacío que contenía las semillas GMPCPP con otros 50 μL de BRB80-TDT caliente, pipetee hacia arriba y hacia abajo, y agregue este tampón al tubo centrífugo de 500 μL que contiene la mezcla.
- Antes de girar, marque el borde del tubo de la centrífuga para indicar dónde estará el pellet (el pellet será demasiado pequeño para verlo). Girar durante 10 min a 244.900 x g a 30 °C12.
- Pipetee cuidadosamente el sobrenadante y deseche. Resuspend el pellet en 100 μL de BRB80-TDT caliente. Toca para mezclar.
- Girar durante 10 min a 244.900 x g a 30 °C, alineando el marcado con el rotor para peletizar en el mismo lugar.
- Retire el sobrenadante y vuelva a suspender el pellet en 16 μL de BRB80-TDT caliente. Transfiera la solución de microtúbulos a un tubo de microcentrífuga limpio de 0,6 ml. Proteger de la luz y mantener a temperatura ambiente o por encima de ella.
NOTA: Después de la polimerización, mantenga los microtúbulos a temperatura ambiente o por encima de ella. Si se enfrían, se despolimerizarán. Incubar a 28 °C para mayor estabilidad.
- Comprobación de microtúbulos mediante microscopía TIRF
- Pipetear una mezcla de 4,5 μL de BRB80-TDT y 1 μL de solución de microtúbulos (paso 3.2.9) en un portaobjetos de microscopio. Cubra con un cubrebocas de 18 x 18 mm y selle los bordes con esmalte de uñas transparente o una mezcla 1:1:1 de vaselina, lanolina y parafina (sellador de valap), que es sólida a temperatura ambiente y líquida a 95 °C.
- Coloque el objetivo TIRF debajo del cobertor (consulte el paso 4 para conocer los ajustes recomendados del microscopio) y visualice los microtúbulos recién polimerizados en la longitud de onda adecuada para la tubulina marcada fluorescentemente en la mezcla Bright (Tabla 3), para determinar qué dilución de microtúbulos usar en los próximos experimentos.
| Reactivo | Mezcla brillante (μL) | Orden de adición | Mezcla brillante + biotina (μL) | Orden de adición |
| Tubulina fluorescente, 10 mg/ml | 2 | 6 | 2 | 7 |
| Biotina-tubulina, 10 mg/ml | 0 | N/A | 2 | 6 |
| Tubulina sin etiquetar, 10 mg/ml | 20 | 5 | 18 | 5 |
| GMPCPP, 10 mM | 30 | 4 | 30 | 4 |
| TDT, 0,2 M | 0.7 | 3 | 0.7 | 3 |
| 5X BRB80 | 26.4 | 2 | 26.4 | 2 |
| agua estéril | 52.9 | 1 | 52.9 | 1 |
| Volumen total (μL) | 132 | 132 |
Tabla 3: Mezcla de semillas GMPCPP. Componentes de las semillas de microtúbulos GMPCPP, incluido el volumen y el orden de adición. Preparar alícuotas de 5 μL y conservar hasta 1 año a -80 °C.
4. Ajustes del microscopio
- Temperatura: Ajuste la temperatura del microscopio a 28 °C para ver los microtúbulos dinámicos.
- Filtros: Utilice la mejor combinación de cubos de filtro y filtros de emisión, dependiendo de los canales fluorescentes a fotografiar. Para visualizar longitudes de onda de 488 nm, 560 nm y 647 nm en el mismo experimento, utilice un conjunto de banda cuádruple láser de 405/488/560/647 nm, junto con filtros de emisión para las longitudes de onda designadas.
- Alinear láseres: Asegúrese de que los rayos láser utilizados en el experimento estén alineados. Determinar empíricamente la intensidad del láser para el experimento, de modo que todas las proteínas fluorescentes puedan ser fotografiadas con la mayor relación señal-ruido posible, pero no sufran un fotoblanqueo significativo en el transcurso del experimento.
- Objetivo: Utilice papel de lente para limpiar un objetivo 100x con 70% de etanol. Antes de tomar imágenes, agregue una gota de aceite de inmersión de microscopio al objetivo.
- Configurar una secuencia de imágenes
- Para un experimento con microtúbulos biotinilados marcados con fluoróforos de 647 nm, microtúbulos no biotinilados marcados con fluoróforos de 560 nm y tubulina soluble, y proteína de interés marcada con GFP, imagen durante 20 min. Imagine los canales de 560 nm y 488 nm cada 10 s y el canal de 647 nm cada 30 s.
- Para capturar una imagen de referencia de haces antes de la adición de tubulina soluble y MAP, configure una secuencia con una imagen cada una en longitudes de onda de 560 nm y 647 nm.
5. Generar haces de microtúbulos inmovilizados en la superficie
NOTA: Para los siguientes pasos, fluya todas las soluciones en una cámara de flujo mediante pipeteo en un lado abierto, mientras coloca un papel de filtro contra el otro lado. Proteja la cámara de imágenes de la luz para reducir el fotoblanqueo de proteínas marcadas fluorescentemente. Pegue con cinta adhesiva la cámara de imágenes preparada en un soporte deslizante (Figura 1G, H). Siga los pasos de la Tabla 4, que corresponden al protocolo steps 5.2-6.4.
- Prepare la mezcla de tubulina soluble de acuerdo con la Tabla 5 y manténgala en hielo.
NOTA: La tubulina soluble siempre debe colocarse sobre hielo para evitar la polimerización. Prepare una mezcla de tubulina soluble fresca aproximadamente cada 2 h, o cuando los microtúbulos ya no estén polimerizando. - Para inmovilizar los microtúbulos a través de un enlace biotina-neutravidina-biotina, primero fluya en la solución de Neutravidina (NA) hasta que se llene la cámara (~ 7.5 μL) e incube durante 5 min.
- Lavar con 10 μL de MB-frío.
- Flujo en 7,5 μL de la proteína bloqueadora κ-caseína (KC) e incubar durante 2 min.
- Lavar con 10 μL de MB-warm para preparar la cámara para la introducción de microtúbulos.
- Diluir el stock de microtúbulos biotinilados (según las observaciones de la etapa 3.3.2) en 1X BRB80-TDT y añadir 1 μL de esta dilución a 9 μL de MB-caliente. Fluya la mezcla en la cámara e incube durante 10 min. Use una mayor concentración de microtúbulos para más haces.
- Lave los microtúbulos no inmovilizados con 10 μL de MB-warm.
- Fluya 7.5 μL de KC caliente en la cámara e incube durante 2 min.
- Durante la incubación, prepare una solución de 2 nM de la proteína reticulante PRC1 en KC caliente. Fluya 10 μL de esta solución en la cámara de flujo e incube durante 5 min.
NOTA: PrC1 recombinante se expresa y purifica a partir de células bacterianas como se describió anteriormente13. - Para hacer haces, fluya 10 μL de microtúbulos no biotinilados en la cámara e incube durante 10 min. PRC1 reticulará los microtúbulos biotinilados no biotinilados e inmovilizados15,16 (Figura 3).
NOTA: Consulte el paso 6.1 para preparar la mezcla de ensayo durante el tiempo de incubación. - Lave la cámara dos veces con 10 μL de MB-warm. Los microtúbulos unidos son estables durante unos 20 minutos desde este punto.
| Paso | Reactivo | Volumen (μL) | Tiempo de incubación (minutos) |
| 1 | Neutravidina | 7.5 | 5 |
| 2 | MB-frío | 10 | - |
| 3 | κ-caseína | 7.5 | 2 |
| 4 | MB-cálido | 10 | - |
| 5 | Microtúbulos biotinilados (diluidos en MB-warm) | 10 | 10 |
| 6 | MB-cálido | 10 | - |
| 7 | κ-caseína tibia | 7.5 | 2 |
| 8 | 2 nM PRC1 diluido en κ-caseína | 10 | 5 |
| 9 | Microtúbulos no biotinilados | 10 | 10 |
| 10 | MB-cálido x 2 | 10 | - |
| 11 | Mezcla de ensayos | 10 | - |
| Las semillas adheridas son estables durante unos 20 minutos en este punto. | |||
Tabla 4: Pasos del ensayo. Lista de reactivos añadidos a la cámara de imagen, con indicación del tiempo de lavado (-) o incubación.
| Reactivo | Volumen (μL) |
| Tubulina reciclada, 10 mg/ml | 10 |
| MB-Frío | 10.3 |
| MBMC | 13.7 |
| BRB80-TDT | 3.4 |
| GTP, 10 mM | 6.7 |
| ATP, 10 mM (Si usa kinesinas) | 6.7 |
| Tubulina marcada fluorescentemente, 10 mg/ml | 1 (Resuspend tubulina liofilizada etiquetada en FRÍO BRB80-TDT) |
Tabla 5: Componentes solubles de la mezcla de tubulina. Mezclar al inicio del experimento y mantener en hielo.

Figura 3: Esquema de adición de componentes de ensayo para hacer e imagen de haces marcados fluorescentemente y microtúbulos individuales. Las semillas biotiniladas se muestran en azul, semillas no biotiniladas y tubulina soluble en rojo, PRC1 en negro y proteína de interés en cian. Los números de paso en la figura corresponden a los de la Tabla 4. El panel correspondiente al paso 9 muestra un haz preformado (abajo a la izquierda); El paso 11 muestra un haz recién formado (arriba a la izquierda). Creado con BioRender.com. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
6. Dinámica de microtúbulos de imagen
- Durante los 10 min de tiempo de incubación en el paso 5.10, preparar 10 μL de mezcla de ensayo que contenga proteínas de interés, tubulina soluble, nucleótidos, eliminadores de oxígeno14 y antioxidantes según la Tabla 6. Mantenga la mezcla en hielo.
- Cargue la cámara de imagen preparada, pegada al soporte deslizante, en el objetivo TIRF 100x. Utilice los canales de 560 nm y 647 nm para encontrar un campo de visión que contenga un número y una densidad óptimos de microtúbulos y haces individuales.
NOTA: Si tanto los microtúbulos biotinilados como los no biotinilados están marcados con el mismo fluoróforo, los análisis de escaneo en línea de las intensidades de fluorescencia pueden distinguir entre microtúbulos individuales y haces. - Una vez que se identifica un campo de visión, tome una imagen de referencia.
- Fluya cuidadosamente en la mezcla de ensayo sin perturbar la cámara de imágenes.
- Selle los extremos abiertos de la cámara con sellador valap.
- Inicie la secuencia de imágenes como se describe en el paso 4.5.1.
| Reactivo | Volumen (μL) |
| Mezcla de tubulina soluble | 4 |
| OSF | 1 |
| Trolox (si se usan microtúbulos marcados con un fluoróforo fácilmente fotoblanqueante) | 1 |
| ATP, 10 mM (Si usa kinesinas) | 1 |
| PRC1 (o reticulador de elección) | 1 |
| Proteínas de interés | X |
| MB-frío | 2-X |
Tabla 6: Componentes de la mezcla de ensayos. Mezcle, fluya hacia la cámara de imágenes y visualice la dinámica de los microtúbulos, en 30 minutos.
Resultados
El experimento descrito anteriormente se realizó utilizando microtúbulos biotinilados marcados con fluoróforos de 647 nm, microtúbulos no biotinilados marcados con fluoróforos de 560 nm y mezcla de tubulina soluble marcada con fluoróforos de 560 nm. Los microtúbulos fueron reticulados por la proteína reticulante PRC1 (marcada con GFP). Después de generar haces inmovilizados en la superficie y microtúbulos individuales (paso 5.11), la cámara de imágenes se montó en un objetivo de aceite TIRF 100X 1.49 NA y se...
Discusión
El experimento descrito aquí amplía significativamente el alcance y la complejidad de los ensayos convencionales de reconstitución de microtúbulos, que tradicionalmente se realizan en microtúbulos individuales o en un tipo de matriz. El ensayo actual proporciona un método para cuantificar y comparar simultáneamente la actividad reguladora de MAP en dos poblaciones, a saber, microtúbulos individuales y haces reticulados. Además, este ensayo permite el examen de dos tipos de haces: los que están preformados a par...
Divulgaciones
Los autores no declaran intereses contrapuestos.
Agradecimientos
Este trabajo fue apoyado por una subvención de los NIH (no. 1DP2GM126894-01), y por fondos de Pew Charitable Trusts y Smith Family Foundation a R.S. Los autores agradecen al Dr. Shuo Jiang por su contribución al desarrollo y optimización de los protocolos.
Materiales
| Name | Company | Catalog Number | Comments |
| (±)-6-Hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (Trolox) | Sigma Aldrich | 238813 | |
| 1,4-piperazinediethanesulfonic acid (PIPES) | Sigma Aldrich | P6757 | |
| 18x18 mm #1.5 coverslips | Electron Microscopy Sciences | 63787 | |
| 2-Mercaptoethanol (BME) | Sigma Aldrich | M-6250 | |
| 24x60 mm #1.5 coverslips | Electron Microscopy Sciences | 63793 | |
| 405/488/560/647 nm Laser Quad Band | Chroma | TRF89901-NK | |
| Acetone | Sigma Aldrich | 320110 | |
| Adenosine 5'-triphosphate disodium salt hydrate (ATP) | Sigma Aldrich | A7699-5G | |
| Avidin, NeutrAvidin® Biotin-binding Protein (Molecular Probes®) | Thermo Fischer Scientific | A2666 | |
| Bath sonicator: Branson 2800 Cleaner | Branson | CPX2800H | |
| Beckman Coulter Polycarbonate Thickwall Tubes, 11 x 34 mm | Beckman-Coulter | 343778 | |
| Beckman Coulter Polycarbonate Thickwall Tubes, 8 x 34 mm | Beckman-Coulter | 343776 | |
| Biotin-PEG-SVA, MW 5,000 | Laysan Bio | #Biotin-PEG-SVA-5000 | |
| Bovine Serum Albumin (BSA) | Sigma Aldrich | 2905 | |
| Catalase | Sigma Aldrich | C40 | |
| Corning LSE Mini Microcentrifuge, AC100-240V | Corning | 6670 | |
| Delicate Task Wipes | Kimtech | 34120 | |
| Dithiothreitol (DTT) | GoldBio | DTT10 | |
| Emission filter | Chroma | ET610/75m | |
| Ethanol (200-proof) | Decon Labs | 2705 | |
| Ethylene glycol tetraacetic acid (EGTA) | Sigma Aldrich | 3777 | |
| Glucose Oxidase | Sigma Aldrich | G2133 | |
| GMPCPP | Jena Bioscience | NU-405 | |
| Guanosine 5'-triphosphate sodium salt hydrate (GTP) | Sigma Aldrich | G8877 | |
| Hellmanex III detergent | Sigma Aldrich | Z805939 | |
| Immersion oil, Type A | Fisher Scientific | 77010 | |
| Kappa-casein | Sigma Aldrich | C0406 | |
| Lanolin | Fisher Scientific | S25376 | |
| Lens Cleaning Tissue | ThorLabs | MC-5 | |
| Magnesium Chloride (MgCl2) | Sigma Aldrich | M9272 | |
| Methylcellulose | Sigma Aldrich | M0512 | |
| Microfuge 16 Benchtop Centrifuge | Beckman-Coulter | A46474 | |
| Microscope Slides, Diamond White Glass, 25 x 75mm, 90° Ground Edges, WHITE Frosted | Globe Scientific | 1380-50W | |
| mPEG-Succinimidyl Valerate, MW 5,000 | Laysan Bio | #NH2-PEG-VA-5K | |
| Optima™ Max-XP Tabletop Ultracentrifuge | Beckman-Coulter | 393315 | |
| Paraffin | Fisher Scientific | P31-500 | |
| PELCO Reverse (self-closing), Fine Tweezers | Ted Pella | 5377-NM | |
| Petrolatum, White | Fisher Scientific | 18-605-050 | |
| Plasma Cleaner, 115V | Harrick Plasma | PDC-001 | |
| Potassium Hydroxide (KOH) | Sigma Aldrich | 221473 | |
| Sodium bicarbonate | Sigma Aldrich | S6014 | |
| Sucrose | Sigma Aldrich | S7903 | |
| Thermal-Lok 1-Position Dry Heat Bath | USA Scientific | 2510-1101 | |
| Thermal-Lok Block for 1.5 and 2.0 mL Tubes | USA Scientific | 2520-0000 | |
| Thermo Scientific™ Pierce™ Bond-Breaker™ TCEP Solution, Neutral pH; 500mM | Thermo Fischer Scientific | PI-77720 | |
| TIRF 100X NA 1.49 Oil Objective | Nikon | CFI Apochromat TIRF 100XC Oil | |
| TIRF microscope | Nikon | Eclipse Ti | |
| TLA 120.1 rotor | Beckman-Coulter | 362224 | |
| TLA 120.2 rotor | Beckman-Coulter | 357656 | |
| Tubulin protein (>99% pure): porcine brain | Cytoskeleton | T240 | |
| Tubulin Protein (Biotin): Porcine Brain | Cytoskeleton | T333P | |
| Tubulin protein (fluorescent HiLyte 647): porcine brain | Cytoskeleton | TL670M | |
| Tubulin protein (X-rhodamine): bovine brain | Cytoskeleton | TL620M | |
| VECTABOND® Reagent, Tissue Section Adhesion | Vector Biolabs | SP-1800-7 | |
| VWR® Personal-Sized Incubator, 120V, 50/60Hz, 0.6A | VWR | 97025-630 |
Referencias
- Subramanian, R., Kapoor, T. M. Building complexity: insights into self-organized assembly of microtubule-based architectures. Developmental Cell. 23 (5), 874-885 (2012).
- Baas, P. W., Rao, A. N., Matamoros, A. J., Leo, L. Stability properties of neuronal microtubules. Cytoskeleton (Hoboken). 73 (9), 442-460 (2016).
- Bitan, A., Rosenbaum, I., Abdu, U. Stable and dynamic microtubules coordinately determine and maintain Drosophila bristle shape. Development. 139 (11), 1987-1996 (2012).
- Foe, V. E., von Dassow, G. Stable and dynamic microtubules coordinately shape the myosin activation zone during cytokinetic furrow formation. The Journal of Cell Biology. 183 (3), 457-470 (2008).
- Pous, C., et al. Functional specialization of stable and dynamic microtubules in protein traffic in WIF-B cells. The Journal of Cell Biology. 142 (1), 153-165 (1998).
- Uehara, R., Goshima, G. Functional central spindle assembly requires de novo microtubule generation in the interchromosomal region during anaphase. The Journal of Cell Biology. 191 (2), 259-267 (2010).
- Mitchison, T., Kirschner, M. Dynamic instability of microtubule growth. Nature. 312 (5991), 237-242 (1984).
- Bieling, P., et al. Reconstitution of a microtubule plus-end tracking system in vitro. Nature. 450 (7172), 1100-1105 (2007).
- Bieling, P., Telley, I. A., Surrey, T. A minimal midzone protein module controls formation and length of antiparallel microtubule overlaps. Cell. 142 (3), 420-432 (2010).
- Shimamoto, Y., Forth, S., Kapoor, T. M. Measuring pushing and braking forces generated by ensembles of Kinesin-5 crosslinking two microtubules. Developmental Cell. 34 (6), 669-681 (2015).
- Mani, N., Jiang, S., Neary, A. E., Wijeratne, S. S., Subramanian, R. Differential regulation of single microtubules and bundles by a three-protein module. Nature Chemical Biology. 17 (9), 964-974 (2021).
- Hyman, A. A., Salser, S., Drechsel, D., Unwin, N., Mitchison, T. J. Role of GTP hydrolysis in microtubule dynamics: information from a slowly hydrolyzable analogue, GMPCPP. Molecular Biology of the Cell. 3 (10), 1155-1167 (1992).
- Subramanian, R., et al. Insights into antiparallel microtubule crosslinking by PRC1, a conserved nonmotor microtubule binding protein. Cell. 142 (3), 433-443 (2010).
- Rasnik, I., McKinney, S. A., Ha, T. Nonblinking and long-lasting single-molecule fluorescence imaging. Nature Methods. 3 (11), 891-893 (2006).
- Wijeratne, S., Subramanian, R. Geometry of antiparallel microtubule bundles regulates relative sliding and stalling by PRC1 and Kif4A. eLife. 7, 32595 (2018).
- Mani, N., Wijeratne, S. S., Subramanian, R. Micron-scale geometrical features of microtubules as regulators of microtubule organization. eLife. 10, 63880 (2021).
- Freal, A., et al. Feedback-driven assembly of the axon initial segment. Neuron. 104 (2), 305-321 (2019).
- Ledbetter, M., Porter, K. A "microtubule" in plant cell fine structure. The Journal of Cell Biology. 19 (1), 239-250 (1963).
- Wijeratne, S. S., Marchan, M. F., Tresback, J. S., Subramanian, R. Atomic force microscopy reveals distinct protofilament-scale structural dynamics in depolymerizing microtubule arrays. Proceedings of the National Academy of Sciences of the United States of America. , 119 (2022).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados