
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Visualizzazione simultanea della dinamica di microtubuli reticolati e singoli in vitro mediante microscopia TIRF
* Questi autori hanno contribuito in egual misura
In questo articolo
Riepilogo
Qui, viene presentato un test di ricostituzione in vitro basato sulla microscopia TIRF per quantificare e confrontare contemporaneamente la dinamica di due popolazioni di microtubuli. Viene descritto un metodo per visualizzare simultaneamente l'attività collettiva di più proteine associate a microtubuli su fasci di microtubuli reticolati e singoli microtubuli.
Abstract
I microtubuli sono polimeri di eterodimeri αβ-tubulina che si organizzano in strutture distinte nelle cellule. Le architetture e le reti basate su microtubuli spesso contengono sottoinsiemi di array di microtubuli che differiscono nelle loro proprietà dinamiche. Ad esempio, nelle cellule in divisione, fasci stabili di microtubuli reticolati coesistono in prossimità di microtubuli dinamici non reticolati. Gli studi di ricostituzione in vitro basati sulla microscopia TIRF consentono la visualizzazione simultanea della dinamica di questi diversi array di microtubuli. In questo test, una camera di imaging è assemblata con microtubuli immobilizzati in superficie, che sono presenti come singoli filamenti o organizzati in fasci reticolati. L'introduzione di tubulina, nucleotidi e regolatori proteici consente la visualizzazione diretta delle proteine associate e delle proprietà dinamiche di microtubuli singoli e reticolati. Inoltre, i cambiamenti che si verificano quando i singoli microtubuli dinamici si organizzano in fasci possono essere monitorati in tempo reale. Il metodo qui descritto consente una valutazione sistematica dell'attività e della localizzazione delle singole proteine, nonché degli effetti sinergici dei regolatori proteici su due diversi sottoinsiemi di microtubuli in condizioni sperimentali identiche, fornendo così intuizioni meccanicistiche inaccessibili con altri metodi.
Introduzione
I microtubuli sono biopolimeri che formano scaffold strutturali essenziali per molteplici processi cellulari, che vanno dal trasporto intracellulare e dal posizionamento degli organelli alla divisione cellulare e all'allungamento. Per eseguire queste diverse funzioni, i singoli microtubuli sono organizzati in array di dimensioni micron, come fusi mitotici, assonemi ciliari, fasci neuronali, array interfase e array corticali vegetali. Un motivo architettonico onnipresente trovato in queste strutture è un fascio di microtubuli reticolati lungo le loro lunghezze1. Una caratteristica intrigante di diverse strutture a base di microtubuli è la coesistenza di microtubuli raggruppati e singoli microtubuli non reticolati in stretta vicinanza spaziale. Queste sottopopolazioni di microtubuli possono mostrare dinamiche di polimerizzazione nettamente diverse l'una dall'altra, come necessario per il loro corretto funzionamento2,3,4,5. Ad esempio, all'interno del fuso mitotico, fasci reticolati stabili e singoli microtubuli dinamici sono presenti all'interno di una regione su scala micron al centro cellulare6. Studiare come vengono specificate le proprietà dinamiche delle popolazioni di microtubuli coesistenti è, quindi, fondamentale per comprendere l'assemblaggio e la funzione delle strutture basate sui microtubuli.
I microtubuli sono polimeri dinamici che si alternano tra le fasi di polimerizzazione e depolimerizzazione, passando da una fase all'altra in eventi noti come catastrofe e salvataggio7. La dinamica dei microtubuli cellulari è regolata da una miriade di Microtubule Associated Proteins (MAP) che modulano i tassi di polimerizzazione e depolimerizzazione dei microtubuli e le frequenze degli eventi catastrofici e di soccorso. È difficile studiare l'attività delle MAP su array spazialmente prossimali nelle cellule, a causa dei limiti della risoluzione spaziale nella microscopia ottica, specialmente nelle regioni ad alta densità di microtubuli. Inoltre, la presenza di più MAP nella stessa regione cellulare ostacola le interpretazioni degli studi biologici cellulari. I saggi di ricostituzione in vitro, eseguiti in combinazione con la microscopia TIRF (Total Internal Reflection Fluorescence), aggirano le sfide dell'esame dei meccanismi con cui specifici sottoinsiemi di MAP regolano la dinamica degli array di microtubuli cellulari prossimali. Qui, la dinamica dei microtubuli assemblati in vitro viene esaminata in presenza di una o più MAP ricombinanti in condizioni controllate8,9,10. Tuttavia, i saggi di ricostituzione convenzionali vengono tipicamente eseguiti su singoli microtubuli o su un tipo di array, precludendo la visualizzazione di popolazioni coesistenti.
Qui presentiamo saggi di ricostituzione in vitro che consentono la visualizzazione simultanea di due popolazioni di microtubuli nelle stesse condizioni di soluzione11. Descriviamo un metodo per visualizzare simultaneamente l'attività collettiva di più MAP su singoli microtubuli e su fasci di microtubuli reticolati dalla proteina PRC1 associata al fuso mitotico. La proteina PRC1 si lega preferenzialmente alla sovrapposizione tra microtubuli antiparalleli, reticolandoli9. In breve, questo protocollo consiste nelle seguenti fasi: (i) preparazione di soluzioni stock e reagenti, (ii) pulizia e trattamento superficiale dei coverslip utilizzati per creare la camera di imaging per esperimenti di microscopia, (iii) preparazione di "semi" di microtubuli stabili da cui viene avviata la polimerizzazione durante l'esperimento, (iv) specifica delle impostazioni del microscopio TIRF per visualizzare la dinamica dei microtubuli, (v) immobilizzazione dei semi di microtubuli e generazione di fasci di microtubuli reticolati nella camera di imaging e (vi) visualizzazione della dinamica dei microtubuli nella camera di imaging attraverso la microscopia TIRF, previa aggiunta di tubulina solubile, MAP e nucleotidi. Questi saggi consentono la valutazione qualitativa e l'esame quantitativo della localizzazione MAP e del loro effetto sulla dinamica di due popolazioni di microtubuli. Inoltre, facilitano la valutazione degli effetti sinergici di più MAP su queste popolazioni di microtubuli, in una vasta gamma di condizioni sperimentali.
Protocollo
1. Preparare i reagenti
- Preparare tamponi e reagenti come indicato nelle Tabelle 1 e Tabella 2. Durante l'esperimento, mantenere tutte le soluzioni sul ghiaccio, se non diversamente specificato.
| Soluzione | Componenti | Durata di archiviazione consigliata | Note | ||
| 5X BRB80 | 400 mM K-PIPES, 5 mM MgCl2, 5 mM EGTA, pH 6,8 con KOH, filtro sterilizzato | fino a 2 anni | Conservare a 4 °C | ||
| 1X BRB80 | 80 mM K-PIPES, 1 mM MgCl2, 1 mM EGTA, pH 6,8 | fino a 2 anni | Conservare a 4 °C | ||
| BRB80-DTT | 1X BRB80, 1 mM DTT | fino a 2 giorni | |||
| Tampone di analisi | 80 mM K-PIPES, 3 mM MgCl2, 1 mM EGTA, pH 6,8, 5% saccarosio (OR 1X BRB80, 5% saccarosio, 2 mM MgCl2) | fino a 1 anno | Conservare a 4 °C | ||
| Buffer master (MB) | Tampone di analisi, 5mM TCEP | 1 settimana | Prepararsi il giorno dell'esperimento; Separare in due tubi: MB-caldo a temperatura ambiente e MB-freddo su ghiaccio; includere 1 mM DTT se si utilizzano coloranti fluorescenti | ||
| Master Buffer con metilcellulosa (MBMC) | 1X BRB80, 0,8% metilcellulosa, 5 mM TCEP, 5 mM MgCl2 | 1 settimana | Prepararsi il giorno dell'esperimento; includere 1 mM DTT se si utilizzano coloranti fluorescenti | ||
| Tampone di diluizione proteica (DB) | MB, 1 mg/mL di albumina sierica bovina (BSA), 1 μM di ATP | 1 giorno, su ghiaccio | Prepararsi il giorno dell'esperimento; includere 1 mM DTT se si utilizzano coloranti fluorescenti | ||
| Miscela di scavenging di ossigeno (OSM) | MB, 389 μg/mL catalasi, 4,44 mg/mL glucosio ossidasi, 15,9 mM 2-mercaptoetanolo (BME) | 1 giorno, su ghiaccio | Preparati il giorno dell'esperimento | ||
| Oxygen Scavenging Final (OSF) | MB, 350 μg/mL catalasi, 4mg/mL glucosio ossidasi, 14,3 mM BME, 15 mg/mL glucosio | utilizzare entro 30 min | Preparare immediatamente prima dell'uso aggiungendo 1 μL di glucosio a 9 μL di OSM | ||
Tabella 1: Elenco dei buffer utilizzati in questo protocollo e dei relativi componenti. Vedere la colonna "Durata di archiviazione consigliata" per indicazioni su quanto in anticipo è possibile preparare ciascun buffer.
| Reagente | Concentrazione di stoccaggio | Solvente di stoccaggio | Temperatura di stoccaggio | Concentrazione di lavoro | Concentrazione finale | Durata di archiviazione consigliata | Note | |||||||||
| Neutravidina (NA) | 5 mg/ml | 1X BRB80 | -80°C | 0,2 mg/ml | 0,2 mg/ml | fino a 1 anno | Utilizzato per immobilizzare i microtubuli attraverso un legame biotina-neutravidina-biotina; conservare in piccole aliquote | |||||||||
| Kappa-caseina (KC) | 5 mg/ml | 1X BRB80 | -80°C | 0,5 mg/mL | 0,5 mg/mL | fino a 2 anni | Utilizzato per bloccare la superficie della camera di imaging; Conservare in piccole aliquote; Il giorno dell'esperimento, mettere da parte un piccolo volume a temperatura ambiente | |||||||||
| Albumina sierica bovina (BSA) | 50 mg/ml | 1X BRB80 | -20°C | 1 mg/mL (in DB) | N/D | fino a 2 anni | conservare in piccole aliquote | |||||||||
| Catalasi | 3,5 mg/ml | 1X BRB80 | -80°C | 350 μg/mL (in OSF) | 35 μg/ml | fino a 2 anni | componente della miscela di scavenging di ossigeno; conservare in piccole aliquote | |||||||||
| Glucosio ossidasi | 40 mg/ml | 1X BRB80 | -80°C | 4 mg/mL (in OSF) | 0,4 mg/ml | fino a 2 anni | componente della miscela di scavenging di ossigeno; conservare in piccole aliquote | |||||||||
| Tubulina | Liofilizzato | N/D | 4°C | 10 mg/ml | 2,12 mg/mL (in miscela di tubuline) | fino a 1 anno | Una volta che la tubulina è in soluzione, tenerla fredda per evitare la polimerizzazione. | |||||||||
| Adenosina trifosfato (ATP) | 100 metri | acqua ultrapura | -20°C | 10 mM | 1 mM | 6 mesi | Preparare la soluzione in acqua sterilizzata con filtro, regolare il pH a ~ 7,0 e congelare in piccole aliquote. | |||||||||
| Guanosina Trifosfato (GTP) | 100 metri | acqua ultrapura | -20°C | 10 mM | 1,29 mM (in miscela di tubuline) | 6 mesi | Preparare la soluzione in acqua sterilizzata con filtro, regolare il pH a ~ 7,0 e congelare in piccole aliquote. | |||||||||
| Guanosina-5'-[(α,β)-metileno] trifosfato (GMPCPP) | 10 mM | acqua ultrapura | -20°C | 10 μM | 0,5 μM | 6 mesi | ||||||||||
| Ditiotreitolo (DTT) | 1 M | acqua sterile | -20°C | 1 mM | N/D | fino a 2 anni | ||||||||||
| Tris(2-carbossietil) fosfina (TCEP) | 0,5 M | acqua sterilizzata con filtro | Temperatura ambiente | 5 mM | N/D | fino a 2 anni | ||||||||||
| Metilcellulosa | 1% | acqua sterile | Temperatura ambiente | 0,8% (in MBMC) | 0,21% (in miscela di tubuline) | fino a 1 anno | Sciogliere la metilcellulosa aggiungendola lentamente all'acqua quasi bollente. Lasciare raffreddare mescolando continuamente. | |||||||||
| Beta-mercaptoetanolo (BME) | 143 metri quadrati | acqua sterile | Temperatura ambiente | 14,3 mM (in OSF) | 1,43 mM | fino a 5 anni | 143 mM è una diluizione 1:100 del BME di stock | |||||||||
| Glucosio | 150 mg/ml | 1X BRB80 | -80°C | 15 mg/mL (in OSF) | 1,5 mg/ml | fino a 2 anni | Aggiungere a OSM immediatamente prima dell'uso | |||||||||
| Acido (±)-6-idrossi-2,5,7,8-tetrametilcromano-2-carbossilico (Trolox) | 10mM | 1X BRB80 | -80°C | 10 mM | 1 mM | fino a 1 anno | Non si dissolve completamente. Aggiungere un po 'di NaOH, mescolare per ~ 4 ore e filtrare sterilizzare prima dell'uso | |||||||||
| mPEG-Succinimidyl Valerate, MW 5.000 | polvere | N/D | -20°C | 333 mg/ml (in bicarbonato di sodio 0,1 M) | 324 mg/ml (in bicarbonato di sodio 0,1 M) | 6 mesi | Preparare aliquote ~ 34 mg, contrassegnando ogni tubo con un peso esatto di polvere. Passare il gas azoto sul solido, sigillare i tubi con parafilm e conservare a -20 ° C in un contenitore con essiccante. | |||||||||
| Biotina-PEG-SVA, MW 5.000 | polvere | N/D | -20°C | 111 mg/ml (in bicarbonato di sodio 0,1 M) | 3,24 mg/mL (in 0,1 M di bicarbonato di sodio) | 6 mesi | Preparare aliquote ~ 3 mg, contrassegnando ogni tubo con un peso esatto di polvere. Passare il gas azoto sul solido, sigillare i tubi con parafilm e conservare a -20 ° C in un contenitore con essiccante. | |||||||||
Tabella 2: Elenco dei reagenti utilizzati in questo protocollo. Sono incluse le condizioni e le concentrazioni di conservazione raccomandate, le concentrazioni di lavoro delle soluzioni stock utilizzate durante l'esperimento e la concentrazione finale nella camera di imaging. Ulteriori note sono riportate nella colonna all'estrema destra.
2. Preparare i vetrini Biotin-PEG
NOTA: Preparare le camere di imaging il più vicino possibile all'inizio di un esperimento e con non più di 2 settimane di anticipo.
- Coperture pulite
- Posizionare un numero uguale di coverslip #1.5 da 24 x 60 mm e 18 x 18 mm #18 mm (Figura 1F) rispettivamente nei barattoli di colorazione dei vetrini e nei rack per il lavaggio delle diapositive (Figura 1A,B). Posizionare le scaffalature per il lavaggio delle diapositive contenenti 18 coverslip da 18 x 18 mm in un becher da 100 ml.
- Risciacquare tutte le coperture 5-6 volte in acqua ultrapura (resistività 18,2 MΩ-cm) e rimuovere il liquido in eccesso dopo ogni risciacquo con una punta della pipetta attaccata a un tubo a vuoto (Figura 1C).
- Riempi i bicchieri e i barattoli contenenti i coperchi con acqua ultrapura, sigilla con parafilm e sonicare per 10 minuti.
- Riempire due bicchieri da 150 ml con etanolo a prova di 200. Usando una pinzetta, immergere ogni coverslip in un becher pieno di etanolo e poi l'altro.
- Utilizzando una pinzetta, trasferire i coperchi al rack di asciugatura a scorrimento (Figura 1D), asciugarli sotto il flusso di gas di azoto e incubare a 37 °C fino a completa asciugatura (~15 min).
- Posizionare le coperture essiccate in un unico strato all'interno del detergente al plasma. Formare il sigillo sottovuoto, quindi impostare il livello di radiofrequenza (RF) del pulitore al plasma su ~ 8 MHz.
- Una volta generato il plasma, lasciare le coperture nel detergente al plasma per 5 minuti. Spegnere il pulitore al plasma e rilasciare lentamente il vuoto.
- Una volta rilasciato il sigillo sottovuoto, capovolgere i coverslip e ripetere la pulizia al plasma per 5 minuti per l'altro lato dei coverslips.
- Alternativa alla pulizia al plasma: Al posto dei passaggi 2.1.2-2.1.3, sonicare le coperture in una soluzione calda di detergente al 2% (in acqua ultrapura) per 10 minuti. Quindi, lavare accuratamente i coverslip con acqua ultrapura e sonicare in acqua ultrapura 2-3 volte (10 minuti ciascuno). Quindi, lavare in etanolo e asciugare come nei passaggi 2.1.4-2.1.5. Saltare i passaggi 2.1.6-2.1.8.
- Trattamento biotina-PEG
- Immediatamente prima dell'uso, sciogliere 400 μL di 3-aminopropiltrietossisilano in 40 ml di acetone. Usando una pinzetta, sposta i coperchi puliti al plasma nel rack di lavaggio delle diapositive e nei barattoli di colorazione delle diapositive. Immergere le coperture in soluzione di 3-aminopropiltrietossisilano e incubare per 5 min12,13.
- Lavare tutte le coperture 5-6 volte con acqua ultrapura.
- Trasferire i coperchi allo stendino, asciugarli sotto un flusso di gas di azoto e incubare a 37 °C fino a completa asciugatura (~20 min).
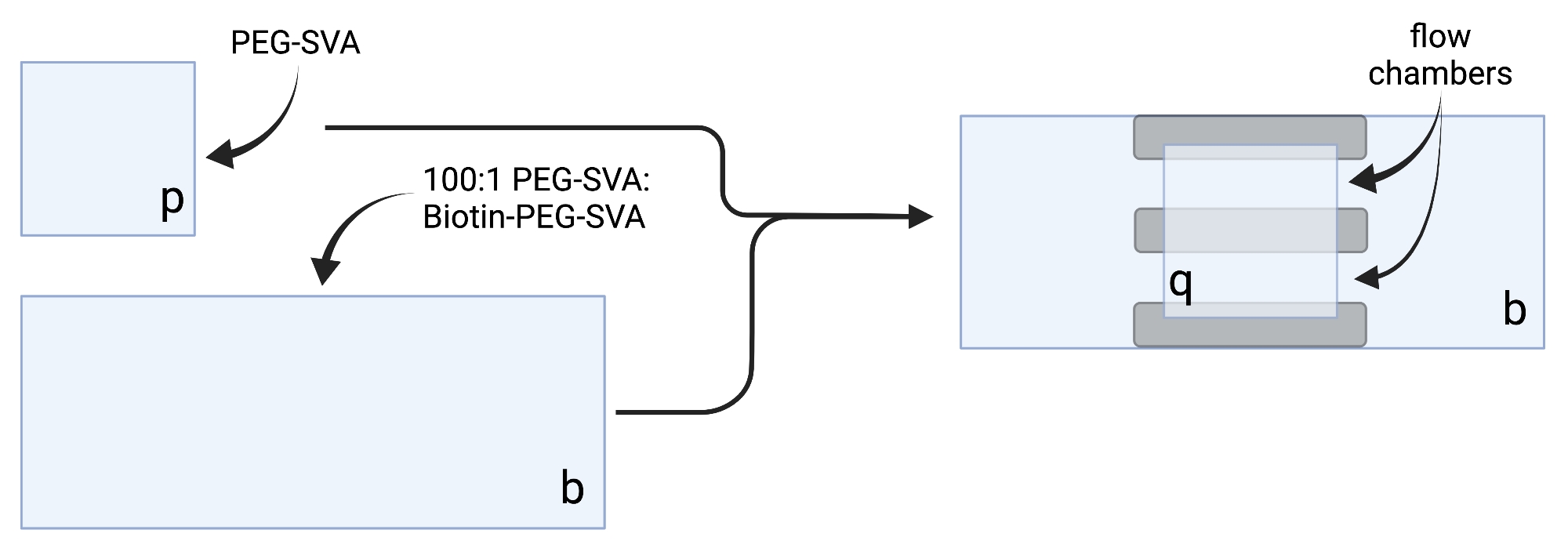
- Posare le coperture essiccate su salviette delicate ed etichettare ogni coverslip su un angolo, ad esempio "p" su ogni coverslip di 18 x 18 mm e "b" su ogni coverslip di 24 x 60 mm (vedi Figura 2).
- Il giorno dell'esperimento, preparare una soluzione fresca di bicarbonato di sodio 0,1 M sciogliendo 0,84 mg di NaHCO3 in 10 ml di acqua ultrapura.
- Portare aliquote di mPEG-Succinimidyl Valerate (PEG-SVA) e Biotin-PEG-SVA a temperatura ambiente, immediatamente prima dell'uso. Cfr. note sulla preparazione aliquota del polietilenglicole (PEG) nella tabella 2.
NOTA: Lavorare rapidamente perché l'emivita di idrolisi della frazione di Succinimidyl Valerate (SVA) è di ~ 30 min. - Aggiungere 102 μL di 0,1 M NaHCO3 a 34 mg di PEG-SVA, ruotare in una microcentrifuga da banco a 2.656 x g per 20 s, quindi mescolare pipettando su e giù. Sciogliere 3 mg di Biotina-PEG-SVA in 27 μL di 0,1 M NaHCO3 mediante pipettaggio su e giù. Regolare i volumi di diluizione in base al peso esatto del PEG indicato sui tubi (vedere Tabella 2).
- Preparare una miscela PEG:biotina-PEG 100:1 p/p combinando 75 μL di soluzione PEG-SVA e 2,25 μL di soluzione di Biotina-PEG-SVA per 20 coverslip, 100 μL e 3 μL per 30 coverslip, o 125 μL e 3,75 μL per 40 coverslips.
- Costruire una camera di idratazione posizionando asciugamani di carta bagnati sotto il rack della punta sul fondo di una scatola di punta vuota da 10 μL (Figura 1E). Ciò impedirà l'evaporazione delle soluzioni PEG.
- Pipetta 6 μL di miscela PEG-SVA:biotina-PEG 100:1 al centro di un coperchio di 24 x 60 mm sul lato etichettato. Posiziona un altro coverslip di 24 x 60 mm sopra il primo coverslip in modo tale che la coppia formi una forma a X, con i lati etichettati "b" uno di fronte all'altro. Posizionare la coppia su un rack di punta vuoto nella camera di idratazione e ripetere per i restanti 24 x 60 mm coverslips.
- Pipetta 6 μL di PEG-SVA sul centro di un coperchio di 18 x 18 mm sul lato etichettato. Posiziona un altro coverslip di 18 x 18 mm sopra il primo coverslip, con i lati etichettati "p" uno di fronte all'altro. Posizionare la coppia su un rack di punta vuoto nella camera di idratazione e ripetere per i restanti 18 x 18 mm coverslips.
- Chiudere la camera di idratazione e incubare per 3 ore o durante la notte.
- Separare le coppie di coverslips e risciacquare in acqua ultrapura.
- Asciugare i coperchi con un flusso di azoto e metterli in un incubatore a 37 °C per asciugare completamente.
- Per costruire la camera di imaging, incollare tre strisce di nastro biadesivo su un coperchio di 24 x 60 mm sul lato etichettato "b". All'altro lato delle strisce di nastro, attaccare un coperchio di 18 x 18 mm con il lato etichettato "p" rivolto verso il coperchio più grande. Questo forma due camere di flusso per esperimenti di microscopia, con superfici trattate l'una di fronte all'altra (Figura 2 e Figura 1G).

Figura 1: Apparecchiature per il trattamento delle coperture e la preparazione della camera di imaging. (A) barattoli di colorazione dei vetrini per i coperchi da 24 x 60 mm, (B) scaffalature per il lavaggio delle diapositive per 18 x 18 mm, (C) set-set-set per vuoto, (D) stendino-stendino, (E) camera di idratazione, (F) coverslips, (G) camera di imaging, (H) supporto per diapositive. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2: Schema per la preparazione di camere di imaging utilizzando nastro biadesivo (grigio) e coverslip trattati con PEG/Biotina-PEG. Creato con BioRender.com. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
3. Polimerizzare i microtubuli
- Preparare i semi GMPCPP
NOTA: Preparare i semi GMPCPP in una cella frigorifera, mantenendo tutti i reagenti, le punte e i tubi a 4 °C. I semi GMPCPP possono essere preparati in anticipo e conservati a -80 °C per un massimo di 1 anno. Posizionare il rotore ultracentrifuga ad angolo fisso, contenente tubi centrifuga da 1 mL, in ultracentrifuga e impostare la temperatura a 4 °C.- Risospese tubulina liofilizzata (Tabella 2) a ~ 10 mg / mL in 1X BRB80, immediatamente prima dell'uso.
- Mescolare i componenti dei semi GMPCPP come descritto nella Tabella 3.
NOTA: Mantenere tutti i componenti della tubulina sul ghiaccio il più possibile per ridurre al minimo la polimerizzazione della tubulina solubile. - Chiarire la miscela in un rotore ultracentrifuga ad angolo fisso a 352.700 x g per 5 minuti a 4 °C.
- Separare il surnatante in aliquote da 5 μL, congelarle a scatto in azoto liquido e conservarle a -80 °C.
- Polimerizzare i semi il giorno dell'esperimento
- Caldo 1-2 mL di BRB80-DTT (Tabella 1) a 37 °C.
- Introdurre sul ghiaccio un'aliquota di 5 μL di semi GMPCPP (-80 °C) dal punto 3.1.4 e sciogliere immediatamente in 20 μL di BRB80-DTT calda. Girare a 2.000 x g per 5 s a temperatura ambiente e toccare per mescolare.
NOTA: Il volume di diluizione iniziale può variare tra 13 μL e 21 μL ed è determinato empiricamente per ogni lotto di semi. Se i semi non riescono a polimerizzare, risolvere i problemi integrando il tampone di diluizione iniziale (fase 3.2.2) con 0,5 μM GMPCPP. - Proteggere dalla luce e incubare a 37 °C per 30-45 min.
NOTA: La lunghezza dei microtubuli dipende dalla durata dell'incubazione. Per i microtubuli corti, il tempo di incubazione può essere breve come 15 min. Per i microtubuli lunghi, il tempo di incubazione può durare fino a 2 ore. I microtubuli biotinilati tendono a richiedere tempi di incubazione più lunghi rispetto ai microtubuli non biotinilati. - Posizionare il rotore ultracentrifuga ad angolo fisso, contenente tubi centrifuga da 500 μL, in ultracentrifuga e preriscaldato a 30 °C.
- Dopo l'incubazione, aggiungere 50 μL di BRB80-DTT caldo (fase 3.2.1) ai semi GMPCPP polimerizzati e trasferire la miscela in un tubo centrifugo da 500 μL. Lavare il tubo vuoto che conteneva i semi GMPCPP con altri 50 μL di BRB80-DTT caldo, pipetta su e giù e aggiungere questo tampone al tubo centrifugo da 500 μL contenente la miscela.
- Prima di girare, segnare il bordo del tubo della centrifuga per indicare dove sarà il pellet (il pellet sarà troppo piccolo per essere visto). Ruotare per 10 minuti a 244.900 x g a 30 °C12.
- Pipettare con cura il surnatante e scartare. Risospesare il pellet in 100 μL di BRB80-DTT caldo. Tocca per mescolare.
- Ruotare per 10 minuti a 244.900 x g a 30 °C, allineando la marcatura con il rotore al pellet nello stesso punto.
- Rimuovere il surnatante e risospesciare il pellet in 16 μL di BRB80-DTT caldo. Trasferire la soluzione di microtubulo in un tubo microcentrifuga pulito da 0,6 mL. Proteggere dalla luce e mantenere a temperatura ambiente o superiore.
NOTA: Dopo la polimerizzazione, mantenere i microtubuli a temperatura ambiente pari o superiore. Se si raffreddano, si depolimerizzeranno. Incubare a 28 °C per una maggiore stabilità.
- Controllare i microtubuli tramite microscopia TIRF
- Pipettare una miscela di 4,5 μL di BRB80-DTT e 1 μL di soluzione di microtubuli (fase 3.2.9) su un vetrino per microscopio. Coprire con un coperchio di 18 x 18 mm e sigillare i bordi con smalto trasparente o una miscela 1:1:1 di petrolato, lanolina e paraffina (sigillante valap), che è solido a temperatura ambiente e liquido a 95 °C.
- Posizionare l'obiettivo TIRF sotto il coverslip (vedere il passaggio 4 per le impostazioni consigliate del microscopio) e visualizzare i microtubuli appena polimerizzati alla lunghezza d'onda appropriata per la tubulina marcata fluorescentemente nella miscela Bright (Tabella 3), per determinare quale diluizione di microtubuli utilizzare nei prossimi esperimenti.
| Reagente | Miscela brillante (μL) | Ordine di aggiunta | Miscela brillante + biotina (μL) | Ordine di aggiunta |
| Tubulina fluorescente, 10 mg/mL | 2 | 6 | 2 | 7 |
| Biotina-tubulina, 10 mg/mL | 0 | N/D | 2 | 6 |
| Tubulina non etichettata, 10 mg/mL | 20 | 5 | 18 | 5 |
| GMPCPP, 10 mM | 30 | 4 | 30 | 4 |
| DTT, 0,2 M | 0.7 | 3 | 0.7 | 3 |
| 5X BRB80 | 26.4 | 2 | 26.4 | 2 |
| acqua sterile | 52.9 | 1 | 52.9 | 1 |
| Volume totale (μL) | 132 | 132 |
Tabella 3: Miscela di semi GMPCPP. Componenti dei semi di microtubuli GMPCPP, compreso il volume e l'ordine di aggiunta. Preparare aliquote da 5 μL e conservare fino a 1 anno a -80 °C.
4. Impostazioni del microscopio
- Temperatura: impostare la temperatura del microscopio a 28 °C per visualizzare i microtubuli dinamici.
- Filtri: utilizzare la migliore combinazione di cubi filtranti e filtri di emissione, a seconda dei canali fluorescenti da visualizzare. Per visualizzare lunghezze d'onda di 488 nm, 560 nm e 647 nm nello stesso esperimento, utilizzare un set laser a banda quadrupla 405/488/560/647 nm, accoppiato con filtri di emissione per le lunghezze d'onda designate.
- Allinea laser: assicurarsi che i raggi laser utilizzati nell'esperimento siano allineati. Determinare empiricamente l'intensità del laser per l'esperimento, in modo tale che tutte le proteine fluorescenti possano essere visualizzate con il più alto rapporto segnale-rumore possibile, ma non subiscano un significativo fotosbiancamento nel corso dell'esperimento.
- Obiettivo: utilizzare la carta per lenti per pulire un obiettivo 100x con il 70% di etanolo. Prima dell'imaging, aggiungere una goccia di olio per immersione al microscopio all'obiettivo.
- Impostare una sequenza di imaging
- Per un esperimento con microtubuli biotinilati marcati con fluoroforo da 647 nm, microtubuli non biotinilati marcati con fluoroforo a 560 nm e tubulina solubile e proteina di interesse marcata GFP, immagine per 20 min. Immagine dei canali a 560 nm e 488 nm ogni 10 s e del canale a 647 nm ogni 30 s.
- Per acquisire un'immagine di riferimento di fasci prima dell'aggiunta di tubulina solubile e MAP, impostare una sequenza con un'immagine ciascuna in lunghezze d'onda di 560 nm e 647 nm.
5. Genera fasci di microtubuli immobilizzati in superficie
NOTA: per i passaggi seguenti, far scorrere tutte le soluzioni in una camera di flusso mediante pipettaggio in un lato aperto, mentre si posiziona una carta da filtro contro l'altro lato. Proteggere la camera di imaging dalla luce per ridurre il fotosbiancamento delle proteine marcate fluorescentemente. Fissare la camera di imaging preparata su un supporto per diapositive (Figura 1G,H). Seguire i passaggi nella Tabella 4, che corrispondono agli steps di protocollo 5.2-6.4.
- Preparare la miscela solubile di tubuline secondo la Tabella 5 e tenerla sul ghiaccio.
NOTA: La tubulina solubile deve essere sempre posta su ghiaccio per evitare la polimerizzazione. Preparare una miscela di tubuline solubili fresche circa ogni 2 ore o quando i microtubuli non polimerizzano più. - Per immobilizzare i microtubuli tramite un legame biotina-neutravidina-biotina, prima fluire nella soluzione di Neutravidin (NA) fino a riempire la camera (~7,5 μL) e incubare per 5 min.
- Lavare con 10 μL di MB-freddo.
- Flusso in 7,5 μL della proteina bloccante κ-caseina (KC) e incubare per 2 min.
- Lavare con 10 μL di MB-caldo per preparare la camera per l'introduzione di microtubuli.
- Diluire lo stock di microtubuli biotinilati (secondo le osservazioni nel passaggio 3.3.2) in 1X BRB80-DTT e aggiungere 1 μL di questa diluizione a 9 μL di MB-warm. Far scorrere la miscela nella camera e incubare per 10 minuti. Utilizzare una maggiore concentrazione di microtubuli per più fasci.
- Lavare via i microtubuli non immobilizzati con 10 μL di MB-caldo.
- Flusso 7,5 μL di KC caldo nella camera e incubazione per 2 minuti.
- Durante l'incubazione, preparare una soluzione da 2 nM della proteina reticolante PRC1 in KC caldo. Flusso 10 μL di questa soluzione nella camera di flusso e incubare per 5 minuti.
NOTA: la PRC1 ricombinante è espressa e purificata dalle cellule batteriche come descritto in precedenza13. - Per creare fasci, il flusso di 10 μL di microtubuli non biotinilati nella camera e l'incubazione per 10 minuti. PRC1 collegherà i microtubuli biotinilati non biotinilati e immobilizzati15,16 (Figura 3).
NOTA: Vedere il passaggio 6.1 per la preparazione della miscela di dosaggio durante il periodo di incubazione. - Lavare la camera due volte con 10 μL di MB-warm. I microtubuli attaccati sono stabili per circa 20 minuti da questo punto.
| Passo | Reagente | Volume (μL) | Tempo di incubazione (minuti) |
| 1 | Neutravidina | 7.5 | 5 |
| 2 | MB-freddo | 10 | - |
| 3 | κ-caseina | 7.5 | 2 |
| 4 | MB-caldo | 10 | - |
| 5 | Microtubulo biotinilato (diluito in MB-warm) | 10 | 10 |
| 6 | MB-caldo | 10 | - |
| 7 | Κ-caseina calda | 7.5 | 2 |
| 8 | 2 nM PRC1 diluito in κ-caseina | 10 | 5 |
| 9 | Microtubuli non biotinilati | 10 | 10 |
| 10 | MB-caldo x 2 | 10 | - |
| 11 | Miscela di saggi | 10 | - |
| I semi attaccati sono stabili per circa 20 minuti a questo punto | |||
Tabella 4: Fasi del test. Elenco dei reagenti aggiunti alla camera di imaging, con indicazione del tempo di lavaggio (-) o di incubazione.
| Reagente | Volume (μL) |
| Tubulina riciclata, 10 mg/mL | 10 |
| MB-Freddo | 10.3 |
| MBMC · | 13.7 |
| BRB80-DTT | 3.4 |
| GTP, 10 mM | 6.7 |
| ATP, 10 mm (Se si utilizzano kinesine) | 6.7 |
| Tubulina marcata fluorescentemente, 10 mg/ml | 1 (Tubulina liofilizzata resuspend in BRB80-DTT fredda) |
Tabella 5: Componenti solubili della miscela di tubuline. Mescolare all'inizio dell'esperimento e mantenere sul ghiaccio.

Figura 3: Schema di aggiunta di componenti di analisi per creare e visualizzare fasci etichettati fluorescentemente e singoli microtubuli. I semi biotinilati sono mostrati in semi blu, non biotinilati e tubulina solubile in rosso, PRC1 in nero e proteine di interesse nel ciano. I numeri dei passi in figura corrispondono a quelli della tabella 4. Il pannello corrispondente al passaggio 9 mostra un fascio preformato (in basso a sinistra); il passaggio 11 mostra un bundle appena formato (in alto a sinistra). Creato con BioRender.com. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
6. Dinamica dei microtubuli dell'immagine
- Durante il tempo di incubazione di 10 minuti nella fase 5.10, preparare 10 μL di miscela di saggi contenente proteine di interesse, tubulina solubile, nucleotidi, spazzini di ossigeno14 e antiossidanti secondo la Tabella 6. Mantenere il mix su ghiaccio.
- Caricare la camera di imaging preparata, nastrata al supporto del vetrino, sull'obiettivo TIRF 100x. Utilizzare i canali a 560 nm e 647 nm per trovare un campo visivo che contenga un numero e una densità ottimali di singoli microtubuli e fasci.
NOTA: Se entrambi i microtubuli biotinilati e non biotinilati sono etichettati con lo stesso fluoroforo, le analisi line-scan delle intensità di fluorescenza possono distinguere tra singoli microtubuli e fasci. - Una volta identificato un campo visivo, scatta un'immagine di riferimento.
- Fluire con attenzione nella miscela di analisi senza disturbare la camera di imaging.
- Sigillare le estremità aperte della camera con sigillante valap.
- Avviare la sequenza di imaging come descritto nel passaggio 4.5.1.
| Reagente | Volume (μL) |
| Miscela solubile di tubulina | 4 |
| OSF | 1 |
| Trolox (se si utilizzano microtubuli etichettati con un fluoroforo facilmente fotosbiancante) | 1 |
| ATP, 10 mm (Se si utilizzano kinesine) | 1 |
| PRC1 (o reticolante a scelta) | 1 |
| Proteine di interesse | X |
| MB-freddo | 2-X |
Tabella 6: Componenti della miscela di analisi. Mescola, fluisci nella camera di imaging e visualizza la dinamica dei microtubuli, entro 30 minuti.
Risultati
L'esperimento sopra descritto è stato eseguito utilizzando microtubuli biotinilati marcati con fluoroforo da 647 nm, microtubuli non biotinilati marcati con fluoroforo da 560 nm e mix di tubulina solubile marcata con fluoroforo da 560 nm. I microtubuli sono stati reticolati dalla proteina reticolante PRC1 (marcata GFP). Dopo aver generato fasci immobilizzati in superficie e singoli microtubuli (fase 5.11), la camera di imaging è stata montata su un obiettivo di olio TIRF 100X 1,49 NA e visualizzata nei canali di fluore...
Discussione
L'esperimento qui descritto espande significativamente la portata e la complessità dei saggi convenzionali di ricostituzione dei microtubuli, che vengono tradizionalmente eseguiti su singoli microtubuli o su un tipo di array. L'attuale test fornisce un metodo per quantificare e confrontare simultaneamente l'attività map regolatoria su due popolazioni, vale a dire singoli microtubuli e fasci reticolati. Inoltre, questo test consente l'esame di due tipi di fasci: quelli che sono preformati da semi stabili prima dell'iniz...
Divulgazioni
Gli autori non dichiarano interessi concorrenti.
Riconoscimenti
Questo lavoro è stato sostenuto da una sovvenzione del NIH (n. 1DP2GM126894-01) e da fondi del Pew Charitable Trusts e della Smith Family Foundation a R.S. Gli autori ringraziano il Dr. Shuo Jiang per il suo contributo allo sviluppo e all'ottimizzazione dei protocolli.
Materiali
| Name | Company | Catalog Number | Comments |
| (±)-6-Hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (Trolox) | Sigma Aldrich | 238813 | |
| 1,4-piperazinediethanesulfonic acid (PIPES) | Sigma Aldrich | P6757 | |
| 18x18 mm #1.5 coverslips | Electron Microscopy Sciences | 63787 | |
| 2-Mercaptoethanol (BME) | Sigma Aldrich | M-6250 | |
| 24x60 mm #1.5 coverslips | Electron Microscopy Sciences | 63793 | |
| 405/488/560/647 nm Laser Quad Band | Chroma | TRF89901-NK | |
| Acetone | Sigma Aldrich | 320110 | |
| Adenosine 5'-triphosphate disodium salt hydrate (ATP) | Sigma Aldrich | A7699-5G | |
| Avidin, NeutrAvidin® Biotin-binding Protein (Molecular Probes®) | Thermo Fischer Scientific | A2666 | |
| Bath sonicator: Branson 2800 Cleaner | Branson | CPX2800H | |
| Beckman Coulter Polycarbonate Thickwall Tubes, 11 x 34 mm | Beckman-Coulter | 343778 | |
| Beckman Coulter Polycarbonate Thickwall Tubes, 8 x 34 mm | Beckman-Coulter | 343776 | |
| Biotin-PEG-SVA, MW 5,000 | Laysan Bio | #Biotin-PEG-SVA-5000 | |
| Bovine Serum Albumin (BSA) | Sigma Aldrich | 2905 | |
| Catalase | Sigma Aldrich | C40 | |
| Corning LSE Mini Microcentrifuge, AC100-240V | Corning | 6670 | |
| Delicate Task Wipes | Kimtech | 34120 | |
| Dithiothreitol (DTT) | GoldBio | DTT10 | |
| Emission filter | Chroma | ET610/75m | |
| Ethanol (200-proof) | Decon Labs | 2705 | |
| Ethylene glycol tetraacetic acid (EGTA) | Sigma Aldrich | 3777 | |
| Glucose Oxidase | Sigma Aldrich | G2133 | |
| GMPCPP | Jena Bioscience | NU-405 | |
| Guanosine 5'-triphosphate sodium salt hydrate (GTP) | Sigma Aldrich | G8877 | |
| Hellmanex III detergent | Sigma Aldrich | Z805939 | |
| Immersion oil, Type A | Fisher Scientific | 77010 | |
| Kappa-casein | Sigma Aldrich | C0406 | |
| Lanolin | Fisher Scientific | S25376 | |
| Lens Cleaning Tissue | ThorLabs | MC-5 | |
| Magnesium Chloride (MgCl2) | Sigma Aldrich | M9272 | |
| Methylcellulose | Sigma Aldrich | M0512 | |
| Microfuge 16 Benchtop Centrifuge | Beckman-Coulter | A46474 | |
| Microscope Slides, Diamond White Glass, 25 x 75mm, 90° Ground Edges, WHITE Frosted | Globe Scientific | 1380-50W | |
| mPEG-Succinimidyl Valerate, MW 5,000 | Laysan Bio | #NH2-PEG-VA-5K | |
| Optima™ Max-XP Tabletop Ultracentrifuge | Beckman-Coulter | 393315 | |
| Paraffin | Fisher Scientific | P31-500 | |
| PELCO Reverse (self-closing), Fine Tweezers | Ted Pella | 5377-NM | |
| Petrolatum, White | Fisher Scientific | 18-605-050 | |
| Plasma Cleaner, 115V | Harrick Plasma | PDC-001 | |
| Potassium Hydroxide (KOH) | Sigma Aldrich | 221473 | |
| Sodium bicarbonate | Sigma Aldrich | S6014 | |
| Sucrose | Sigma Aldrich | S7903 | |
| Thermal-Lok 1-Position Dry Heat Bath | USA Scientific | 2510-1101 | |
| Thermal-Lok Block for 1.5 and 2.0 mL Tubes | USA Scientific | 2520-0000 | |
| Thermo Scientific™ Pierce™ Bond-Breaker™ TCEP Solution, Neutral pH; 500mM | Thermo Fischer Scientific | PI-77720 | |
| TIRF 100X NA 1.49 Oil Objective | Nikon | CFI Apochromat TIRF 100XC Oil | |
| TIRF microscope | Nikon | Eclipse Ti | |
| TLA 120.1 rotor | Beckman-Coulter | 362224 | |
| TLA 120.2 rotor | Beckman-Coulter | 357656 | |
| Tubulin protein (>99% pure): porcine brain | Cytoskeleton | T240 | |
| Tubulin Protein (Biotin): Porcine Brain | Cytoskeleton | T333P | |
| Tubulin protein (fluorescent HiLyte 647): porcine brain | Cytoskeleton | TL670M | |
| Tubulin protein (X-rhodamine): bovine brain | Cytoskeleton | TL620M | |
| VECTABOND® Reagent, Tissue Section Adhesion | Vector Biolabs | SP-1800-7 | |
| VWR® Personal-Sized Incubator, 120V, 50/60Hz, 0.6A | VWR | 97025-630 |
Riferimenti
- Subramanian, R., Kapoor, T. M. Building complexity: insights into self-organized assembly of microtubule-based architectures. Developmental Cell. 23 (5), 874-885 (2012).
- Baas, P. W., Rao, A. N., Matamoros, A. J., Leo, L. Stability properties of neuronal microtubules. Cytoskeleton (Hoboken). 73 (9), 442-460 (2016).
- Bitan, A., Rosenbaum, I., Abdu, U. Stable and dynamic microtubules coordinately determine and maintain Drosophila bristle shape. Development. 139 (11), 1987-1996 (2012).
- Foe, V. E., von Dassow, G. Stable and dynamic microtubules coordinately shape the myosin activation zone during cytokinetic furrow formation. The Journal of Cell Biology. 183 (3), 457-470 (2008).
- Pous, C., et al. Functional specialization of stable and dynamic microtubules in protein traffic in WIF-B cells. The Journal of Cell Biology. 142 (1), 153-165 (1998).
- Uehara, R., Goshima, G. Functional central spindle assembly requires de novo microtubule generation in the interchromosomal region during anaphase. The Journal of Cell Biology. 191 (2), 259-267 (2010).
- Mitchison, T., Kirschner, M. Dynamic instability of microtubule growth. Nature. 312 (5991), 237-242 (1984).
- Bieling, P., et al. Reconstitution of a microtubule plus-end tracking system in vitro. Nature. 450 (7172), 1100-1105 (2007).
- Bieling, P., Telley, I. A., Surrey, T. A minimal midzone protein module controls formation and length of antiparallel microtubule overlaps. Cell. 142 (3), 420-432 (2010).
- Shimamoto, Y., Forth, S., Kapoor, T. M. Measuring pushing and braking forces generated by ensembles of Kinesin-5 crosslinking two microtubules. Developmental Cell. 34 (6), 669-681 (2015).
- Mani, N., Jiang, S., Neary, A. E., Wijeratne, S. S., Subramanian, R. Differential regulation of single microtubules and bundles by a three-protein module. Nature Chemical Biology. 17 (9), 964-974 (2021).
- Hyman, A. A., Salser, S., Drechsel, D., Unwin, N., Mitchison, T. J. Role of GTP hydrolysis in microtubule dynamics: information from a slowly hydrolyzable analogue, GMPCPP. Molecular Biology of the Cell. 3 (10), 1155-1167 (1992).
- Subramanian, R., et al. Insights into antiparallel microtubule crosslinking by PRC1, a conserved nonmotor microtubule binding protein. Cell. 142 (3), 433-443 (2010).
- Rasnik, I., McKinney, S. A., Ha, T. Nonblinking and long-lasting single-molecule fluorescence imaging. Nature Methods. 3 (11), 891-893 (2006).
- Wijeratne, S., Subramanian, R. Geometry of antiparallel microtubule bundles regulates relative sliding and stalling by PRC1 and Kif4A. eLife. 7, 32595 (2018).
- Mani, N., Wijeratne, S. S., Subramanian, R. Micron-scale geometrical features of microtubules as regulators of microtubule organization. eLife. 10, 63880 (2021).
- Freal, A., et al. Feedback-driven assembly of the axon initial segment. Neuron. 104 (2), 305-321 (2019).
- Ledbetter, M., Porter, K. A "microtubule" in plant cell fine structure. The Journal of Cell Biology. 19 (1), 239-250 (1963).
- Wijeratne, S. S., Marchan, M. F., Tresback, J. S., Subramanian, R. Atomic force microscopy reveals distinct protofilament-scale structural dynamics in depolymerizing microtubule arrays. Proceedings of the National Academy of Sciences of the United States of America. , 119 (2022).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati