
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Visualisation simultanée de la dynamique des microtubules réticulés et simples in vitro par microscopie TIRF
* Ces auteurs ont contribué à parts égales
Dans cet article
Résumé
Ici, un essai de reconstitution in vitro basé sur la microscopie TIRF est présenté pour quantifier et comparer simultanément la dynamique de deux populations de microtubules. Une méthode est décrite pour visualiser simultanément l’activité collective de plusieurs protéines associées aux microtubules sur des faisceaux de microtubules réticulés et des microtubules simples.
Résumé
Les microtubules sont des polymères d’hétérodimères d’αβ-tubuline qui s’organisent en structures distinctes dans les cellules. Les architectures et les réseaux basés sur des microtubules contiennent souvent des sous-ensembles de réseaux de microtubules qui diffèrent par leurs propriétés dynamiques. Par exemple, dans les cellules en division, des faisceaux stables de microtubules réticulés coexistent à proximité de microtubules dynamiques non réticulés. Les études de reconstitution in vitro basées sur la microscopie TIRF permettent la visualisation simultanée de la dynamique de ces différents réseaux de microtubules. Dans ce test, une chambre d’imagerie est assemblée avec des microtubules immobilisés en surface, qui sont soit présents sous forme de filaments simples, soit organisés en faisceaux réticulés. L’introduction de la tubuline, des nucléotides et des régulateurs de protéines permet une visualisation directe des protéines associées et des propriétés dynamiques des microtubules simples et réticulés. De plus, les changements qui se produisent lorsque des microtubules uniques dynamiques s’organisent en faisceaux peuvent être surveillés en temps réel. La méthode décrite ici permet une évaluation systématique de l’activité et de la localisation de protéines individuelles, ainsi que des effets synergiques des régulateurs de protéines sur deux sous-ensembles de microtubules différents dans des conditions expérimentales identiques, fournissant ainsi des informations mécanistes inaccessibles par d’autres méthodes.
Introduction
Les microtubules sont des biopolymères qui forment des échafaudages structurels essentiels à de multiples processus cellulaires, allant du transport intracellulaire et du positionnement des organites à la division cellulaire et à l’allongement. Pour exécuter ces diverses fonctions, les microtubules individuels sont organisés en réseaux de la taille d’un micron, tels que les fuseaux mitotiques, les axonèmes ciliaires, les faisceaux neuronaux, les réseaux interphasiques et les réseaux corticaux végétaux. Un motif architectural omniprésent que l’on retrouve dans ces structures est un faisceau de microtubules réticulés sur toute leur longueur1. Une caractéristique intrigante de plusieurs structures à base de microtubules est la coexistence de microtubules groupés et de microtubules simples non réticulés à proximité spatiale étroite. Ces sous-populations de microtubules peuvent présenter une dynamique de polymérisation très différente les unes des autres, selon les besoins pour leur bon fonctionnement2,3,4,5. Par exemple, dans le fuseau mitotique, des faisceaux réticulés stables et des microtubules simples dynamiques sont présents dans une région à l’échelle du micron au centre cellulaire6. L’étude de la façon dont les propriétés dynamiques des populations de microtubules coexistantes sont spécifiées est donc essentielle pour comprendre l’assemblage et la fonction des structures à base de microtubules.
Les microtubules sont des polymères dynamiques qui alternent entre les phases de polymérisation et de dépolymérisation, passant d’une phase à l’autre dans des événements connus sous le nom de catastrophe et de sauvetage7. La dynamique des microtubules cellulaires est régulée par une myriade de protéines associées aux microtubules (MAP) qui modulent les taux de polymérisation et de dépolymérisation des microtubules et les fréquences des catastrophes et des événements de sauvetage. Il est difficile d’étudier l’activité des MAP sur les réseaux proximaux spatiaux dans les cellules, en raison des limites de la résolution spatiale en microscopie optique, en particulier dans les régions à haute densité de microtubules. De plus, la présence de plusieurs MAP dans la même région cellulaire entrave l’interprétation des études biologiques cellulaires. Les essais de reconstitution in vitro, effectués conjointement avec la microscopie à fluorescence par réflexion interne totale (TIRF), contournent les défis liés à l’examen des mécanismes par lesquels des sous-ensembles spécifiques de MAP régulent la dynamique des réseaux de microtubules cellulaires proximaux. Ici, la dynamique des microtubules assemblés in vitro est examinée en présence d’un ou plusieurs MAP recombinants dans des conditions contrôlées8,9,10. Cependant, les essais de reconstitution conventionnels sont généralement effectués sur des microtubules simples ou sur un type de réseau, empêchant la visualisation de populations coexistantes.
Nous présentons ici des essais de reconstitution in vitro qui permettent la visualisation simultanée de deux populations de microtubules dans les mêmes conditions de solution11. Nous décrivons une méthode permettant de visualiser simultanément l’activité collective de plusieurs MAP sur des microtubules simples et sur des faisceaux de microtubules réticulés par la protéine mitotique associée au fuseau PRC1. La protéine PRC1 se lie préférentiellement au chevauchement entre les microtubules anti-parallèles, les réticulant9. En bref, ce protocole comprend les étapes suivantes: (i) préparation de solutions mères et de réactifs, (ii) nettoyage et traitement de surface des couvercles utilisés pour créer la chambre d’imagerie pour les expériences de microscopie, (iii) préparation de « graines » de microtubules stables à partir desquelles la polymérisation est initiée au cours de l’expérience, (iv) spécification des paramètres du microscope TIRF pour visualiser la dynamique des microtubules, (v) immobilisation des graines de microtubules et génération de faisceaux de microtubules réticulés dans la chambre d’imagerie, et (vi) visualisation de la dynamique des microtubules dans la chambre d’imagerie par microscopie TIRF, après addition de tubuline soluble, de MAP et de nucléotides. Ces essais permettent l’évaluation qualitative et l’examen quantitatif de la localisation de la MAP et de son effet sur la dynamique de deux populations de microtubules. En outre, ils facilitent l’évaluation des effets synergiques de plusieurs MAP sur ces populations de microtubules, dans un large éventail de conditions expérimentales.
Access restricted. Please log in or start a trial to view this content.
Protocole
1. Préparez les réactifs
- Préparer les tampons et les réactifs comme indiqué dans les tableaux 1 et 2. Pendant l’expérience, conservez toutes les solutions sur la glace, sauf indication contraire.
| Solution | Composants | Durée de stockage recommandée | Notes | ||
| 5X BRB80 | 400 mM K-PIPES, 5 mM MgCl2, 5 mM EGTA, pH 6,8 avec KOH, stérilisation du filtre | jusqu’à 2 ans | Conserver à 4 °C | ||
| 1X BRB80 | 80 mM K-PIPES, 1 mM MgCl2, 1 mM EGTA, pH 6,8 | jusqu’à 2 ans | Conserver à 4 °C | ||
| BRB80-TNT | 1X BRB80, 1 mM TNT | jusqu’à 2 jours | |||
| Tampon d’essai | 80 mM K-PIPES, 3 mM MgCl2, 1 mM EGTA, pH 6,8, 5% saccharose (OR 1X BRB80, 5% saccharose, 2 mM MgCl2) | jusqu’à 1 an | Conserver à 4 °C | ||
| Mémoire tampon principale (Mo) | Tampon d’essai, 5mM TCEP | 1 semaine | Préparez-vous le jour de l’expérience; Séparer en deux tubes: MB-chaud à température ambiante et MB-froid sur glace; inclure 1 mM TNT si vous utilisez des colorants fluorescents | ||
| Tampon maître avec méthylcellulose (MBMC) | 1X BRB80, 0,8% méthylcellulose, 5 mM TCEP, 5 mM MgCl2 | 1 semaine | Préparez-vous le jour de l’expérience; inclure 1 mM TNT si vous utilisez des colorants fluorescents | ||
| Tampon de dilution des protéines (DB) | MB, 1 mg/mL d’albumine sérique bovine (BSA), 1 μM d’ATP | 1 jour, sur glace | Préparez-vous le jour de l’expérience; inclure 1 mM TNT si vous utilisez des colorants fluorescents | ||
| Mélange de récupération d’oxygène (OSM) | MB, 389 μg/mL catalase, 4,44 mg/mL de glucose oxydase, 15,9 mM de 2-mercaptoéthanol (BME) | 1 jour, sur glace | Préparez-vous le jour de l’expérience | ||
| Finale de récupération de l’oxygène (OSF) | MB, 350 μg/mL catalase, 4 mg/mL de glucose oxydase, 14,3 mM BME, 15 mg/mL de glucose | utiliser dans les 30 minutes | Préparer immédiatement avant utilisation en ajoutant 1 μL de glucose à 9 μL d’OSM | ||
Tableau 1 : Liste des tampons utilisés dans ce protocole et de leurs composants. Reportez-vous à la colonne « Durée de stockage recommandée » pour obtenir des conseils sur la durée de préparation de chaque tampon.
| Réactif | Concentration de stockage | Solvant de stockage | Température de stockage | Concentration de travail | Concentration finale | Durée de stockage recommandée | Notes | |||||||||
| Neutravidine (NA) | 5 mg/mL | 1X BRB80 | -80°C | 0,2 mg/mL | 0,2 mg/mL | jusqu’à 1 an | Utilisé pour immobiliser les microtubules via un lien biotine-neutravidine-biotine; stocker dans de petites aliquotes | |||||||||
| Kappa-caséine (KC) | 5 mg/mL | 1X BRB80 | -80°C | 0,5 mg/mL | 0,5 mg/mL | jusqu’à 2 ans | Utilisé pour bloquer la surface de la chambre d’imagerie; Conserver dans de petites aliquotes; Le jour de l’expérience, réserver un petit volume à température ambiante | |||||||||
| Albumine sérique bovine (BSA) | 50 mg/mL | 1X BRB80 | -20°C | 1 mg/mL (en DB) | N/A | jusqu’à 2 ans | stocker dans de petites aliquotes | |||||||||
| Catalase | 3,5 mg/mL | 1X BRB80 | -80°C | 350 μg/mL (en OSF) | 35 μg/mL | jusqu’à 2 ans | composant du mélange de récupération d’oxygène; stocker dans de petites aliquotes | |||||||||
| Glucose oxydase | 40 mg/mL | 1X BRB80 | -80°C | 4 mg/mL (dans OSF) | 0,4 mg/mL | jusqu’à 2 ans | composant du mélange de récupération d’oxygène; stocker dans de petites aliquotes | |||||||||
| Tubuline | Lyophilisé | N/A | 4°C | 10 mg/mL | 2,12 mg/mL (dans un mélange de tubuline) | jusqu’à 1 an | Une fois que la tubuline est en solution, gardez-la au froid pour éviter la polymérisation. | |||||||||
| Adénosine triphosphate (ATP) | 100 mM | eau ultrapure | -20°C | 10 mM | 1 mM | 6 mois | Préparer la solution dans de l’eau stérilisée par filtre, ajuster le pH à ~7,0 et congeler dans de petites aliquotes. | |||||||||
| Guanosine Triphosphate (GTP) | 100 mM | eau ultrapure | -20°C | 10 mM | 1,29 mM (dans un mélange de tubuline) | 6 mois | Préparer la solution dans de l’eau stérilisée par filtre, ajuster le pH à ~7,0 et congeler dans de petites aliquotes. | |||||||||
| Guanosine-5'-[(α,β)-méthyléno] triphosphate (GMPCPP) | 10 mM | eau ultrapure | -20°C | 10 μM | 0,5 μM | 6 mois | ||||||||||
| Dithiothréitol (TNT) | 1 M | eau stérile | -20°C | 1 mM | N/A | jusqu’à 2 ans | ||||||||||
| Tris(2-carboxyéthyl) phosphine (TCEP) | 0,5 M | eau stérilisée par filtre | Température ambiante | 5 mM | N/A | jusqu’à 2 ans | ||||||||||
| Méthylcellulose | 1% | eau stérile | Température ambiante | 0,8 % (en MBMC) | 0,21 % (dans le mélange de tubuline) | jusqu’à 1 an | Dissoudre la méthylcellulose en l’ajoutant lentement à de l’eau presque bouillante. Laisser refroidir en remuant continuellement. | |||||||||
| Bêta-mercaptoéthanol (BME) | 143 mM | eau stérile | Température ambiante | 14,3 mM (en OSF) | 1,43 mM | jusqu’à 5 ans | 143 mM est une dilution 1:100 du stock BME | |||||||||
| Glucose | 150 mg/mL | 1X BRB80 | -80°C | 15 mg/mL (en FSI) | 1,5 mg/mL | jusqu’à 2 ans | Ajouter à OSM immédiatement avant utilisation | |||||||||
| Acide (±)-6-hydroxy-2,5,7,8-tétraméthyl chromane-2-carboxylique (Trolox) | 10mM | 1X BRB80 | -80°C | 10 mM | 1 mM | jusqu’à 1 an | Ne se dissout pas complètement. Ajouter un peu de NaOH, remuer pendant environ 4 heures et filtrer stériliser avant utilisation | |||||||||
| Valérate de mPEG-Succinimidyl, MW 5 000 | poudre | N/A | -20°C | 333 mg/mL (dans 0,1 M de bicarbonate de sodium) | 324 mg/mL (dans 0,1 M de bicarbonate de sodium) | 6 mois | Préparez environ 34 mg d’aliquotes, en marquant chaque tube avec un poids exact de poudre. Passer l’azote gazeux sur le solide, sceller les tubes avec un parafilm et conserver à -20 °C dans un récipient contenant du dessiccant. | |||||||||
| Biotine-PEG-SVA, MW 5 000 | poudre | N/A | -20°C | 111 mg/mL (dans 0,1 M de bicarbonate de sodium) | 3,24 mg/mL (dans 0,1 M de bicarbonate de sodium) | 6 mois | Préparez environ 3 mg d’aliquotes, en marquant chaque tube avec un poids exact de poudre. Passer l’azote gazeux sur le solide, sceller les tubes avec un parafilm et conserver à -20 °C dans un récipient contenant du dessiccant. | |||||||||
Tableau 2 : Liste des réactifs utilisés dans ce protocole. Sont inclus les conditions et concentrations de stockage recommandées, les concentrations de travail des solutions mères utilisées pendant l’expérience et la concentration finale dans la chambre d’imagerie. Des notes supplémentaires sont données dans la colonne d’extrême droite.
2. Préparez les lames Biotin-PEG
REMARQUE: Préparez les chambres d’imagerie aussi près que possible du début d’une expérience et pas plus de 2 semaines à l’avance.
- Couvercles propres
- Placez un nombre égal de 24 x 60 mm et de 18 x 18 mm #1.5 (Figure 1F) dans des bocaux à glissière et des supports de lavage de glissière, respectivement (Figure 1A,B). Placez les supports de lavage à glissière contenant des couvercles de 18 x 18 mm dans un bécher de 100 ml.
- Rincez tous les couvercles 5 à 6 fois à l’eau ultrapure (résistivité de 18,2 MΩ-cm) et retirez l’excès de liquide après chaque rinçage avec une pointe de pipette fixée à un tube à vide (Figure 1C).
- Remplissez les béchers et les pots à glissière contenant les couvercles avec de l’eau ultrapure, scellez avec un parafilm et soniquez pendant 10 min.
- Remplissez deux béchers de 150 ml avec de l’éthanol résistant à 200 mL. À l’aide d’une pince à épiler, trempez chaque couvercle dans un bécher rempli d’éthanol, puis dans l’autre.
- À l’aide d’une pince à épiler, transférer les couvercles sur le séchoir à glissière (Figure 1D), les sécher sous un flux d’azote gazeux et les incuber à 37 °C jusqu’à ce qu’ils soient complètement secs (~15 min).
- Placez les couvercles séchés en une seule couche à l’intérieur du nettoyeur plasma. Formez un joint sous vide, puis réglez le niveau de radiofréquence (RF) du nettoyeur plasma sur ~ 8 MHz.
- Une fois le plasma généré, laissez les couvercles dans le nettoyeur plasma pendant 5 min. Éteignez le nettoyeur plasma et relâchez l’aspirateur lentement.
- Une fois le joint sous vide relâché, retournez les couvercles et répétez le nettoyage au plasma pendant 5 minutes pour l’autre côté des couvercles.
- Alternative au nettoyage au plasma: À la place des étapes 2.1.2-2.1.3, les couvercles de sonicate dans une solution chaude de détergent à 2% (dans de l’eau ultrapure) pendant 10 min. Ensuite, lavez soigneusement les couvercles avec de l’eau ultrapure et soniquez dans de l’eau ultrapure 2 à 3 fois (10 minutes chacun). Ensuite, laver à l’éthanol et sécher comme dans les étapes 2.1.4-2.1.5. Ignorez les étapes 2.1.6 à 2.1.8.
- Traitement Biotine-PEG
- Immédiatement avant utilisation, dissoudre 400 μL de 3-Aminopropyltriethoxysilane dans 40 mL d’acétone. À l’aide d’une pince à épiler, déplacez les couvercles nettoyés au plasma dans le rack de lavage à glissière et les pots à taches. Immerger les couvercles dans une solution de 3-aminopropyltriéthoxysilane et incuber pendant 5 min12,13.
- Lavez tous les couvercles 5 à 6 fois avec de l’eau ultrapure.
- Transférer les couvercles sur le séchoir à glissière, les sécher sous un flux d’azote gazeux et incuber à 37 °C jusqu’à ce qu’ils soient complètement secs (~20 min).
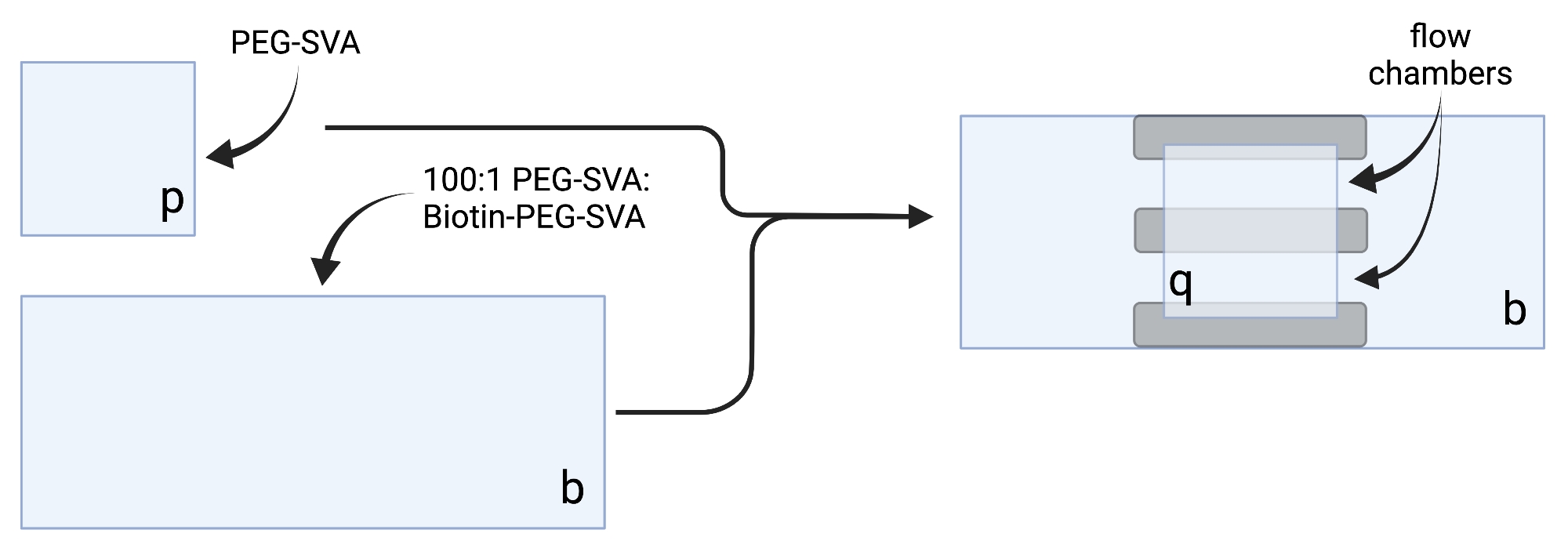
- Posez les couvercles séchés sur des lingettes de travail délicates et étiquetez chaque couvercle sur un coin, par exemple, « p » sur chaque couvercle de 18 x 18 mm et « b » sur chaque couvercle de 24 x 60 mm (voir figure 2).
- Le jour de l’expérience, préparer une solution fraîche de bicarbonate de sodium de 0,1 M en dissolvant 0,84 mg de NaHCO3 dans 10 mL d’eau ultrapure.
- Porter les aliquotes de valérate de mPEG-Succinimidyl (PEG-SVA) et de Biotine-PEG-SVA à température ambiante, immédiatement avant utilisation. Voir les notes sur la préparation de l’aliquote de polyéthylène glycol (PEG) dans le tableau 2.
REMARQUE: Travailler rapidement car la demi-vie d’hydrolyse de la fraction valérate de succinimidyle (SVA) est d’environ 30 min. - Ajouter 102 μL de 0,1 M de NaHCO3 à 34 mg de PEG-SVA, faire tourner dans une microcentrifugeuse de paillasse à 2 656 x g pendant 20 s, puis mélanger en pipetant de haut en bas. Dissoudre 3 mg de Biotine-PEG-SVA dans 27 μL de 0,1 M NaHCO3 en pipetant de haut en bas. Ajuster les volumes de dilution en fonction du poids exact de PEG indiqué sur les tubes (voir tableau 2).
- Préparer 100:1 p/p de mélange PEG:biotine-PEG en combinant 75 μL de solution PEG-SVA et 2,25 μL de solution Biotine-PEG-SVA pour 20 couvercles, 100 μL et 3 μL pour 30 couvercles, ou 125 μL et 3,75 μL pour 40 couvercles.
- Construisez une chambre d’hydratation en plaçant des serviettes en papier humides sous le porte-embouts au fond d’une boîte à embouts vide de 10 μL (Figure 1E). Cela empêchera l’évaporation des solutions PEG.
- Pipette 6 μL de mélange PEG-SVA:biotine-PEG 100:1 sur le centre d’un couvercle de 24 x 60 mm sur le côté étiqueté. Placez un autre couvercle de 24 x 60 mm sur le dessus du premier couvercle de sorte que la paire forme une forme en X, avec les côtés étiquetés « b » l’un en face de l’autre. Placez la paire sur un porte-embouts vide dans la chambre d’hydratation et répétez l’opération pour les couvercles restants de 24 x 60 mm.
- Pipette 6 μL de PEG-SVA sur le centre d’un couvercle de 18 x 18 mm sur le côté étiqueté. Placez un autre couvercle de 18 x 18 mm sur le dessus du premier couvercle, avec les côtés étiquetés « p » l’un en face de l’autre. Placez la paire sur une grille à embouts vide dans la chambre d’hydratation et répétez l’opération pour les couvercles restants de 18 x 18 mm.
- Fermez la chambre d’hydratation et incubez pendant 3 h ou toute la nuit.
- Séparez les paires de couvercles et rincez à l’eau ultrapure.
- Sécher les couvercles avec un flux d’azote et les placer dans un incubateur à 37 °C pour les sécher complètement.
- Pour construire la chambre d’imagerie, collez trois bandes de ruban adhésif double face sur un couvercle de 24 x 60 mm sur le côté étiqueté « b ». De l’autre côté des bandes de ruban adhésif, fixez un couvercle de 18 x 18 mm avec son côté étiqueté « p » face au plus grand couvercle. Cela forme deux chambres d’écoulement pour les expériences de microscopie, avec des surfaces traitées se faisant face (Figure 2 et Figure 1G).

Figure 1: Équipement pour le traitement des couvercles et la préparation de la chambre d’imagerie. (A) pots de coloration par glissière pour 24 x 60 mm de couvercles, (B) racks de lavage de glissières pour 18 x 18 mm de couvercles, (C) installation sous vide, (D) séchoir à glissière, (E) chambre d’hydratation, (F) couvercles, (G) chambre d’imagerie, (H) porte-glissières. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 2 : Schéma pour la préparation des chambres d’imagerie à l’aide de ruban adhésif double face (gris) et de couvercles traités PEG/Biotine-PEG. Créé avec BioRender.com. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
3. Polymériser les microtubules
- Préparer les semences GMPCPP
REMARQUE: Préparez les graines de GMPCPP dans une chambre froide, en gardant tous les réactifs, embouts et tubes à 4 ° C. Les graines de GMPCPP peuvent être préparées à l’avance et stockées à -80 ° C pendant 1 an maximum. Placer le rotor d’ultracentrifugation à angle fixe, contenant des tubes centrifuges de 1 mL, dans l’ultracentrifugeuse et régler la température à 4 °C.- Remettre en suspension la tubuline lyophilisée (tableau 2) à ~10 mg/mL dans 1X BRB80, immédiatement avant utilisation.
- Mélanger les composants des semences GMPCPP comme décrit dans le tableau 3.
REMARQUE: Gardez tous les composants de la tubuline sur la glace autant que possible pour minimiser la polymérisation de la tubuline soluble. - Clarifier le mélange dans un rotor ultracentrifuge à angle fixe à 352 700 x g pendant 5 min à 4 °C.
- Séparez le surnageant en aliquotes de 5 μL, congelez-les dans de l’azote liquide et conservez-les à -80 °C.
- Polymériser les graines le jour de l’expérience
- Chaud 1-2 mL de BRB80-DTT (Tableau 1) à 37 °C.
- Placer 5 μL d’aliquote de graines de GMPCPP (-80 °C) de l’étape 3.1.4 sur de la glace et dissoudre immédiatement dans 20 μL de BRB80-DTT chaud. Tourner à 2 000 x g pendant 5 s à température ambiante et tapoter pour mélanger.
REMARQUE : Le volume de dilution initial peut varier entre 13 μL et 21 μL et est déterminé empiriquement pour chaque lot de semences. Si les graines ne parviennent pas à polymériser, dépannez en complétant le tampon de dilution initial (étape 3.2.2) avec 0,5 μM gmPCPP. - Protéger de la lumière et incuber à 37 °C pendant 30-45 min.
REMARQUE: La longueur des microtubules dépend de la durée de l’incubation. Pour les microtubules courts, le temps d’incubation peut être aussi court que 15 min. Pour les microtubules longs, le temps d’incubation peut aller jusqu’à 2 h. Les microtubules biotinylés ont tendance à nécessiter des temps d’incubation plus longs que les microtubules non biotinylés. - Placer le rotor d’ultracentrifugation à angle fixe, contenant des tubes centrifuges de 500 μL, dans une ultracentrifugeuse et préchauffer à 30 °C.
- Après incubation, ajouter 50 μL de BRB80-DTT chaud (étape 3.2.1) aux graines de GMPCPP polymérisées et transférer le mélange dans un tube de centrifugeuse de 500 μL. Lavez le tube vide qui contenait les graines de GMPCPP avec 50 μL de BRB80-DTT chaud, pipette de haut en bas, et ajoutez ce tampon au tube de centrifugeuse de 500 μL contenant le mélange.
- Avant de tourner, marquez le bord du tube de centrifugeuse pour indiquer où se trouvera la pastille (la pastille sera trop petite pour être vue). Tourner pendant 10 min à 244 900 x g à 30 °C12.
- Pipettez soigneusement le surnageant et jetez-le. Remettez en suspension la pastille dans 100 μL de BRB80-DTT chaud. Appuyez pour mélanger.
- Tourner pendant 10 min à 244 900 x g à 30 °C, en alignant le marquage avec le rotor pour granuler au même endroit.
- Retirez le surnageant et remettez en suspension la pastille dans 16 μL de BRB80-DTT chaud. Transférer la solution de microtubule dans un tube de microcentrifugation propre de 0,6 mL. Protéger de la lumière et maintenir à température ambiante ou au-dessus.
REMARQUE: Après la polymérisation, maintenir les microtubules à température ambiante ou au-dessus. S’ils ont froid, ils vont se dépolymeriser. Incuber à 28 °C pour plus de stabilité.
- Vérifier les microtubules par microscopie TIRF
- Pipeter un mélange de 4,5 μL de BRB80-DTT et de 1 μL de solution de microtubules (étape 3.2.9) sur une lame de microscope. Couvrir avec un couvercle de 18 x 18 mm et sceller les bords avec un vernis à ongles transparent ou un mélange 1:1:1 de vaseline, de lanoline et de paraffine (scellant valap), qui est solide à température ambiante et liquide à 95 °C.
- Placez l’objectif TIRF sous le couvercle (voir l’étape 4 pour les réglages recommandés au microscope) et visualisez les microtubules nouvellement polymérisés à la longueur d’onde appropriée pour la tubuline marquée par fluorescence dans le mélange Bright (tableau 3), afin de déterminer quelle dilution des microtubules utiliser dans les expériences à venir.
| Réactif | Mélange lumineux (μL) | Ordre d’addition | Mélange brillant + biotine (μL) | Ordre d’addition |
| Tubuline fluorescente, 10 mg/mL | 2 | 6 | 2 | 7 |
| Biotine-tubuline, 10 mg/mL | 0 | N/A | 2 | 6 |
| Tubuline non étiquetée, 10 mg/mL | 20 | 5 | 18 | 5 |
| GMPCPP, 10 mM | 30 | 4 | 30 | 4 |
| TNT, 0,2 M | 0.7 | 3 | 0.7 | 3 |
| 5X BRB80 | 26.4 | 2 | 26.4 | 2 |
| eau stérile | 52.9 | 1 | 52.9 | 1 |
| Volume total (μL) | 132 | 132 |
Tableau 3 : Mélange de semences gmpcpp. Composants des graines de microtubules GMPCPP, y compris le volume et l’ordre d’addition. Préparer des aliquotes de 5 μL et conserver jusqu’à 1 an à -80 °C.
4. Réglages du microscope
- Température : Réglez la température du microscope à 28 °C pour afficher les microtubules dynamiques.
- Filtres: Utilisez la meilleure combinaison de cubes de filtre et de filtres d’émission, en fonction des canaux fluorescents à imager. Pour visualiser des longueurs d’onde de 488 nm, 560 nm et 647 nm dans la même expérience, utilisez un ensemble laser quadribande 405/488/560/647 nm, couplé à des filtres d’émission pour les longueurs d’onde désignées.
- Aligner les lasers : Assurez-vous que les faisceaux laser utilisés dans l’expérience sont alignés. Déterminer empiriquement l’intensité du laser pour l’expérience, de sorte que toutes les protéines fluorescentes puissent être imagées avec le rapport signal/bruit le plus élevé possible, mais ne subissent pas de photoblanchiment significatif au cours de l’expérience.
- Objectif : Utilisez du papier pour nettoyer un objectif 100x avec 70 % d’éthanol. Avant l’imagerie, ajoutez une goutte d’huile d’immersion au microscope à l’objectif.
- Configurer une séquence d’imagerie
- Pour une expérience avec des microtubules biotinylés marqués au fluorophore de 647 nm, des microtubules non biotinylés marqués au fluorophore de 560 nm et de la tubuline soluble, et une protéine d’intérêt marquée GFP, image pendant 20 min. Imagez les canaux 560 nm et 488 nm toutes les 10 s et le canal 647 nm toutes les 30 s.
- Pour capturer une image de référence des faisceaux avant l’ajout de tubuline soluble et de MAP, établissez une séquence avec une image chacune dans des longueurs d’onde de 560 nm et 647 nm.
5. Générer des faisceaux de microtubules immobilisés en surface
REMARQUE: Pour les étapes suivantes, versez toutes les solutions dans une chambre d’écoulement en pipetant un côté ouvert, tout en plaçant un papier filtre contre l’autre côté. Protéger la chambre d’imagerie de la lumière pour réduire le photoblanchiment des protéines marquées par fluorescence. Collez la chambre d’imagerie préparée sur un support de diapositive (Figure 1G,H). Suivez les étapes du tableau 4, qui correspondent aux protocoles steps 5.2-6.4.
- Préparez le mélange de tubuline soluble conformément au tableau 5 et conservez-le sur la glace.
REMARQUE: La tubuline soluble doit toujours être placée sur de la glace pour éviter la polymérisation. Préparez un mélange de tubuline soluble frais environ toutes les 2 h, ou lorsque les microtubules ne sont plus polymérisés. - Pour immobiliser les microtubules via une liaison biotine-neutravidine-biotine, d’abord s’écouler dans une solution de neutravidine (NA) jusqu’à ce que la chambre soit remplie (~7,5 μL) et incuber pendant 5 min.
- Laver avec 10 μL de MB-froid.
- Faire circuler dans 7,5 μL la protéine bloquante κ-caséine (KC) et incuber pendant 2 min.
- Laver avec 10 μL de MB-chaud pour préparer la chambre à l’introduction de microtubules.
- Diluer le stock de microtubules biotinylés (selon les observations de l’étape 3.3.2) dans 1X BRB80-DTT et ajouter 1 μL de cette dilution à 9 μL de MB-chaud. Verser le mélange dans la chambre et incuber pendant 10 min. Utilisez une concentration plus élevée de microtubules pour plus de faisceaux.
- Laver les microtubules non immobilisés avec 10 μL de MB-chaud.
- Faire couler 7,5 μL de KC chaud dans la chambre et incuber pendant 2 min.
- Pendant l’incubation, préparer une solution de 2 nM de la protéine de réticulation PRC1 dans du KC chaud. Faire couler 10 μL de cette solution dans la chambre d’écoulement et incuber pendant 5 min.
REMARQUE: Le PRC1 recombinant est exprimé et purifié à partir de cellules bactériennes comme décrit précédemment13. - Pour former des faisceaux, faire circuler 10 μL de microtubules non biotinylés dans la chambre et incuber pendant 10 min. PRC1 va réticuler les microtubules biotinylés non biotinylés et immobilisés15,16 (Figure 3).
REMARQUE : Voir l’étape 6.1 pour préparer le mélange d’essai pendant la période d’incubation. - Lavez la chambre deux fois avec 10 μL de MB-warm. Les microtubules attachés sont stables pendant environ 20 minutes à partir de ce point.
| Pas | Réactif | Volume (μL) | Temps d’incubation (minutes) |
| 1 | Neutravidin | 7.5 | 5 |
| 2 | MB-froid | 10 | - |
| 3 | κ-caséine | 7.5 | 2 |
| 4 | MB-chaud | 10 | - |
| 5 | Microtubule biotinylé (dilué dans MB-warm) | 10 | 10 |
| 6 | MB-chaud | 10 | - |
| 7 | κ-caséine chaude | 7.5 | 2 |
| 8 | 2 nM PRC1 dilué dans de la κ-caséine | 10 | 5 |
| 9 | Microtubule non biotinylé | 10 | 10 |
| 10 | MB-chaud x 2 | 10 | - |
| 11 | Mélange d’essai | 10 | - |
| Les graines attachées sont stables pendant environ 20 minutes à ce stade | |||
Tableau 4 : Étapes du test. Liste des réactifs ajoutés à la chambre d’imagerie, avec indication du temps de lavage (-) ou d’incubation.
| Réactif | Volume (μL) |
| Tubuline recyclée, 10 mg/mL | 10 |
| MB-Froid | 10.3 |
| Le | 13.7 |
| BRB80-TNT | 3.4 |
| GTP, 10 mM | 6.7 |
| ATP, 10 mM (Si vous utilisez des kinésines) | 6.7 |
| Tubuline marquée par fluorescence, 10 mg/mL | 1 (Remettre en suspension de la tubuline marquée lyophilisée dans la TNT À froid BRB80-DTT) |
Tableau 5 : Composants solubles du mélange de tubuline. Mélanger au début de l’expérience et rester sur la glace.

Figure 3 : Schéma de l’ajout de composants d’essai pour fabriquer et imager des faisceaux marqués par fluorescence et des microtubules simples. Les graines biotinylées sont représentées en bleu, les graines non biotinylées et la tubuline soluble en rouge, la PRC1 en noir et les protéines d’intérêt dans le cyan. Les numéros d’échelons figurant dans la figure correspondent à ceux du tableau 4. Le panneau correspondant à l’étape 9 montre un faisceau préformé (en bas à gauche); l’étape 11 montre un faisceau nouvellement formé (en haut à gauche). Créé avec BioRender.com. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
6. Dynamique des microtubules d’image
- Pendant le temps d’incubation de 10 minutes à l’étape 5.10, préparez 10 μL de mélange d’essai contenant des protéines d’intérêt, de la tubuline soluble, des nucléotides, des piégeurs d’oxygène14 et des antioxydants conformément au tableau 6. Gardez le mélange sur la glace.
- Chargez la chambre d’imagerie préparée, scotchée au support de diapositive, sur l’objectif TIRF 100x. Utilisez les canaux 560 nm et 647 nm pour trouver un champ de vision qui contient un nombre et une densité optimaux de microtubules et de faisceaux uniques.
REMARQUE: Si les microtubules biotinylés et non biotinylés sont marqués avec le même fluorophore, les analyses par balayage linéaire des intensités de fluorescence peuvent faire la distinction entre les microtubules simples et les faisceaux. - Une fois qu’un champ de vision est identifié, prenez une image de référence.
- Appliquer soigneusement dans le mélange d’essai sans perturber la chambre d’imagerie.
- Scellez les extrémités ouvertes de la chambre avec du scellant valap.
- Démarrez la séquence d’imagerie comme décrit à l’étape 4.5.1.
| Réactif | Volume (μL) |
| Mélange de tubuline soluble | 4 |
| OSF | 1 |
| Trolox (si vous utilisez des microtubules marqués avec un fluorophore à photoblanchiment facile) | 1 |
| ATP, 10 mM (Si vous utilisez des kinésines) | 1 |
| PRC1 (ou réticulateur de choix) | 1 |
| Protéines d’intérêt | X |
| MB-froid | 2-X |
Tableau 6 : Composants du mélange d’essais. Mélangez, débitez dans la chambre d’imagerie et imagez la dynamique des microtubules, dans les 30 minutes.
Access restricted. Please log in or start a trial to view this content.
Résultats
L’expérience décrite ci-dessus a été réalisée à l’aide de microtubules biotinylés marqués au fluorophore de 647 nm, de microtubules non biotinylés marqués au fluorophore de 560 nm et d’un mélange de tubuline soluble marqué au fluorophore de 560 nm. Les microtubules ont été réticulés par la protéine réticulée PRC1 (marquée GFP). Après la génération de faisceaux immobilisés en surface et de microtubules simples (étape 5.11), la chambre d’imagerie a été montée sur un objectif d’huile T...
Access restricted. Please log in or start a trial to view this content.
Discussion
L’expérience décrite ici élargit considérablement la portée et la complexité des essais conventionnels de reconstitution de microtubules, qui sont traditionnellement effectués sur des microtubules simples ou sur un type de réseau. Le test actuel fournit une méthode pour quantifier et comparer simultanément l’activité map régulatrice sur deux populations, à savoir les microtubules simples et les faisceaux réticulés. De plus, ce test permet d’examiner deux types de faisceaux : ceux qui sont préformés...
Access restricted. Please log in or start a trial to view this content.
Déclarations de divulgation
Les auteurs ne déclarent aucun intérêt concurrent.
Remerciements
Ce travail a été soutenu par une subvention du NIH (n° 1DP2GM126894-01) et par des fonds des Pew Charitable Trusts et de la Smith Family Foundation à R.S. Les auteurs remercient le Dr Shuo Jiang pour sa contribution au développement et à l’optimisation des protocoles.
Access restricted. Please log in or start a trial to view this content.
matériels
| Name | Company | Catalog Number | Comments |
| (±)-6-Hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (Trolox) | Sigma Aldrich | 238813 | |
| 1,4-piperazinediethanesulfonic acid (PIPES) | Sigma Aldrich | P6757 | |
| 18x18 mm #1.5 coverslips | Electron Microscopy Sciences | 63787 | |
| 2-Mercaptoethanol (BME) | Sigma Aldrich | M-6250 | |
| 24x60 mm #1.5 coverslips | Electron Microscopy Sciences | 63793 | |
| 405/488/560/647 nm Laser Quad Band | Chroma | TRF89901-NK | |
| Acetone | Sigma Aldrich | 320110 | |
| Adenosine 5'-triphosphate disodium salt hydrate (ATP) | Sigma Aldrich | A7699-5G | |
| Avidin, NeutrAvidin® Biotin-binding Protein (Molecular Probes®) | Thermo Fischer Scientific | A2666 | |
| Bath sonicator: Branson 2800 Cleaner | Branson | CPX2800H | |
| Beckman Coulter Polycarbonate Thickwall Tubes, 11 x 34 mm | Beckman-Coulter | 343778 | |
| Beckman Coulter Polycarbonate Thickwall Tubes, 8 x 34 mm | Beckman-Coulter | 343776 | |
| Biotin-PEG-SVA, MW 5,000 | Laysan Bio | #Biotin-PEG-SVA-5000 | |
| Bovine Serum Albumin (BSA) | Sigma Aldrich | 2905 | |
| Catalase | Sigma Aldrich | C40 | |
| Corning LSE Mini Microcentrifuge, AC100-240V | Corning | 6670 | |
| Delicate Task Wipes | Kimtech | 34120 | |
| Dithiothreitol (DTT) | GoldBio | DTT10 | |
| Emission filter | Chroma | ET610/75m | |
| Ethanol (200-proof) | Decon Labs | 2705 | |
| Ethylene glycol tetraacetic acid (EGTA) | Sigma Aldrich | 3777 | |
| Glucose Oxidase | Sigma Aldrich | G2133 | |
| GMPCPP | Jena Bioscience | NU-405 | |
| Guanosine 5'-triphosphate sodium salt hydrate (GTP) | Sigma Aldrich | G8877 | |
| Hellmanex III detergent | Sigma Aldrich | Z805939 | |
| Immersion oil, Type A | Fisher Scientific | 77010 | |
| Kappa-casein | Sigma Aldrich | C0406 | |
| Lanolin | Fisher Scientific | S25376 | |
| Lens Cleaning Tissue | ThorLabs | MC-5 | |
| Magnesium Chloride (MgCl2) | Sigma Aldrich | M9272 | |
| Methylcellulose | Sigma Aldrich | M0512 | |
| Microfuge 16 Benchtop Centrifuge | Beckman-Coulter | A46474 | |
| Microscope Slides, Diamond White Glass, 25 x 75mm, 90° Ground Edges, WHITE Frosted | Globe Scientific | 1380-50W | |
| mPEG-Succinimidyl Valerate, MW 5,000 | Laysan Bio | #NH2-PEG-VA-5K | |
| Optima™ Max-XP Tabletop Ultracentrifuge | Beckman-Coulter | 393315 | |
| Paraffin | Fisher Scientific | P31-500 | |
| PELCO Reverse (self-closing), Fine Tweezers | Ted Pella | 5377-NM | |
| Petrolatum, White | Fisher Scientific | 18-605-050 | |
| Plasma Cleaner, 115V | Harrick Plasma | PDC-001 | |
| Potassium Hydroxide (KOH) | Sigma Aldrich | 221473 | |
| Sodium bicarbonate | Sigma Aldrich | S6014 | |
| Sucrose | Sigma Aldrich | S7903 | |
| Thermal-Lok 1-Position Dry Heat Bath | USA Scientific | 2510-1101 | |
| Thermal-Lok Block for 1.5 and 2.0 mL Tubes | USA Scientific | 2520-0000 | |
| Thermo Scientific™ Pierce™ Bond-Breaker™ TCEP Solution, Neutral pH; 500mM | Thermo Fischer Scientific | PI-77720 | |
| TIRF 100X NA 1.49 Oil Objective | Nikon | CFI Apochromat TIRF 100XC Oil | |
| TIRF microscope | Nikon | Eclipse Ti | |
| TLA 120.1 rotor | Beckman-Coulter | 362224 | |
| TLA 120.2 rotor | Beckman-Coulter | 357656 | |
| Tubulin protein (>99% pure): porcine brain | Cytoskeleton | T240 | |
| Tubulin Protein (Biotin): Porcine Brain | Cytoskeleton | T333P | |
| Tubulin protein (fluorescent HiLyte 647): porcine brain | Cytoskeleton | TL670M | |
| Tubulin protein (X-rhodamine): bovine brain | Cytoskeleton | TL620M | |
| VECTABOND® Reagent, Tissue Section Adhesion | Vector Biolabs | SP-1800-7 | |
| VWR® Personal-Sized Incubator, 120V, 50/60Hz, 0.6A | VWR | 97025-630 |
Références
- Subramanian, R., Kapoor, T. M. Building complexity: insights into self-organized assembly of microtubule-based architectures. Developmental Cell. 23 (5), 874-885 (2012).
- Baas, P. W., Rao, A. N., Matamoros, A. J., Leo, L. Stability properties of neuronal microtubules. Cytoskeleton (Hoboken). 73 (9), 442-460 (2016).
- Bitan, A., Rosenbaum, I., Abdu, U. Stable and dynamic microtubules coordinately determine and maintain Drosophila bristle shape. Development. 139 (11), 1987-1996 (2012).
- Foe, V. E., von Dassow, G. Stable and dynamic microtubules coordinately shape the myosin activation zone during cytokinetic furrow formation. The Journal of Cell Biology. 183 (3), 457-470 (2008).
- Pous, C., et al. Functional specialization of stable and dynamic microtubules in protein traffic in WIF-B cells. The Journal of Cell Biology. 142 (1), 153-165 (1998).
- Uehara, R., Goshima, G. Functional central spindle assembly requires de novo microtubule generation in the interchromosomal region during anaphase. The Journal of Cell Biology. 191 (2), 259-267 (2010).
- Mitchison, T., Kirschner, M. Dynamic instability of microtubule growth. Nature. 312 (5991), 237-242 (1984).
- Bieling, P., et al. Reconstitution of a microtubule plus-end tracking system in vitro. Nature. 450 (7172), 1100-1105 (2007).
- Bieling, P., Telley, I. A., Surrey, T. A minimal midzone protein module controls formation and length of antiparallel microtubule overlaps. Cell. 142 (3), 420-432 (2010).
- Shimamoto, Y., Forth, S., Kapoor, T. M. Measuring pushing and braking forces generated by ensembles of Kinesin-5 crosslinking two microtubules. Developmental Cell. 34 (6), 669-681 (2015).
- Mani, N., Jiang, S., Neary, A. E., Wijeratne, S. S., Subramanian, R. Differential regulation of single microtubules and bundles by a three-protein module. Nature Chemical Biology. 17 (9), 964-974 (2021).
- Hyman, A. A., Salser, S., Drechsel, D., Unwin, N., Mitchison, T. J. Role of GTP hydrolysis in microtubule dynamics: information from a slowly hydrolyzable analogue, GMPCPP. Molecular Biology of the Cell. 3 (10), 1155-1167 (1992).
- Subramanian, R., et al. Insights into antiparallel microtubule crosslinking by PRC1, a conserved nonmotor microtubule binding protein. Cell. 142 (3), 433-443 (2010).
- Rasnik, I., McKinney, S. A., Ha, T. Nonblinking and long-lasting single-molecule fluorescence imaging. Nature Methods. 3 (11), 891-893 (2006).
- Wijeratne, S., Subramanian, R. Geometry of antiparallel microtubule bundles regulates relative sliding and stalling by PRC1 and Kif4A. eLife. 7, 32595(2018).
- Mani, N., Wijeratne, S. S., Subramanian, R. Micron-scale geometrical features of microtubules as regulators of microtubule organization. eLife. 10, 63880(2021).
- Freal, A., et al. Feedback-driven assembly of the axon initial segment. Neuron. 104 (2), 305-321 (2019).
- Ledbetter, M., Porter, K. A "microtubule" in plant cell fine structure. The Journal of Cell Biology. 19 (1), 239-250 (1963).
- Wijeratne, S. S., Marchan, M. F., Tresback, J. S., Subramanian, R. Atomic force microscopy reveals distinct protofilament-scale structural dynamics in depolymerizing microtubule arrays. Proceedings of the National Academy of Sciences of the United States of America. , 119(2022).
Access restricted. Please log in or start a trial to view this content.
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.