
このコンテンツを視聴するには、JoVE 購読が必要です。 サインイン又は無料トライアルを申し込む。
Method Article
TIRF顕微鏡による インビトロでの 架橋小管と単一微小管のダイナミクスの同時可視化
要約
ここでは、TIRF顕微鏡ベースの インビトロ 再構成アッセイを提示し、2つの微小管集団のダイナミクスを同時に定量および比較する。架橋された微小管束および単一の微小管上の複数の微小管関連タンパク質の集合的活性を同時に見る方法が記載されている。
要約
微小管は、細胞内で別個の構造に組織化するαβ-チューブリンヘテロ二量体のポリマーである。微小管ベースのアーキテクチャとネットワークには、多くの場合、動的特性が異なる微小管アレイのサブセットが含まれています。例えば、細胞を分裂させる際に、架橋微小管の安定な束は、動的非架橋微小管のすぐ近くに共存する。TIRF顕微鏡ベースの インビトロ 再構成研究は、これらの異なる微小管アレイのダイナミクスの同時可視化を可能にする。このアッセイでは、イメージングチャンバは、単一のフィラメントとして存在するか、または架橋束に組織化された表面固定化微小管で組み立てられる。チューブリン、ヌクレオチド、およびタンパク質調節剤の導入により、関連するタンパク質の直接可視化、および単一および架橋微小管の動的特性の直接可視化が可能になります。さらに、動的な単一の微小管がバンドルに編成されるときに発生する変化をリアルタイムで監視することができます。ここで説明する方法は、個々のタンパク質の活性および局在化の体系的な評価、ならびに同一の実験条件下での2つの異なる微小管サブセットに対するタンパク質調節因子の相乗効果を可能にし、それによって他の方法ではアクセスできない機構的洞察を提供する。
概要
微小管は、細胞内輸送および細胞小器官の位置決めから細胞分裂および伸長に至るまで、複数の細胞プロセスに不可欠な構造足場を形成する生体高分子である。これらの多様な機能を実行するために、個々の微小管は、有糸分裂紡錘体、毛様体軸索、ニューロンバンドル、相間配列、および植物皮質配列などのミクロンサイズの配列に編成される。これらの構造に見られる普遍的な建築モチーフは、その長さに沿って架橋された微小管の束です1。いくつかの微小管ベースの構造の興味深い特徴は、束ねられた微小管と非架橋された単一の微小管が空間的に近接して共存することである。これらの微小管亜集団は、その適切な機能のために必要なように、互いに著しく異なる重合ダイナミクスを示すことができる2,3,4,5。例えば、有糸分裂紡錘体内では、安定な架橋束および動的単一微小管が、細胞中心のミクロンスケール領域内に存在する6。したがって、共存する微小管集団の動的特性がどのように特定されるかを研究することは、微小管ベースの構造の組み立てと機能を理解する上で中心的である。
微小管は、重合と解重合の相を循環する動的なポリマーであり、大惨事と救助として知られるイベントで2つの相を切り替えます7。細胞微小管のダイナミクスは、微小管の重合および解重合の速度、ならびに大惨事および救助事象の頻度を調節する無数の微小管関連タンパク質(MAP)によって調節される。細胞内の空間的に近位アレイ上のMAPの活性を調べることは、光学顕微鏡、特に微小管密度の高い領域における空間分解能の限界のために困難である。さらに、同じ細胞領域に複数のMAPが存在することは、細胞生物学的研究の解釈を妨げる。全内部反射蛍光(TIRF)顕微鏡法と組み合わせて実施されるインビトロ再構成アッセイは、MAPの特定のサブセットが近位細胞微小管アレイのダイナミクスを調節するメカニズムを調べるという課題を回避します。ここで、インビトロで組み立てられた微小管の動態は、制御された条件下で1つ以上の組換えMAPの存在下で調べられる8、9、10。 しかしながら、従来の再構成アッセイは、典型的には、単一の微小管または1つのタイプのアレイ上で行われ、共存する集団の可視化を妨げる。
ここでは、同じ溶液条件下で2つの微小管集団を同時に可視化することを可能にするin vitro 再構成アッセイを紹介します11。我々は、単一の微小管および有糸分裂紡錘体関連タンパク質PRC1によって架橋された微小管束上の複数のMAPの集合活性を同時に見る方法を説明する。タンパク質PRC1は、抗平行微小管間の重なりで優先的に結合し、それらを架橋する9。簡単に言えば、このプロトコルは、(i)原液および試薬の調製、(ii)顕微鏡実験用のイメージングチャンバの作成に使用されるカバースリップの洗浄および表面処理、(iii)実験中に重合が開始される安定した微小管「種子」の調製、(iv)微小管ダイナミクスを視覚化するためのTIRF顕微鏡設定の仕様、(v)微小管種子の固定化および架橋微小管束の生成 画像化チャンバ内において、(vi)TIRF顕微鏡による画像化チャンバ内の微小管ダイナミクスの可視化、可溶性チューブリン、MAP、およびヌクレオチドの添加時。これらのアッセイは、MAP局在化および2つの微小管集団の動態に対するそれらの影響の定性的評価および定量的検査を可能にする。さらに、これらの微小管集団に対する複数のMAPの相乗効果の評価を、幅広い実験条件にわたって容易にする。
プロトコル
1. 試薬の準備
- 表 1 および表 2 に概説されているようにバッファーおよび試薬を調製します。実験中は、特に断りのない限り、すべての溶液を氷上に保管してください。
| 解決 | コンポーネント | 推奨保存期間 | 筆記 | ||
| 5X BRB80 | 400 mM Kパイプ、5 mM MgCl2、5 mM EGTA、KOH付きpH 6.8、フィルター滅菌 | 最長2年間 | 4°Cで保存 | ||
| 1X BRB80 | 80 mM Kパイプ, 1 mM MgCl2, 1 mM EGTA, pH 6.8 | 最長2年間 | 4°Cで保存 | ||
| BRB80-DTT | 1X BRB80, 1 mM DTT | 最長 2 日 | |||
| アッセイバッファー | 80 mM Kパイプ、3 mM MgCl2、1 mM EGTA、pH 6.8、5% スクロース (または 1X BRB80、5% スクロース、2 mM MgCl2) | 最長1年間 | 4°Cで保存 | ||
| マスター バッファー (MB) | アッセイバッファー、5mM TCEP | 1週間 | 実験当日に準備する;2つのチューブに分ける:室温でMB-暖かく、氷上でMB-冷たい。蛍光色素を使用する場合は1 mM DTTを含む | ||
| メチルセルロース(MBMC)を含むマスターバッファー | 1X BRB80, 0.8% メチルセルロース, 5 mM TCEP, 5 mM MgCl2 | 1週間 | 実験当日に準備する;蛍光色素を使用する場合は1 mM DTTを含む | ||
| タンパク質希釈バッファー(DB) | MB, 1 mg/mL ウシ血清アルブミン (BSA), 1 μM ATP | 1日、氷上 | 実験当日に準備する;蛍光色素を使用する場合は1 mM DTTを含む | ||
| 脱酸素ミックス(OSM) | MB, 389 μg/mL カタラーゼ, 4.44 mg/mL グルコースオキシダーゼ, 15.9 mM 2-メルカプトエタノール (BME) | 1日、氷上 | 実験当日の準備 | ||
| 脱酸素ファイナル(OSF) | MB, 350 μg/mL カタラーゼ, 4mg/mL グルコースオキシダーゼ, 14.3 mM BME, 15 mg/mL グルコース | 30分以内に使用 | 使用直前に9 μLのOSMに1 μLのグルコースを加えて調製する | ||
表 1: このプロトコルで使用されるバッファとそのコンポーネントの一覧 各バッファーをどの程度前に準備できるかについてのガイダンスについては、「推奨ストレージ期間」列を参照してください。
| 試薬 | 貯蔵濃度 | 保存溶剤 | 保管温度 | 作業集中力 | 最終濃度 | 推奨保存期間 | 筆記 | |||||||||
| ノイトラアビジン(NA) | 5ミリグラム/ミリリットル | 1X BRB80 | -80°C | 0.2 ミリグラム/ミリリットル | 0.2 ミリグラム/ミリリットル | 最長1年間 | ビオチン - ニュートラアビジン - ビオチン結合を介して微小管を固定化するために使用される。小さなアリコートに保存する | |||||||||
| カッパカゼイン (KC) | 5ミリグラム/ミリリットル | 1X BRB80 | -80°C | 0.5 ミリグラム/mL | 0.5 ミリグラム/mL | 最長2年間 | イメージングチャンバ表面を遮断するために使用される。小さなアリコートに保管する。実験当日は、少量を室温で脇に置いておく | |||||||||
| ウシ血清アルブミン(BSA) | 50 ミリグラム/ミリリットル | 1X BRB80 | -20°C | 1 ミリグラム/mL (単位: DB) | 該当なし | 最長2年間 | 小さなアリコートに保存する | |||||||||
| カタラーゼ | 3.5 ミリグラム/ミリリットル | 1X BRB80 | -80°C | 350 μg/mL (標準解像度: OSF) | 35 μg/mL | 最長2年間 | 脱酸素ミックスの成分;小さなアリコートに保存する | |||||||||
| グルコースオキシダーゼ | 40 ミリグラム/mL | 1X BRB80 | -80°C | 4 mg/mL (OSF 単位) | 0.4 ミリグラム/mL | 最長2年間 | 脱酸素ミックスの成分;小さなアリコートに保存する | |||||||||
| チューブリン | 凍結 乾燥 | 該当なし | 4°C | 10 ミリグラム/ミリリットル | 2.12 mg/mL (チューブリンミックス中) | 最長1年間 | チューブリンが溶液に入ったら、重合を避けるために冷たく保ちます。 | |||||||||
| アデノシン三リン酸(ATP) | 100ミリアンペア月間 | 純水 | -20°C | 10ミリオンメートル | 1 ミリオン | 6ヶ月 | フィルター滅菌水で溶液を調製し、pHを〜7.0に調整し、小さなアリコートで凍結する。 | |||||||||
| グアノシン三リン酸(GTP) | 100ミリアンペア月間 | 純水 | -20°C | 10ミリオンメートル | 1.29 mM (チューブリンミックス中) | 6ヶ月 | フィルター滅菌水で溶液を調製し、pHを〜7.0に調整し、小さなアリコートで凍結する。 | |||||||||
| グアノシン-5'-[(α,β)-メチルエノ]三リン酸(GMPCPP) | 10ミリオンメートル | 純水 | -20°C | 10 μM | 0.5 μM | 6ヶ月 | ||||||||||
| ジチオスレイトール(DTT) | 1 M | 滅菌水 | -20°C | 1 ミリオン | 該当なし | 最長2年間 | ||||||||||
| トリス(2-カルボキシエチル)ホスフィン(TCEP) | 0.5メートル | フィルター滅菌水 | 室温 | 5ミリオン | 該当なし | 最長2年間 | ||||||||||
| メチルセルロース | 1% | 滅菌水 | 室温 | 0.8% (単位: MBMC) | 0.21% (チューブリンミックス中) | 最長1年間 | メチルセルロースを沸騰に近い水にゆっくりと加えて溶解する。連続的に攪拌しながら冷却する。 | |||||||||
| β-メルカプトエタノール(BME) | 143ミリアンペア月間 | 滅菌水 | 室温 | 14.3 mM (OSF 単位) | 1.43 ミリアン ペア月間 | 最長5年間 | 143 mM は、ストック BME の 1:100 希釈です。 | |||||||||
| グルコース | 150 ミリグラム/ミリリットル | 1X BRB80 | -80°C | 15 mg/mL (OSF 単位) | 1.5 ミリグラム/mL | 最長2年間 | 使用直前にOSMに追加する | |||||||||
| (±)-6-ヒドロキシ-2,5,7,8-テトラメチルクロマン-2-カルボン酸(トロロックス) | 10ミリオンメートル | 1X BRB80 | -80°C | 10ミリオンメートル | 1 ミリオン | 最長1年間 | 完全に溶解しない。NaOHを少し加え、約4時間攪拌し、使用前にろ過滅菌する | |||||||||
| mPEG-吉草酸スクシンイミジル、MW 5,000 | 粉 | 該当なし | -20°C | 333 ミリグラム/ミリリットル (0.1 M炭酸水素ナトリウム中) | 324 ミリグラム/ミリリットル (0.1 M炭酸水素ナトリウム中) | 6ヶ月 | 〜34mgのアリコートを準備し、各チューブに正確な量の粉末をマーキングする。固体の上に窒素ガスを通し、パラフィルムでチューブを密封し、乾燥剤の入った容器に-20°Cで保存します。 | |||||||||
| ビオチン-ペグ-SVA、MW 5,000 | 粉 | 該当なし | -20°C | 111 ミリグラム/ミリリットル (0.1 M炭酸水素ナトリウム中) | 3.24 mg/mL (0.1 M 炭酸水素ナトリウム中) | 6ヶ月 | 〜3mgのアリコートを準備し、各チューブに正確な量の粉末をマーキングする。固体の上に窒素ガスを通し、パラフィルムでチューブを密封し、乾燥剤の入った容器に-20°Cで保存します。 | |||||||||
表2:このプロトコルで使用される試薬のリスト。 推奨される保存条件と濃度、実験中に使用した原液の作業濃度、およびイメージングチャンバ内の最終濃度が含まれます。右端の列に追加のメモがあります。
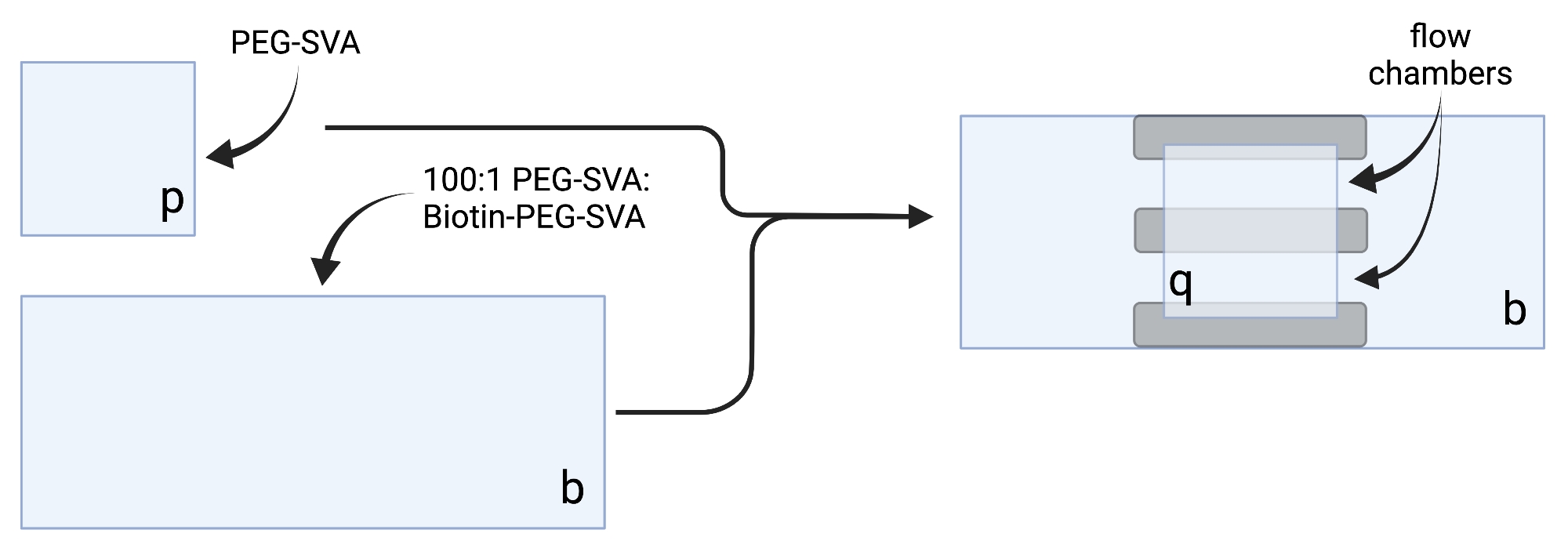
2. ビオチン-PEGスライドの準備
注:イメージングチャンバーは、実験開始のできるだけ近く、2週間以内に準備してください。
- 清潔なカバースリップ
- 同じ数の24 x 60 mmと18 x 18 mm #1.5カバースリップ(図1F)を、それぞれスライド染色ジャーとスライド洗浄ラックに置きます(図1A、B)。18 x 18 mm のカバースリップが入ったスライド洗浄ラックを 100 mL ビーカーに入れます。
- すべてのカバースリップを超純水(抵抗率18.2MΩ-cm)で5~6回すすぎ、真空管に取り付けられたピペットチップで各リンス後に余分な液体を除去します(図1C)。
- カバースリップを含むビーカーとスライド染色瓶に超純水を入れ、パラフィルムで密封し、10分間超音波処理します。
- 2つの150mLビーカーに200プルーフエタノールを充填する。ピンセットを使用して、各カバースリップをエタノールで満たされた1つのビーカーに浸し、次にもう1つのビーカーに浸します。
- ピンセットを使用して、カバースリップをスライド乾燥ラックに移し(図1D)、窒素ガス気流下で乾燥させ、完全に乾燥するまで37°Cでインキュベートします(約15分)。
- 乾燥したカバースリップをプラズマクリーナー内の1つの層に置きます。真空シールを形成し、プラズマクリーナーの無線周波数(RF)レベルを〜8MHzに設定します。
- プラズマが生成されたら、カバースリップをプラズマクリーナーに5分間放置します。プラズマクリーナーの電源を切り、ゆっくりと真空を解除します。
- 真空シールが解除されたら、カバースリップを裏返し、カバースリップの反対側でプラズマ洗浄を5分間繰り返します。
- プラズマ洗浄の代替:ステップ2.1.2-2.1.3の代わりに、超音波処理は、2%洗剤の暖かい溶液(超純水中)中の10分間カバースリップを覆います。その後、カバースリップを超純水で十分に洗浄し、超純水中で超音波処理を2~3回(各10分)行う。次に、エタノールで洗浄し、ステップ2.1.4〜2.1.5のように乾燥させる。手順 2.1.6-2.1.8 をスキップします。
- ビオチン-PEG処理
- 使用直前に、400μLの3-アミノプロピルトリエトキシシランを40mLのアセトンに溶解する。ピンセットを使用して、プラズマ洗浄したカバースリップをスライド洗浄ラックとスライド染色ジャーに入れます。カバースリップを3-アミノプロピルトリエトキシシラン溶液に沈め、5分間インキュベートする12,13。
- すべてのカバースリップを超純水で5〜6回洗ってください。
- カバースリップをスライド式乾燥ラックに移し、窒素ガス気流下で乾燥させ、完全に乾燥するまで37°Cでインキュベートします(〜20分)。
- 乾燥したカバースリップを繊細な作業用ワイプの上に置き、各カバースリップに1つの角にラベルを付けます(例:各18 x 18 mmカバースリップに「p」、各24 x 60 mmカバースリップに「b」)。
- 実験当日、0.84 mgのNaHCO3を10 mLの超純水に溶解して新鮮な0.1 M炭酸水素ナトリウム溶液を調製した。
- 使用直前に、mPEG-吉草酸スクシンイミジル(PEG-SVA)およびビオチン-PEG-SVAのアリコートを室温に持参してください。 表2のポリエチレングリコール(PEG)アリコート調製に関する注記を参照してください。
注:吉草酸スクシンイミジル(SVA)部分の加水分解半減期は約30分であるため、迅速に作業できます。 - 102 μL の 0.1 M NaHCO3 を 34 mg の PEG-SVA に加え、ベンチトップ型微量遠心分離機で 2,656 x g で 20 秒間スピンした後、上下にピペッティングして混合します。ビオチン-PEG-SVA3 mgを27 μLの0.1 M NaHCO3に上下にピペッティングして溶解する。チューブに記されたPEGの正確な重量に従って希釈量を調整する(表2参照)。
- カバースリップ 20 枚に対して 75 μL の PEG-SVA 溶液と 2.25 μL のビオチン-PEG-SVA 溶液を組み合わせて、100:1 w/w の PEG:ビオチン-PEG 混合物を調製し、カバースリップ 30 枚に対して 100 μL および 3 μL、カバースリップ 40 枚に対して 125 μL および 3.75 μL を調製します。
- 空の10μLチップボックスの底にあるチップラックの下に濡れたペーパータオルを置いて、水分補給チャンバーを構築します(図1E)。これにより、PEG溶液の蒸発が防止されます。
- 100:1 PEG-SVA:ビオチン-PEG混合物6 μLを、標識側の24 x 60 mmカバースリップの中央にピペット6 μL。最初のカバースリップの上に別の24 x 60 mmのカバースリップを置き、ペアがX字型を形成し、「b」とラベル付けされた辺が互いに向くようにします。ペアを水和チャンバー内の空のチップラックに置き、残りの24 x 60 mmカバースリップについて繰り返します。
- 標識側の18 x 18 mmカバースリップの中央に6 μLのPEG-SVAをピペットで貼り付けます。最初のカバースリップの上に別の 18 x 18 mm のカバースリップを置き、「p」とラベル付けされた辺を互いに向かわせます。ペアを水和チャンバー内の空のチップラックに置き、残りの18 x 18 mmカバースリップについて繰り返します。
- 水和チャンバーを閉じ、3時間または一晩インキュベートする。
- カバースリップのペアを分離し、超純水ですすいでください。
- カバースリップを窒素気流で乾燥させ、37°Cのインキュベーターに入れて完全に乾燥させます。
- イメージングチャンバを構成するには、「b」とラベル付けされた側面の24 x 60 mmカバースリップに両面テープを3枚貼り付けます。テープストリップの反対側に、18 x 18 mmのカバースリップを取り付け、その側面に「p」とラベルが付いたものを大きなカバースリップに向けます。これにより、顕微鏡実験用の2つのフローチャンバが形成され、処理された表面が互いに対向します(図2 および 図1G)。

図1:カバースリップ処理およびイメージングチャンバの準備のための装置。 (A)24 x 60 mmカバースリップ用のスライド染色ジャー、(B)18 x 18 mmカバースリップ用のスライド洗浄ラック、(C)真空セットアップ、(D)スライド乾燥ラック、(E)水和チャンバ、(F)カバースリップ、(G)イメージングチャンバ、(H)スライドホルダー。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図2:両面テープ(灰色)およびPEG/ビオチン-PEG処理カバースリップを使用したイメージングチャンバの調製のための概略図。 BioRender.com で作成されました。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
3. 微小管の重合
- GMPCPP種子を準備する
注:GMPCPP種子は、すべての試薬、チップ、チューブを4°Cに保ちながら、冷蔵室で調製してください。1mL遠沈管を入れた固定角超遠心ローターを、超遠心機に入れ、温度を4°Cに設定した。- 凍結乾燥チューブリン(表2)を1X BRB80に約10mg/mLに再懸濁し、使用直前に再懸濁する。
- GMPCPP種子の成分を 表3に記載したように混合する。
注:可溶性チューブリンの重合を最小限に抑えるために、すべてのチューブリン成分をできるだけ氷上に保管してください。 - 352,700 x g の固定角度超遠心ローターで4°Cで5分間混合を明確にします。
- 上清を5μLのアリコートに分離し、液体窒素で急速凍結し、-80°Cで保存する。
- 実験当日に種子を重合する
- 1-2 mLのBRB80-DTT(表1)を37°Cに温める。
- ステップ3.1.4のGMPCPP種子(-80°C)の5μLアリコートを氷上に置き、直ちに20μLの温かいBRB80-DTTに溶解する。室温で2,000 x g で5秒間スピンし、タップして混合します。
注:初期希釈量は13 μL~21 μLの間で変化し、種子の各バッチについて経験的に決定される。種子が重合しない場合は、初期希釈バッファー(ステップ3.2.2)に0.5μM GMPCPPを補充してトラブルシューティングを行います。 - 光から保護し、37°Cで30〜45分間インキュベートします。
注:微小管の長さは、インキュベーションの期間に依存する。短い微小管の場合、インキュベーション時間は15分と短くすることができます。長い微小管の場合、インキュベーション時間は2時間にもなります。ビオチン化微小管は、非ビオチン化微小管よりも長いインキュベーション時間を必要とする傾向がある。 - 500 μLの遠沈管を入れた固定角度超遠心ローターを超遠心機に入れ、30°Cに予備加温した。
- インキュベーション後、重合したGMPCPP種子に50 μLの温かいBRB80-DTT(ステップ3.2.1)を加え、混合物を500 μLの遠沈管に移します。GMPCPP種子を含む空のチューブを、別の50μLの温かいBRB80-DTTで洗浄し、ピペットを上下にピペットし、この緩衝液を混合物を含む500μL遠沈管に加える。
- 回転する前に、遠心管の縁に印を付け、ペレットがどこにあるかを示します(ペレットが小さすぎて見えません)。30°Cで244,900 x g で10分間スピン12。
- 上澄み液を慎重にピペットで取り出し、廃棄する。ペレットを100μLの温かいBRB80-DTTに再懸濁する。タップしてミックスします。
- 30°Cで244,900 x g で10分間スピンし、同じ場所にペレットにローターでマーキングを合わせた。
- 上清を除去し、ペレットを16μLの温かいBRB80-DTTに再懸濁する。微小管溶液を清潔な0.6mL微量遠心管に移す。光から保護し、室温以上に保ちます。
注:重合後、微小管を室温以上に保ってください。寒くなると解重合します。添加安定性のために28°Cでインキュベートする。
- TIRF顕微鏡 による 微小管の検査
- 4.5 μLのBRB80-DTTと1 μLの微小管溶液(ステップ3.2.9)の混合物を顕微鏡スライド上にピペットで打ちます。18 x 18 mmのカバースリップで覆い、透明なマニキュアまたはワセリン、ラノリン、パラフィン(バラップシーラント)の1:1:1混合物のいずれかでエッジを密封します。
- TIRF対物レンズをカバースリップの下に置き(推奨される顕微鏡設定についてはステップ4を参照)、Brightミックス中の蛍光標識されたチューブリンに適した波長で新たに重合された微小管を視覚化し(表3)、今後の実験で使用する微小管の希釈を決定します。
| 試薬 | ブライトミックス(μL) | 追加順序 | ブライトミックス+ビオチン(μL) | 追加順序 |
| 蛍光チューブリン, 10 ミリグラム/mL | 2 | 6 | 2 | 7 |
| ビオチン - チューブリン、10 mg / mL | 0 | 該当なし | 2 | 6 |
| ラベルなしチューブリン、10 mg/mL | 20 | 5 | 18 | 5 |
| GMPCPP, 10 mM | 30 | 4 | 30 | 4 |
| DTT、0.2 M | 0.7 | 3 | 0.7 | 3 |
| 5X BRB80 | 26.4 | 2 | 26.4 | 2 |
| 滅菌水 | 52.9 | 1 | 52.9 | 1 |
| 総容量(μL) | 132 | 132 |
表3:GMPCPP種子ミックス。 GMPCPP微小管種子の成分は、体積および添加順序を含む。5 μLのアリコートを調製し、-80°Cで最大1年間保存する。
4. 顕微鏡の設定
- 温度: 顕微鏡の温度を 28 °C に設定して、動的な微小管を表示します。
- フィルター: フィルターキューブと発光フィルターの最適な組み合わせを、画像化する蛍光チャンネルに応じて使用します。同じ実験で 488 nm、560 nm、および 647 nm の波長を視覚化するには、405/488/560/647 nm レーザー クワッド バンド セットと、指定された波長の発光フィルターを組み合わせて使用します。
- レーザーを整列させる: 実験で使用したレーザービームが整列していることを確認します。実験のレーザー強度を経験的に決定し、すべての蛍光タンパク質を可能な限り高いシグナル対ノイズ比で画像化できるが、実験の経時的経過にわたって有意なフォトブリーチングを受けないようにします。
- 目的:レンズペーパーを使用して、70%エタノールで100倍の対物レンズを清掃します。イメージングの前に、顕微鏡浸漬オイルを対物レンズに一滴加えます。
- イメージングシーケンスの設定
- 647 nm蛍光色素標識ビオチン化微小管、560 nm蛍光色素標識非ビオチン化微小管および可溶性チューブリン、および目的のGFP標識タンパク質を用いた実験について、20分間画像化。560 nm チャンネルと 488 nm チャンネルを 10 秒ごとに、647 nm チャンネルを 30 秒ごとに画像化します。
- 可溶性チューブリンおよびMAPを添加する前のバンドルの参照画像をキャプチャするには、560nmおよび647nmの波長でそれぞれ1つの画像を含むシーケンスを設定します。
5. 表面固定化微小管束の生成
注:次の手順では、すべての溶液を一方の開いた側にピペッティングしてフローチャンバに流し、もう一方の側にろ紙を置きます。イメージングチャンバを光から保護し、蛍光標識タンパク質のフォトブリーチングを低減します。作製した撮像チャンバをスライドホルダーにテープで固定する(図1G、H)。 表 4 の手順 (プロトコル steps 5.2-6.4 に対応します)。
- 表5に従って可溶性チューブリンミックスを調製し、氷上に保管する。
注:可溶性チューブリンは、重合を防ぐために常に氷の上に置かれなければならない。新鮮な可溶性チューブリンミックスを約2時間ごと、または微小管が重合しなくなったときに調製する。 - ビオチン-ニュートラアビジン-ビオチン結合 を介して 微小管を固定化するには、まずチャンバーが満たされるまでニュートラアビジン(NA)溶液を流し(約7.5 μL)、5分間インキュベートします。
- 10 μLのMBコールドで洗浄する。
- ブロッキングタンパク質κカゼイン(KC)を7.5 μLに流し、2分間インキュベートする。
- 10μLのMB-warmで洗浄し、微小管の導入のためのチャンバーを調製する。
- ビオチン化微小管のストックを(ステップ3.3.2の観察によると)1X BRB80-DTTで希釈し、この希釈液の1 μLを9 μLのMBウォームに加える。混合物をチャンバーに流し、10分間インキュベートする。より多くのバンドルのために、より高濃度の微小管を使用する。
- 非固定化微小管を10 μLのMB温水で洗い流す。
- 7.5 μLの温かいKCをチャンバーに流し、2分間インキュベートする。
- インキュベーション中に、架橋剤タンパク質PRC1の2 nM溶液を温かいKC中に調製し、この溶液10 μLをフローチャンバーに流し、5分間インキュベートする。
注:組換えPRC1は、前述のように細菌細胞から発現および精製される13。 - バンドルを作るには、10 μLの非ビオチン化微小管をチャンバーに流し込み、10分間インキュベートします。PRC1は、非ビオチン化および固定化ビオチン化微小管を架橋します15,16(図3)。
注:インキュベーション時間中にアッセイミックスを調製するためのステップ6.1を参照されたい。 - チャンバーを10μLのMBウォームで2回洗浄する。付着した微小管は、この時点から約20分間安定している。
| 歩 | 試薬 | 容量 (μL) | インキュベーション時間(分) |
| 1 | ノイトラアビジン | 7.5 | 5 |
| 2 | MBコールド | 10 | - |
| 3 | κ-カゼイン | 7.5 | 2 |
| 4 | MBウォーム | 10 | - |
| 5 | ビオチン化微小管(MB温で希釈) | 10 | 10 |
| 6 | MBウォーム | 10 | - |
| 7 | 暖かいκカゼイン | 7.5 | 2 |
| 8 | κ-カゼインで希釈した2nM PRC1 | 10 | 5 |
| 9 | 非ビオチン化微小管 | 10 | 10 |
| 10 | MB-ウォーム x 2 | 10 | - |
| 11 | アッセイミックス | 10 | - |
| 付着した種子はこの時点で約20分間安定しています | |||
表4:アッセイステップ。 画像化チャンバに添加された試薬のリストは、洗浄(−)またはインキュベーション時間の表示を伴う。
| 試薬 | 容量 (μL) |
| リサイクルチューブリン、10 mg/mL | 10 |
| MB-コールド | 10.3 |
| ティッカー | 13.7 |
| BRB80-DTT | 3.4 |
| GTP、10 mM | 6.7 |
| アトピー、10ミリオン (キネシンを使用する場合) | 6.7 |
| 蛍光標識されたチューブリン、 10 ミリグラム/ミリリットル | 1 (凍結乾燥標識チューブリンを冷蔵BRB80-DTTに再懸濁) |
表5:可溶性チューブリン混合成分。 実験の開始時に混合し、氷の上にとどまります。

図3:蛍光標識されたバンドルおよび単一の微小管を作りそして画像化するためのアッセイ成分の添加の概略図。 ビオチン化種子は青色で示され、非ビオチン化種子および可溶性チューブリンは赤色で示され、PRC1は黒色で、および関心のあるタンパク質はシアンで示されている。図中のステップ番号は、表4のステップ番号に対応する。ステップ9に対応するパネルは、予め形成されたバンドル(左下)を示す。ステップ11は、新たに形成されたバンドル(左上)を示す。BioRender.com で作成されました。この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
6. 画像微小管ダイナミクス
- ステップ5.10の10分間のインキュベーション時間の間に、表6に従って、目的のタンパク質、可溶性チューブリン、ヌクレオチド、脱酸素剤14、および抗酸化剤を含む10 μLのアッセイミックスを調製する。ミックスを氷の上に保管してください。
- 準備したイメージングチャンバをスライドホルダーにテープで固定し、100x TIRF対物レンズにロードします。560 nm および 647 nm チャンネルを使用して、単一の微小管およびバンドルの最適な数と密度を含む視野を見つけます。
注: ビオチン化微小管と非ビオチン化微小管の両方が同じ蛍光色素分子で標識されている場合、蛍光強度のラインスキャン分析により、単一の微小管とバンドルを区別できます。 - 視野が特定されたら、参照画像を撮影します。
- アッセイミックスをイメージングチャンバを乱すことなく慎重に流す。
- チャンバーの開放端をバラップシーラントでシールします。
- ステップ 4.5.1 の説明に従ってイメージングシーケンスを開始します。
| 試薬 | 容量 (μL) |
| 可溶性チューブリンミックス | 4 |
| ティッカー | 1 |
| Trolox(容易にフォトブリーチング蛍光色素分子で標識された微小管を使用する場合) | 1 |
| アトピー、10ミリオン (キネシンを使用する場合) | 1 |
| PRC1(または選択した架橋剤) | 1 |
| 目的のタンパク質 | X |
| MBコールド | 2-X |
表6:アッセイミックス成分。 混合し、イメージングチャンバに流し込み、微小管ダイナミクスを30分以内にイメージングします。
結果
上記の実験は、647 nmフルオロフォア標識ビオチン化微小管、560 nmフルオロフォア標識非ビオチン化微小管、および560 nmフルオロフォア標識可溶性チューブリンミックスを用いて実施した。微小管は、架橋タンパク質PRC1(GFP標識)によって架橋された。表面固定化バンドルおよび単一の微小管が生成された後(ステップ5.11)、イメージングチャンバをTIRF 100X 1.49 NAオイル対物レンズに取り付け、560nm...
ディスカッション
ここで説明する実験は、従来、単一の微小管または1種類のアレイで実施される従来の微小管再構成アッセイの範囲と複雑さを大幅に拡大します。現在のアッセイは、2つの集団、すなわち単一の微小管および架橋束に対する調節MAP活性を同時に定量および比較する方法を提供する。さらに、このアッセイでは、ダイナミクスの開始前に安定な種子から予め形成されたバンドルと、2つの成長末?...
開示事項
著者らは、競合する利害関係を宣言していない。
謝辞
この活動は、NIH(no. 1DP2GM126894-01)からの助成金と、ピュー慈善信託とスミスファミリー財団からR.S.への資金によって支援されました。著者らは、プロトコルの開発と最適化に対するShuo Jiang博士の貢献に感謝する。
資料
| Name | Company | Catalog Number | Comments |
| (±)-6-Hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (Trolox) | Sigma Aldrich | 238813 | |
| 1,4-piperazinediethanesulfonic acid (PIPES) | Sigma Aldrich | P6757 | |
| 18x18 mm #1.5 coverslips | Electron Microscopy Sciences | 63787 | |
| 2-Mercaptoethanol (BME) | Sigma Aldrich | M-6250 | |
| 24x60 mm #1.5 coverslips | Electron Microscopy Sciences | 63793 | |
| 405/488/560/647 nm Laser Quad Band | Chroma | TRF89901-NK | |
| Acetone | Sigma Aldrich | 320110 | |
| Adenosine 5'-triphosphate disodium salt hydrate (ATP) | Sigma Aldrich | A7699-5G | |
| Avidin, NeutrAvidin® Biotin-binding Protein (Molecular Probes®) | Thermo Fischer Scientific | A2666 | |
| Bath sonicator: Branson 2800 Cleaner | Branson | CPX2800H | |
| Beckman Coulter Polycarbonate Thickwall Tubes, 11 x 34 mm | Beckman-Coulter | 343778 | |
| Beckman Coulter Polycarbonate Thickwall Tubes, 8 x 34 mm | Beckman-Coulter | 343776 | |
| Biotin-PEG-SVA, MW 5,000 | Laysan Bio | #Biotin-PEG-SVA-5000 | |
| Bovine Serum Albumin (BSA) | Sigma Aldrich | 2905 | |
| Catalase | Sigma Aldrich | C40 | |
| Corning LSE Mini Microcentrifuge, AC100-240V | Corning | 6670 | |
| Delicate Task Wipes | Kimtech | 34120 | |
| Dithiothreitol (DTT) | GoldBio | DTT10 | |
| Emission filter | Chroma | ET610/75m | |
| Ethanol (200-proof) | Decon Labs | 2705 | |
| Ethylene glycol tetraacetic acid (EGTA) | Sigma Aldrich | 3777 | |
| Glucose Oxidase | Sigma Aldrich | G2133 | |
| GMPCPP | Jena Bioscience | NU-405 | |
| Guanosine 5'-triphosphate sodium salt hydrate (GTP) | Sigma Aldrich | G8877 | |
| Hellmanex III detergent | Sigma Aldrich | Z805939 | |
| Immersion oil, Type A | Fisher Scientific | 77010 | |
| Kappa-casein | Sigma Aldrich | C0406 | |
| Lanolin | Fisher Scientific | S25376 | |
| Lens Cleaning Tissue | ThorLabs | MC-5 | |
| Magnesium Chloride (MgCl2) | Sigma Aldrich | M9272 | |
| Methylcellulose | Sigma Aldrich | M0512 | |
| Microfuge 16 Benchtop Centrifuge | Beckman-Coulter | A46474 | |
| Microscope Slides, Diamond White Glass, 25 x 75mm, 90° Ground Edges, WHITE Frosted | Globe Scientific | 1380-50W | |
| mPEG-Succinimidyl Valerate, MW 5,000 | Laysan Bio | #NH2-PEG-VA-5K | |
| Optima™ Max-XP Tabletop Ultracentrifuge | Beckman-Coulter | 393315 | |
| Paraffin | Fisher Scientific | P31-500 | |
| PELCO Reverse (self-closing), Fine Tweezers | Ted Pella | 5377-NM | |
| Petrolatum, White | Fisher Scientific | 18-605-050 | |
| Plasma Cleaner, 115V | Harrick Plasma | PDC-001 | |
| Potassium Hydroxide (KOH) | Sigma Aldrich | 221473 | |
| Sodium bicarbonate | Sigma Aldrich | S6014 | |
| Sucrose | Sigma Aldrich | S7903 | |
| Thermal-Lok 1-Position Dry Heat Bath | USA Scientific | 2510-1101 | |
| Thermal-Lok Block for 1.5 and 2.0 mL Tubes | USA Scientific | 2520-0000 | |
| Thermo Scientific™ Pierce™ Bond-Breaker™ TCEP Solution, Neutral pH; 500mM | Thermo Fischer Scientific | PI-77720 | |
| TIRF 100X NA 1.49 Oil Objective | Nikon | CFI Apochromat TIRF 100XC Oil | |
| TIRF microscope | Nikon | Eclipse Ti | |
| TLA 120.1 rotor | Beckman-Coulter | 362224 | |
| TLA 120.2 rotor | Beckman-Coulter | 357656 | |
| Tubulin protein (>99% pure): porcine brain | Cytoskeleton | T240 | |
| Tubulin Protein (Biotin): Porcine Brain | Cytoskeleton | T333P | |
| Tubulin protein (fluorescent HiLyte 647): porcine brain | Cytoskeleton | TL670M | |
| Tubulin protein (X-rhodamine): bovine brain | Cytoskeleton | TL620M | |
| VECTABOND® Reagent, Tissue Section Adhesion | Vector Biolabs | SP-1800-7 | |
| VWR® Personal-Sized Incubator, 120V, 50/60Hz, 0.6A | VWR | 97025-630 |
参考文献
- Subramanian, R., Kapoor, T. M. Building complexity: insights into self-organized assembly of microtubule-based architectures. Developmental Cell. 23 (5), 874-885 (2012).
- Baas, P. W., Rao, A. N., Matamoros, A. J., Leo, L. Stability properties of neuronal microtubules. Cytoskeleton (Hoboken). 73 (9), 442-460 (2016).
- Bitan, A., Rosenbaum, I., Abdu, U. Stable and dynamic microtubules coordinately determine and maintain Drosophila bristle shape. Development. 139 (11), 1987-1996 (2012).
- Foe, V. E., von Dassow, G. Stable and dynamic microtubules coordinately shape the myosin activation zone during cytokinetic furrow formation. The Journal of Cell Biology. 183 (3), 457-470 (2008).
- Pous, C., et al. Functional specialization of stable and dynamic microtubules in protein traffic in WIF-B cells. The Journal of Cell Biology. 142 (1), 153-165 (1998).
- Uehara, R., Goshima, G. Functional central spindle assembly requires de novo microtubule generation in the interchromosomal region during anaphase. The Journal of Cell Biology. 191 (2), 259-267 (2010).
- Mitchison, T., Kirschner, M. Dynamic instability of microtubule growth. Nature. 312 (5991), 237-242 (1984).
- Bieling, P., et al. Reconstitution of a microtubule plus-end tracking system in vitro. Nature. 450 (7172), 1100-1105 (2007).
- Bieling, P., Telley, I. A., Surrey, T. A minimal midzone protein module controls formation and length of antiparallel microtubule overlaps. Cell. 142 (3), 420-432 (2010).
- Shimamoto, Y., Forth, S., Kapoor, T. M. Measuring pushing and braking forces generated by ensembles of Kinesin-5 crosslinking two microtubules. Developmental Cell. 34 (6), 669-681 (2015).
- Mani, N., Jiang, S., Neary, A. E., Wijeratne, S. S., Subramanian, R. Differential regulation of single microtubules and bundles by a three-protein module. Nature Chemical Biology. 17 (9), 964-974 (2021).
- Hyman, A. A., Salser, S., Drechsel, D., Unwin, N., Mitchison, T. J. Role of GTP hydrolysis in microtubule dynamics: information from a slowly hydrolyzable analogue, GMPCPP. Molecular Biology of the Cell. 3 (10), 1155-1167 (1992).
- Subramanian, R., et al. Insights into antiparallel microtubule crosslinking by PRC1, a conserved nonmotor microtubule binding protein. Cell. 142 (3), 433-443 (2010).
- Rasnik, I., McKinney, S. A., Ha, T. Nonblinking and long-lasting single-molecule fluorescence imaging. Nature Methods. 3 (11), 891-893 (2006).
- Wijeratne, S., Subramanian, R. Geometry of antiparallel microtubule bundles regulates relative sliding and stalling by PRC1 and Kif4A. eLife. 7, 32595 (2018).
- Mani, N., Wijeratne, S. S., Subramanian, R. Micron-scale geometrical features of microtubules as regulators of microtubule organization. eLife. 10, 63880 (2021).
- Freal, A., et al. Feedback-driven assembly of the axon initial segment. Neuron. 104 (2), 305-321 (2019).
- Ledbetter, M., Porter, K. A "microtubule" in plant cell fine structure. The Journal of Cell Biology. 19 (1), 239-250 (1963).
- Wijeratne, S. S., Marchan, M. F., Tresback, J. S., Subramanian, R. Atomic force microscopy reveals distinct protofilament-scale structural dynamics in depolymerizing microtubule arrays. Proceedings of the National Academy of Sciences of the United States of America. , 119 (2022).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved