
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Visualização Simultânea da Dinâmica dos Microtúbulos Cruzados e Únicos In Vitro pela Microscopia TIRF
* Estes autores contribuíram igualmente
Neste Artigo
Resumo
Aqui, um ensaio de reconstituição in vitro baseado em microscopia TIRF é apresentado para quantificar e comparar simultaneamente a dinâmica de duas populações de microtúbulos. Um método é descrito para visualizar simultaneamente a atividade coletiva de múltiplas proteínas associadas a microtúbulos em feixes de microtúbulos interligados e microtúbulos únicos.
Resumo
Microtúbulos são polímeros de heterodimers αβ-tubulin que se organizam em estruturas distintas nas células. Arquiteturas e redes baseadas em microtúbulos geralmente contêm subconjuntos de matrizes de microtúbulos que diferem em suas propriedades dinâmicas. Por exemplo, na divisão de células, pacotes estáveis de microtúbulos transligados coexistem perto de microtúbulos dinâmicos não transligados. Estudos de reconstituição in vitro baseados em TIRF-microscopia permitem a visualização simultânea da dinâmica dessas diferentes matrizes de microtúbulos. Neste ensaio, uma câmara de imagem é montada com microtúbulos imobilizados pela superfície, que estão presentes como filamentos únicos ou organizados em feixes transligados. A introdução de tubulinas, nucleotídeos e reguladores de proteínas permite a visualização direta de proteínas associadas e de propriedades dinâmicas de microtúbulos unides e interligados. Além disso, mudanças que ocorrem à medida que microtúbulos únicos dinâmicos se organizam em pacotes podem ser monitorados em tempo real. O método descrito aqui permite uma avaliação sistemática da atividade e localização de proteínas individuais, bem como efeitos sinérgicos dos reguladores de proteínas em dois subconjuntos microtúbulos diferentes em condições experimentais idênticas, fornecendo assim insights mecanicistas que são inacessíveis por outros métodos.
Introdução
Microtúbulos são biopolímeros que formam andaimes estruturais essenciais para múltiplos processos celulares, desde transporte intracelular e posicionamento de organela até divisão celular e alongamento. Para executar essas diversas funções, microtúbulos individuais são organizados em matrizes do tamanho de micron, como fusos mitotísticos, axônios ciliares, feixes neuronais, matrizes interfásicas e matrizes corticais vegetais. Um motivo arquitetônico onipresente encontrado nessas estruturas é um feixe de microtúbulos interligados ao longo de seus comprimentos1. Uma característica intrigante de várias estruturas baseadas em microtúbulos é a coexistência de microtúbulos empacotados e microtúbulos únicos não-cruzados nas proximidades espaciais próximas. Essas subpopulações de microtúbulos podem exibir dinâmicas de polimerização totalmente diferentes umas das outras, conforme necessário para sua função adequada2,3,4,5. Por exemplo, dentro do eixo mitotístico, pacotes interligados estáveis e microtúbulos únicos dinâmicos estão presentes dentro de uma região de escala de micron no centro celular6. Estudar como as propriedades dinâmicas das populações de microtúbulos coexistindo são especificadas é, portanto, central para entender a montagem e função das estruturas baseadas em microtúbulos.
Microtúbulos são polímeros dinâmicos que pedalam entre fases de polimerização e despolimerização, alternando entre as duas fases em eventos conhecidos como catástrofe e resgate7. A dinâmica dos microtúbulos celulares é regulada por uma miríade de Proteínas Associadas a Microtúbulos (MAPs) que modulam as taxas de polimerização e despomerização de microtúbulos e as frequências de eventos de catástrofe e resgate. É desafiador investigar a atividade de MAPs em matrizes espacialmente proximais nas células, devido às limitações da resolução espacial na microscopia de luz, especialmente em regiões de alta densidade de microtúbulos. Além disso, a presença de múltiplos MAPs na mesma região celular dificulta interpretações de estudos biológicos celulares. Ensaios de reconstituição in vitro, realizados em conjunto com a microscopia de Fluorescência De Reflexão Interna Total (TIRF), contornam os desafios de examinar mecanismos pelos quais subconjuntos específicos de MAPs regulam a dinâmica das matrizes de microtúbulos celulares proximais. Aqui, a dinâmica dos microtúbulos montados in vitro são examinadas na presença de um ou mais MAPs recombinantes em condições controladas8,9,10. No entanto, os ensaios convencionais de reconstituição são tipicamente realizados em microtúbulos únicos ou em um tipo de matriz, impedindo a visualização de populações coexistindo.
Aqui, apresentamos ensaios de reconstituição in vitro que permitem a visualização simultânea de duas populações de microtúbulos sob as mesmas condições de solução11. Descrevemos um método para visualizar simultaneamente a atividade coletiva de múltiplos MAPs em microtúbulos únicos e em feixes de microtúbulos interligados pela proteína mitomática associada ao fuso PRC1. A proteína PRC1 se liga preferencialmente à sobreposição entre microtúbulos anti-paralelos, cruzando-os9. Resumidamente, este protocolo consiste nas seguintes etapas: (i) preparação de soluções de estoque e reagentes, (ii) limpeza e tratamento superficial de deslizamentos de cobertura usados para criar a câmara de imagem para experimentos de microscopia, (iii) preparação de "sementes" de microtúbulos estáveis a partir das quais a polimerização é iniciada durante o experimento, (iv) especificação das configurações do microscópio TIRF para visualizar a dinâmica dos microtúbulos, (v) a imobilização de sementes de microtúbulos e a geração de feixes de microtúbulos transligados na câmara de imagem, e (vi) visualização da dinâmica do microtúbulo na câmara de imagem através da microscopia TIRF, mediante a adição de tubulina solúvel, MAPs e nucleotídeos. Esses ensaios permitem a avaliação qualitativa e o exame quantitativo da localização do MAP e seu efeito na dinâmica de duas populações microtúbulas. Além disso, facilitam a avaliação dos efeitos sinérgicos de múltiplos MAPs nessas populações de microtúbulos, em uma ampla gama de condições experimentais.
Access restricted. Please log in or start a trial to view this content.
Protocolo
1. Prepare os reagentes
- Prepare buffers e reagentes conforme descrito na Tabela 1 e Tabela 2. Durante o experimento, mantenha todas as soluções no gelo, a menos que seja observado de outra forma.
| Solução | Componentes | Duração recomendada do armazenamento | Anotações | ||
| 5X BRB80 | 400 mM K-PIPES, 5 mM MgCl2, 5 mM EGTA, pH 6.8 com KOH, esterilizar filtro | até 2 anos | Armazenar a 4 °C | ||
| 1X BRB80 | 80 mM K-PIPES, 1 mM MgCl2, 1 mM EGTA, pH 6.8 | até 2 anos | Armazenar a 4 °C | ||
| BRB80-DTT | 1X BRB80, 1 mM DTT | até 2 dias | |||
| Tampão de ensaio | 80 mM K-PIPES, 3 mM MgCl2, 1 mM EGTA, pH 6.8, 5% de sacarose (OR 1X BRB80, 5% de sacarose, 2 mM MgCl2) | até 1 ano | Armazenar a 4 °C | ||
| Buffer mestre (MB) | Tampão de ensaio, 5mM TCEP | 1 semana | Prepare-se no dia da experiência; Separe-se em dois tubos: MB-quente à temperatura ambiente e MB-frio no gelo; incluir 1 mM DTT se usar corantes fluorescentes | ||
| Buffer mestre com metilcelulose (MBMC) | 1X BRB80, 0,8% metilcelulose, 5 mM TCEP, 5 mM MgCl2 | 1 semana | Prepare-se no dia da experiência; incluir 1 mM DTT se usar corantes fluorescentes | ||
| Tampão de diluição de proteínas (DB) | MB, 1 mg/mL Bovine Serum Albumin (BSA), 1 μM ATP | 1 dia, no gelo | Prepare-se no dia da experiência; incluir 1 mM DTT se usar corantes fluorescentes | ||
| Mistura de limpeza de oxigênio (OSM) | MB, 389 μg/mL catalase, 4,44 mg/mL de glicose oxidase, 15,9 mM 2-mercaptoethanol (BME) | 1 dia, no gelo | Prepare-se no dia da experiência | ||
| Final de Limpeza de Oxigênio (OSF) | MB, 350 μg/mL catalase, 4mg/mL glicose oxidase, 14,3 mM BME, 15 mg/mL de glicose | usar dentro de 30 min | Prepare-se imediatamente antes do uso adicionando 1 μL de glicose a 9 μL de OSM | ||
Tabela 1: Lista de buffers usados neste protocolo e seus componentes. Consulte a coluna "Duração do armazenamento recomendado" para obter orientações sobre o quão longe cada buffer pode ser preparado.
| Reagente | Concentração de armazenamento | Solvente de armazenamento | Temperatura de armazenamento | Concentração de Trabalho | Concentração Final | Duração recomendada do armazenamento | Anotações | |||||||||
| Neutravidin (NA) | 5 mg/mL | 1X BRB80 | -80°C | 0,2 mg/mL | 0,2 mg/mL | até 1 ano | Usado para imobilizar microtúbulos através de uma ligação biotina-neutravidina-biotina; armazenar em pequenas alíquotas | |||||||||
| Kappa-casein (KC) | 5 mg/mL | 1X BRB80 | -80°C | 0,5 mg/mL | 0,5 mg/mL | até 2 anos | Usado para bloquear a superfície da câmara de imagem; Armazenar em pequenas alíquotas; No dia da experiência, reserve um pequeno volume à temperatura ambiente | |||||||||
| Albumina de soro bovino (BSA) | 50 mg/mL | 1X BRB80 | -20°C | 1 mg/mL (em DB) | N/A | até 2 anos | armazenar em pequenas alíquotas | |||||||||
| Catalase | 3,5 mg/mL | 1X BRB80 | -80°C | 350 μg/mL (em OSF) | 35 μg/mL | até 2 anos | componente da mistura de limpeza de oxigênio; armazenar em pequenas alíquotas | |||||||||
| Glicose oxidase | 40 mg/mL | 1X BRB80 | -80°C | 4 mg/mL (em OSF) | 0,4 mg/mL | até 2 anos | componente da mistura de limpeza de oxigênio; armazenar em pequenas alíquotas | |||||||||
| Tubulina | Liofilizado | N/A | 4°C | 10 mg/mL | 2,12 mg/mL (na mistura de tubulina) | até 1 ano | Uma vez que a tubulina esteja em solução, mantenha-a fria para evitar a polimerização. | |||||||||
| Adenosine Triphosphate (ATP) | 100 mM | água ultrapura | -20°C | 10 mM | 1 mM | 6 meses | Prepare a solução em água esterilizada por filtro, ajuste o pH para ~7.0 e congele em pequenas alíquotas. | |||||||||
| Triptofato de Guanosina (GTP) | 100 mM | água ultrapura | -20°C | 10 mM | 1,29 mM (em mix de tubulina) | 6 meses | Prepare a solução em água esterilizada por filtro, ajuste o pH para ~7.0 e congele em pequenas alíquotas. | |||||||||
| Guanosine-5'-[α,β)-metileno] triphosfato (GMPCPP) | 10 mM | água ultrapura | -20°C | 10 μM | 0,5 μM | 6 meses | ||||||||||
| Dithiothreitol (DTT) | 1 M | água estéril | -20°C | 1 mM | N/A | até 2 anos | ||||||||||
| Fosfina tris (2 carboxyethyl) (TCEP) | 0,5 M | água esterilizada por filtro | Temperatura ambiente | 5 mM | N/A | até 2 anos | ||||||||||
| Metilcelulose | 1% | água estéril | Temperatura ambiente | 0,8% (em MBMC) | 0,21% (na mistura de tubulina) | até 1 ano | Dissolver a metilcelulose adicionando-a lentamente à água quase fervente. Deixe esfriar enquanto mexe continuamente. | |||||||||
| Beta-mercaptoetanol (BME) | 143 mM | água estéril | Temperatura ambiente | 14,3 mM (em OSF) | 1,43 mM | até 5 anos | 143 mM é uma diluição de 1:100 do estoque BME | |||||||||
| Glicose | 150 mg/mL | 1X BRB80 | -80°C | 15 mg/mL (em OSF) | 1,5 mg/mL | até 2 anos | Adicione ao OSM imediatamente antes do uso | |||||||||
| (±)-6-Hidroxi-2,5,7,8-tetrametil cromotano-2-carboxílico ácido (Trolox) | 10mM | 1X BRB80 | -80°C | 10 mM | 1 mM | até 1 ano | Não se dissolve totalmente. Adicione um pouco de NaOH, mexa por ~4 horas e o filtro esterilize antes de usar | |||||||||
| mPEG-Succinimidyl Valerate, MW 5.000 | pó | N/A | -20°C | 333 mg/mL (em 0,1 M bicarbonato de sódio) | 324 mg/mL (em 0,1 M bicarbonato de sódio) | 6 meses | Prepare ~34 mg aliquots, marcando cada tubo com um peso exato de pó. Passe gás nitrogênio sobre o sólido, tubos de vedação com parafilme, e armazene a -20°C em um recipiente com dessecante. | |||||||||
| Biotin-PEG-SVA, MW 5.000 | pó | N/A | -20°C | 111 mg/mL (em 0,1 M bicarbonato de sódio) | 3,24 mg/mL (em 0,1 M bicarbonato de sódio) | 6 meses | Prepare ~3 mg de alíquotas, marcando cada tubo com um peso exato de pó. Passe gás nitrogênio sobre o sólido, tubos de vedação com parafilme, e armazene a -20°C em um recipiente com dessecante. | |||||||||
Tabela 2: Lista de reagentes utilizados neste protocolo. Incluem-se as condições e concentrações recomendadas de armazenamento, concentrações de trabalho de soluções de estoque utilizadas durante o experimento e concentração final na câmara de imagem. Notas adicionais são dadas na coluna de extrema-direita.
2. Prepare slides biotin-PEG
NOTA: Prepare as câmaras de imagem o mais perto possível do início de um experimento, e não mais de 2 semanas de antecedência.
- Tampas limpas
- Posicione um número igual de 24 x 60 mm e 18 x 18 mm #1,5 deslizamentos (Figura 1F) em frascos de manchas de slides e racks de lavagem de slides, respectivamente (Figura 1A,B). Coloque os racks de lavagem de slides contendo tampas de 18 x 18 mm em um béquer de 100 mL.
- Enxágüe todas as tampas 5-6 vezes em água ultrauso (18,2 MΩ-cm resistividade) e remova o excesso de líquido após cada enxágue com uma ponta de pipeta presa a um tubo de vácuo (Figura 1C).
- Encha béquers e frascos de manchas de slides contendo as tampas com água ultrapura, vedação com parafilme e sonicato por 10 minutos.
- Encha dois béquers de 150 mL com etanol à prova de 200. Usando pinças, mergulhe cada tampa em um béquer cheio de etanol, e depois o outro.
- Usando pinças, transfira as tampas para o rack de secagem de slides (Figura 1D), seque-as sob o fluxo de gás nitrogênio e incubar a 37 °C até secar completamente (~15 min).
- Coloque as tampas secas em uma única camada dentro do limpador de plasma. Forme o selo de vácuo e, em seguida, defina o nível de Radio Frequency (RF) do limpador de plasma para ~8 MHz.
- Uma vez que o plasma é gerado, deixe tampas no limpador de plasma por 5 minutos. Desligue o limpador de plasma e solte o vácuo lentamente.
- Uma vez que o selo de vácuo é liberado, vire as tampas e repita a limpeza do plasma por 5 minutos para o outro lado das tampas.
- Alternativa à limpeza plasmática: No lugar das etapas 2.1.2-2.1.3, as tampas sonicadas em uma solução quente de 2% de detergente (em água ultrauso) por 10 minutos. Em seguida, lave minuciosamente as tampas com água ultrauso e sonicato em água ultrauso 2-3 vezes (10 min cada). Em seguida, lave no etanol e seque como nas etapas 2.1.4-2.1.5. Pule as etapas 2.1.6-2.1.8.
- Tratamento biotina-PEG
- Imediatamente antes do uso, dissolva 400 μL de 3-Aminopropyltriethoxysilane em 40 mL de acetona. Usando pinças, mova tampas limpas com plasma para o rack de lavagem de slides e frascos de coloração de slides. Submergir tampas em 3-Aminopropyltriethoxysilane solução e incubar por 5 min12,13.
- Lave todas as tampas 5-6 vezes com água ultrapurada.
- Transfira as manchas para o rack de secagem de slides, seque-as sob um fluxo de gás nitrogênio e incubar a 37 °C até secar completamente (~20 min).
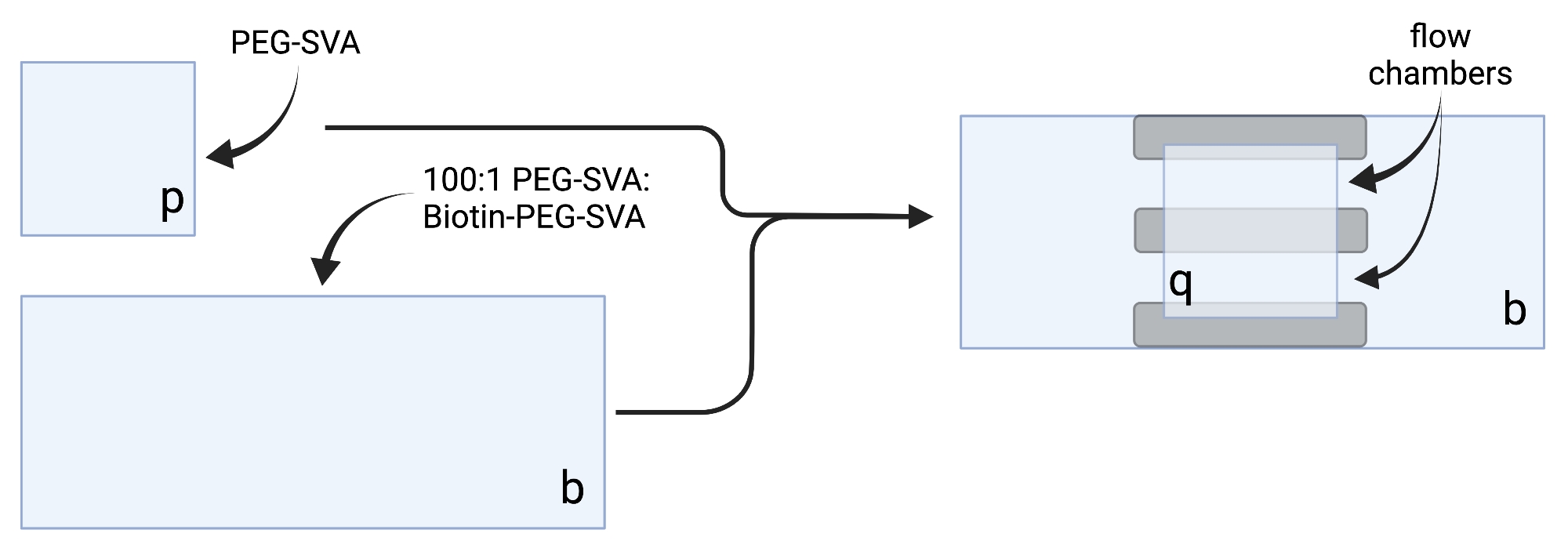
- Coloque as tampas secas em lenços de tarefa delicados e rotule cada mancha de cobertura em um canto, por exemplo, 'p' em cada deslizamento de cobertura de 18 x 18 mm e 'b' em cada deslizamento de cobertura de 24 x 60 mm (ver Figura 2).
- No dia do experimento, prepare uma solução fresca de bicarbonato de sódio de 0,1 M dissolvendo 0,84 mg de NaHCO3 em 10 mL de água ultrapura.
- Leve as alíquotas de mPEG-Succinimidyl Valerate (PEG-SVA) e Biotin-PEG-SVA à temperatura ambiente, imediatamente antes do uso. Consulte notas sobre a preparação de alíquotas de Polietileno Glycol (PEG) na Tabela 2.
NOTA: Trabalhe rapidamente porque a meia-vida hidrolise do Valerate de Succinimidyl (SVA) moiety é ~30 min. - Adicione 102 μL de 0,1 M NaHCO3 a 34 mg de PEG-SVA, gire em uma microcentrifuagem de bancada a 2.656 x g por 20 s e misture por pipetação para cima e para baixo. Dissolva 3 mg de Biotin-PEG-SVA em 27 μL de 0,1 M NaHCO3 por tubulação para cima e para baixo. Ajuste os volumes de diluição de acordo com o peso exato do PEG observado nos tubos (ver Tabela 2).
- Prepare 100:1 w/w PEG:biotin-PEG mistura combinando 75 μL de solução PEG-SVA e 2,25 μL de solução Biotin-PEG-SVA para 20 tampas, 100 μL e 3 μL para 30 tampas, ou 125 μL e 3,75 μL para 40 tampas.
- Construa uma câmara de hidratação colocando toalhas de papel molhadas sob o rack de ponta na parte inferior de uma caixa de ponta vazia de 10 μL (Figura 1E). Isso evitará a evaporação das soluções PEG.
- Pipeta 6 μL de 100:1 PEG-SVA:biotin-PEG mistura no centro de uma tampa de 24 x 60 mm no lado rotulado. Coloque outra mancha de cobertura de 24 x 60 mm em cima do primeiro deslizamento de cobertura, de modo que o par forme uma forma X, com os lados rotulados como 'b' voltados um para o outro. Coloque o par em um rack de ponta vazio na câmara de hidratação e repita para as tampas restantes de 24 x 60 mm.
- Pipeta 6 μL de PEG-SVA no centro de uma tampa de 18 x 18 mm no lado rotulado. Coloque outra mancha de cobertura de 18 x 18 mm em cima do primeiro deslizamento de cobertura, com os lados rotulados como 'p' voltados um para o outro. Coloque o par em um rack de ponta vazio na câmara de hidratação e repita para as tampas restantes de 18 x 18 mm.
- Feche a câmara de hidratação e incubar por 3h ou durante a noite.
- Separe os pares de tampas e enxágue em água ultrauso.
- As tampas secas sequem com um fluxo de nitrogênio e coloque-as em uma incubadora de 37 °C para secar totalmente.
- Para construir a câmara de imagem, coloque três tiras de fita dupla face em um deslizamento de cobertura de 24 x 60 mm na lateral rotulada como 'b'. Para o outro lado das tiras de fita, conecte um deslizamento de tampa de 18 x 18 mm com seu lado rotulado 'p' voltado para o deslizamento de cobertura maior. Isso forma duas câmaras de fluxo para experimentos de microscopia, com superfícies tratadas voltadas umas para as outras (Figura 2 e Figura 1G).

Figura 1: Equipamento para tratamento de deslizamento de cobertura e preparação de câmara de imagem. (A) frascos de coloração de slides para tampas de 24 x 60 mm, (B) racks de lavagem de slides para 18 x 18 mm de cobertura, (C) configuração a vácuo, (D) rack de secagem de slides, (E) câmara de hidratação, (F) tampas, (G) câmara de imagem, (H) suporte de slides( H). Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: Esquema para preparação de câmaras de imagem utilizando fita dupla face (cinza) e manchas tratadas PEG/Biotin-PEG. Criado com BioRender.com. Clique aqui para ver uma versão maior desta figura.
{kind=link}
3. Microtúbulos polimerizais
- Preparar sementes GMPCPP
NOTA: Prepare as sementes GMPCPP em uma sala fria, mantendo todos os reagentes, pontas e tubos a 4 °C. As sementes GMPCPP podem ser preparadas com antecedência e armazenadas a -80 °C por até 1 ano. Coloque rotor ultracentrifuuge de ângulo fixo, contendo tubos de centrífugas de 1 mL, em ultracentrifuugação e afina a temperatura para 4 °C.- Tubulina liofilizada resuspend (Tabela 2) a ~10 mg/mL em 1X BRB80, imediatamente antes do uso.
- Misture componentes de sementes GMPCPP conforme descrito na Tabela 3.
NOTA: Mantenha todos os componentes da tubulina no gelo o máximo possível para minimizar a polimerização da tubulina solúvel. - Esclareça a mistura em um rotor ultracentrífuga de ângulo fixo a 352.700 x g por 5 min a 4 °C.
- Separe o supernatante em alíquotas de 5 μL, congele-as em nitrogênio líquido e armazene-as a -80 °C.
- Sementes polimerizam no dia do experimento
- Quente de 1-2 mL de BRB80-DTT (Tabela 1) a 37 °C.
- Coloque 5 μL aliquot de sementes GMPCPP (-80 °C) a partir da etapa 3.1.4 no gelo e imediatamente dissolva em 20 μL de BRB80-DTT quente. Gire a 2.000 x g por 5 s em temperatura ambiente e toque para misturar.
NOTA: O volume inicial de diluição pode variar entre 13 μL e 21 μL e é empiricamente determinado para cada lote de sementes. Se as sementes não polimerizarem, solucionar problemas suplementando o tampão de diluição inicial (etapa 3.2.2) com 0,5 μM GMPCPP. - Proteja contra a luz e incubar a 37 °C por 30-45 min.
NOTA: O comprimento dos microtúbulos depende da duração da incubação. Para microtúbulos curtos, o tempo de incubação pode ser tão curto quanto 15 min. Para microtúbulos longos, o tempo de incubação pode ser de até 2 h. Microtúbulos biotinilados tendem a exigir tempos de incubação mais longos do que microtúbulos não biotinilados. - Coloque rotor de ultracentrifuagem de ângulo fixo, contendo tubos de centrífugas de 500 μL, em ultracentrifuuge e pré-aquecido a 30 °C.
- Após a incubação, adicione 50 μL de BRB80-DTT quente (etapa 3.2.1) às sementes GMPCPP polimerizadas e transfira a mistura para um tubo de centrífuga de 500 μL. Lave o tubo vazio que continha as sementes GMPCPP com mais 50 μL de BRB80-DTT quente, pipeta para cima e para baixo, e adicione este tampão ao tubo de centrífuga de 500 μL contendo a mistura.
- Antes de girar, marque a borda do tubo de centrífuga para indicar onde a pelota estará (a pelota será muito pequena para ver). Gire por 10 min a 244.900 x g a 30 °C12.
- Pipeta cuidadosamente o supernaste e descarte. Resuspenque a pelota em 100 μL de BRB80-DTT quente. Toque para misturar.
- Gire por 10 min a 244.900 x g a 30 °C, alinhando a marcação com o rotor à pelota no mesmo lugar.
- Remova o supernatante e resuspenque a pelota em 16 μL de BRB80-DTT quente. Transfira a solução de microtúbulo para um tubo de microcentrífugo limpo de 0,6 mL. Proteja-se contra a luz e mantenha-se a temperatura ambiente ou acima.
NOTA: Após a polimerização, mantenha os microtúbulos a temperatura ambiente ou acima. Se ficarem frios, despolimerizarão. Incubar a 28 °C para maior estabilidade.
- Verifique microtúbulos via microscopia DE TIRF
- Pipeta uma mistura de 4,5 μL de BRB80-DTT e 1 μL de solução de microtúbulo (passo 3.2.9) em um slide de microscópio. Cubra com uma tampa de 18 x 18 mm e sele as bordas com esmalte transparente ou uma mistura de 1:1:1 de petrolatum, lanolina e parafina (selante valap), que é sólido à temperatura ambiente e líquido a 95 °C.
- Posicione o objetivo tirf abaixo do deslizamento de tampas (ver passo 4 para configurações recomendadas de microscópio) e visualize os microtúbulos recém-polimerizados no comprimento de onda apropriado para a tubulina fluorescentemente rotulada na mistura Bright (Tabela 3), para determinar qual diluição de microtúbulos usar nos próximos experimentos.
| Reagente | Mistura brilhante (μL) | Ordem de adição | Mix brilhante + biotina (μL) | Ordem de adição |
| Tubulina fluorescente, 10 mg/mL | 2 | 6 | 2 | 7 |
| Biotin-tubulin, 10 mg/mL | 0 | N/A | 2 | 6 |
| Tubulina sem rótulo, 10 mg/mL | 20 | 5 | 18 | 5 |
| GMPCPP, 10 mM | 30 | 4 | 30 | 4 |
| DTT, 0,2 M | 0.7 | 3 | 0.7 | 3 |
| 5X BRB80 | 26.4 | 2 | 26.4 | 2 |
| água estéril | 52.9 | 1 | 52.9 | 1 |
| Volume Total (μL) | 132 | 132 |
Tabela 3: Mistura de sementes GMPCPP. Componentes das sementes de microtúbulos GMPCPP, incluindo volume e ordem de adição. Prepare 5 alíquotas μL e armazene por até 1 ano a -80 °C.
4. Configurações do microscópio
- Temperatura: Coloque a temperatura do microscópio em 28 °C para visualizar microtúbulos dinâmicos.
- Filtros: Use a melhor combinação de cubos de filtro e filtros de emissão, dependendo dos canais fluorescentes a serem imagens. Para visualizar 488 nm, 560 nm e 647 nm comprimentos de onda no mesmo experimento, use um conjunto de banda quad-mor laser de 405/488/560/647 nm, juntamente com filtros de emissão para os comprimentos de onda designados.
- Alinhe lasers: Certifique-se de que os raios laser usados no experimento estejam alinhados. Determine a intensidade laser para o experimento empiricamente, de tal forma que todas as proteínas fluorescentes possam ser imagens com a maior relação sinal-ruído possível, mas não submetam fotobleaching significativo ao longo do tempo do experimento.
- Objetivo: Use papel de lente para limpar um objetivo de 100x com 70% de etanol. Antes da imagem, adicione uma gota de óleo de imersão de microscópio ao objetivo.
- Configure uma sequência de imagens
- Para um experimento com microtúbulos biotinilados rotulados por 647 nm, microtubulos biotinilados rotulados por 647 nm, 560 nm de microtúbulos não biotinilados e tubulina solúvel, e proteína de interesse rotulada por GFP, imagem por 20 minutos. Imagem dos canais de 560 nm e 488 nm a cada 10 s, e o canal de 647 nm a cada 30 s.
- Para capturar uma imagem de referência de feixes antes da adição de tubulina solúvel e MAPs, configure uma sequência com uma imagem cada em comprimentos de onda de 560 nm e 647 nm.
5. Gerar feixes de microtúbulos imobilizados pela superfície
NOTA: Para as etapas a seguir, flua todas as soluções para uma câmara de fluxo, pipetando para um lado aberto, ao mesmo tempo em que coloca um papel filtro contra o outro lado. Proteja a câmara de imagem da luz para reduzir o fotobleaching de proteínas fluorescentes rotuladas. Fita a câmara de imagem preparada em um suporte de slides (Figura 1G,H). Siga os passos da Tabela 4, que correspondem aos protocolos steps 5.2-6.4.
- Prepare a mistura de tubulina solúvel de acordo com a Tabela 5 e mantenha-a no gelo.
NOTA: A tubulina solúvel deve ser sempre colocada no gelo para evitar a polimerização. Prepare uma mistura de tubulina solúvel fresca aproximadamente a cada 2 h, ou quando os microtúbulos não estiverem mais polimerizando. - Para imobilizar microtúbulos através de uma ligação biotina-neutravidina-biotina, primeiro fluxo na solução Neutravidin (NA) até que a câmara seja preenchida (~7,5 μL) e incubar por 5 min.
- Lave com 10 μL de MB-frio.
- Flua em 7,5 μL da proteína de bloqueio κ-casein (KC) e incubar por 2 min.
- Lave com 10 μL de MB-warm para preparar a câmara para a introdução de microtúbulos.
- Diluir o estoque de microtúbulos biotinilados (de acordo com observações na etapa 3.3.2) em 1X BRB80-DTT e adicionar 1 μL desta diluição a 9 μL de MB-warm. Escoar a mistura na câmara e incubar por 10 minutos. Use uma maior concentração de microtúbulos para mais pacotes.
- Lave microtúbulos não imobilizados com 10 μL de MB-warm.
- Flua 7,5 μL de KC quente na câmara e incubar por 2 min.
- Durante a incubação, prepare uma solução de 2 nM da proteína crosslinker PRC1 em KC quente Fluxo 10 μL desta solução para a câmara de fluxo e incubar por 5 min.
NOTA: O PRC1 recombinante é expresso e purificado a partir de células bacterianas como descrito anteriormente13. - Para fazer pacotes, flua 10 μL de microtúbulos não biotinilados na câmara e incubará por 10 min. A RPC1 cruzará o microtúbulo biotinilado e imobilizado não biotinilado15,16 (Figura 3).
NOTA: Consulte a etapa 6.1 para preparar a mistura de ensaio durante o tempo de incubação. - Lave a câmara duas vezes com 10 μL de MB-warm. Os microtúbulos anexados estão estáveis por cerca de 20 minutos a partir deste ponto.
| Passo | Reagente | Volume (μL) | Tempo de incubação (minutos) |
| 1 | Neutravidin | 7.5 | 5 |
| 2 | MB-frio | 10 | - |
| 3 | κ-casein | 7.5 | 2 |
| 4 | MB-quente | 10 | - |
| 5 | Microtúbulo biotinilado (diluído em MB-warm) | 10 | 10 |
| 6 | MB-quente | 10 | - |
| 7 | Κ-caseína quente | 7.5 | 2 |
| 8 | 2 nM PRC1 diluído em κ-casein | 10 | 5 |
| 9 | Microtúbulo não biotinilado | 10 | 10 |
| 10 | MB-quente x 2 | 10 | - |
| 11 | Mistura de ensaio | 10 | - |
| As sementes anexadas estão estáveis por cerca de 20 minutos neste ponto | |||
Tabela 4: Passos de ensaio. Lista de reagentes adicionados à câmara de imagem, com indicação de lavagem (-) ou tempo de incubação.
| Reagente | Volume (μL) |
| Tubulina reciclada, 10 mg/mL | 10 |
| MB-Frio | 10.3 |
| MBMC | 13.7 |
| BRB80-DTT | 3.4 |
| GTP, 10 mM | 6.7 |
| ATP, 10 mM (Se usar quinase) | 6.7 |
| Tubulina fluorescentemente rotulada, 10 mg/mL | 1 (Tubulina rotulada liofilizada resuspend em BRB80-DTT frio) |
Tabela 5: Componentes solúveis da mistura de tubulina. Misture no início do experimento e mantenha no gelo.

Figura 3: Esquema de adição de componentes de ensaio para fazer e imagem fluorescente rotulados pacotes e microtúbulos únicos. As sementes biotiniladas são mostradas em sementes azuis, não biotiniladas e tubulina solúvel em vermelho, PRC1 em preto e proteína de interesse em ciano. Os números de passos correspondem aos da Tabela 4. Painel correspondente à etapa 9 mostra um feixe pré-formado (inferior esquerdo); passo 11 mostra um pacote recém-formado (superior esquerdo). Criado com BioRender.com. Clique aqui para ver uma versão maior desta figura.
{kind=link}
6. Dinâmica do microtúbulo de imagem
- Durante o tempo de incubação de 10 min na etapa 5.10, prepare 10 μL de mistura de ensaio contendo proteínas de interesse, tubulina solúvel, nucleotídeos, catadores de oxigênio14 e antioxidantes de acordo com a Tabela 6. Mantenha a mistura no gelo.
- Carregue a câmara de imagem preparada, gravada no suporte de slides, no objetivo 100x TIRF. Use os canais de 560 nm e 647 nm para encontrar um campo de visão que contenha um número e densidade ideal de microtúbulos e feixes únicos.
NOTA: Se os microtúbulos biotinilados e não biotinilados forem rotulados com o mesmo fluoróforo, as análises de varredura de linha das intensidades de fluorescência podem distinguir entre microtúbulos e feixes únicos. - Uma vez identificado um campo de visão, pegue uma imagem de referência.
- Flua cuidadosamente na mistura de ensaios sem perturbar a câmara de imagem.
- Sele as extremidades abertas da câmara com selante valap.
- Inicie a sequência de imagens conforme descrito na etapa 4.5.1.
| Reagente | Volume (μL) |
| Mistura de tubulina solúvel | 4 |
| OSF | 1 |
| Trolox (se usar microtúbulos rotulados com um fluorohore prontamente fotobleaching) | 1 |
| ATP, 10 mM (Se usar quinase) | 1 |
| PRC1 (ou crosslinker de escolha) | 1 |
| Proteínas de interesse | X |
| MB-frio | 2-X |
Tabela 6: Os componentes da mistura de ensaios. Misture, flua em câmara de imagem e dinâmica de microtúbulos de imagem, dentro de 30 minutos.
Access restricted. Please log in or start a trial to view this content.
Resultados
O experimento descrito acima foi realizado usando microtubulos biotinilados biotinilados rotulados por 647 nm, microtubulas não biotiniladas com rótulo fluoreto, e mistura de tubulina solúvel com rótulo fluorólmico de 560 nm. Os microtúbulos foram cruzados pela proteína crosslinker PRC1 (rotulada por GFP). Após a gerada por feixes imobilizados e microtúbulos únicos (etapa 5.11), a câmara de imagem foi montada em um objetivo de óleo TIRF 100X 1.49 NA e visualizada nos canais de fluorescência de 560 nm e 647 n...
Access restricted. Please log in or start a trial to view this content.
Discussão
O experimento descrito aqui expande significativamente o escopo e a complexidade dos ensaios convencionais de reconstituição da microtúbula, que são tradicionalmente realizados em microtúbulos únicos ou em um tipo de matriz. O ensaio atual fornece um método para quantificar e comparar simultaneamente a atividade de MAP regulatório em duas populações, ou seja, microtúbulos únicos e pacotes interligados. Além disso, este ensaio permite o exame de dois tipos de feixes: aqueles que são pré-formados a partir de...
Access restricted. Please log in or start a trial to view this content.
Divulgações
Os autores não declaram interesses concorrentes.
Agradecimentos
Este trabalho foi apoiado por uma subvenção do NIH (nº 1DP2GM126894-01), e por fundos da Pew Charitable Trusts e da Smith Family Foundation para r.S. Os autores agradecem ao Dr. Shuo Jiang por sua contribuição para o desenvolvimento e otimização dos protocolos.
Access restricted. Please log in or start a trial to view this content.
Materiais
| Name | Company | Catalog Number | Comments |
| (±)-6-Hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (Trolox) | Sigma Aldrich | 238813 | |
| 1,4-piperazinediethanesulfonic acid (PIPES) | Sigma Aldrich | P6757 | |
| 18x18 mm #1.5 coverslips | Electron Microscopy Sciences | 63787 | |
| 2-Mercaptoethanol (BME) | Sigma Aldrich | M-6250 | |
| 24x60 mm #1.5 coverslips | Electron Microscopy Sciences | 63793 | |
| 405/488/560/647 nm Laser Quad Band | Chroma | TRF89901-NK | |
| Acetone | Sigma Aldrich | 320110 | |
| Adenosine 5'-triphosphate disodium salt hydrate (ATP) | Sigma Aldrich | A7699-5G | |
| Avidin, NeutrAvidin® Biotin-binding Protein (Molecular Probes®) | Thermo Fischer Scientific | A2666 | |
| Bath sonicator: Branson 2800 Cleaner | Branson | CPX2800H | |
| Beckman Coulter Polycarbonate Thickwall Tubes, 11 x 34 mm | Beckman-Coulter | 343778 | |
| Beckman Coulter Polycarbonate Thickwall Tubes, 8 x 34 mm | Beckman-Coulter | 343776 | |
| Biotin-PEG-SVA, MW 5,000 | Laysan Bio | #Biotin-PEG-SVA-5000 | |
| Bovine Serum Albumin (BSA) | Sigma Aldrich | 2905 | |
| Catalase | Sigma Aldrich | C40 | |
| Corning LSE Mini Microcentrifuge, AC100-240V | Corning | 6670 | |
| Delicate Task Wipes | Kimtech | 34120 | |
| Dithiothreitol (DTT) | GoldBio | DTT10 | |
| Emission filter | Chroma | ET610/75m | |
| Ethanol (200-proof) | Decon Labs | 2705 | |
| Ethylene glycol tetraacetic acid (EGTA) | Sigma Aldrich | 3777 | |
| Glucose Oxidase | Sigma Aldrich | G2133 | |
| GMPCPP | Jena Bioscience | NU-405 | |
| Guanosine 5'-triphosphate sodium salt hydrate (GTP) | Sigma Aldrich | G8877 | |
| Hellmanex III detergent | Sigma Aldrich | Z805939 | |
| Immersion oil, Type A | Fisher Scientific | 77010 | |
| Kappa-casein | Sigma Aldrich | C0406 | |
| Lanolin | Fisher Scientific | S25376 | |
| Lens Cleaning Tissue | ThorLabs | MC-5 | |
| Magnesium Chloride (MgCl2) | Sigma Aldrich | M9272 | |
| Methylcellulose | Sigma Aldrich | M0512 | |
| Microfuge 16 Benchtop Centrifuge | Beckman-Coulter | A46474 | |
| Microscope Slides, Diamond White Glass, 25 x 75mm, 90° Ground Edges, WHITE Frosted | Globe Scientific | 1380-50W | |
| mPEG-Succinimidyl Valerate, MW 5,000 | Laysan Bio | #NH2-PEG-VA-5K | |
| Optima™ Max-XP Tabletop Ultracentrifuge | Beckman-Coulter | 393315 | |
| Paraffin | Fisher Scientific | P31-500 | |
| PELCO Reverse (self-closing), Fine Tweezers | Ted Pella | 5377-NM | |
| Petrolatum, White | Fisher Scientific | 18-605-050 | |
| Plasma Cleaner, 115V | Harrick Plasma | PDC-001 | |
| Potassium Hydroxide (KOH) | Sigma Aldrich | 221473 | |
| Sodium bicarbonate | Sigma Aldrich | S6014 | |
| Sucrose | Sigma Aldrich | S7903 | |
| Thermal-Lok 1-Position Dry Heat Bath | USA Scientific | 2510-1101 | |
| Thermal-Lok Block for 1.5 and 2.0 mL Tubes | USA Scientific | 2520-0000 | |
| Thermo Scientific™ Pierce™ Bond-Breaker™ TCEP Solution, Neutral pH; 500mM | Thermo Fischer Scientific | PI-77720 | |
| TIRF 100X NA 1.49 Oil Objective | Nikon | CFI Apochromat TIRF 100XC Oil | |
| TIRF microscope | Nikon | Eclipse Ti | |
| TLA 120.1 rotor | Beckman-Coulter | 362224 | |
| TLA 120.2 rotor | Beckman-Coulter | 357656 | |
| Tubulin protein (>99% pure): porcine brain | Cytoskeleton | T240 | |
| Tubulin Protein (Biotin): Porcine Brain | Cytoskeleton | T333P | |
| Tubulin protein (fluorescent HiLyte 647): porcine brain | Cytoskeleton | TL670M | |
| Tubulin protein (X-rhodamine): bovine brain | Cytoskeleton | TL620M | |
| VECTABOND® Reagent, Tissue Section Adhesion | Vector Biolabs | SP-1800-7 | |
| VWR® Personal-Sized Incubator, 120V, 50/60Hz, 0.6A | VWR | 97025-630 |
Referências
- Subramanian, R., Kapoor, T. M. Building complexity: insights into self-organized assembly of microtubule-based architectures. Developmental Cell. 23 (5), 874-885 (2012).
- Baas, P. W., Rao, A. N., Matamoros, A. J., Leo, L. Stability properties of neuronal microtubules. Cytoskeleton (Hoboken). 73 (9), 442-460 (2016).
- Bitan, A., Rosenbaum, I., Abdu, U. Stable and dynamic microtubules coordinately determine and maintain Drosophila bristle shape. Development. 139 (11), 1987-1996 (2012).
- Foe, V. E., von Dassow, G. Stable and dynamic microtubules coordinately shape the myosin activation zone during cytokinetic furrow formation. The Journal of Cell Biology. 183 (3), 457-470 (2008).
- Pous, C., et al. Functional specialization of stable and dynamic microtubules in protein traffic in WIF-B cells. The Journal of Cell Biology. 142 (1), 153-165 (1998).
- Uehara, R., Goshima, G. Functional central spindle assembly requires de novo microtubule generation in the interchromosomal region during anaphase. The Journal of Cell Biology. 191 (2), 259-267 (2010).
- Mitchison, T., Kirschner, M. Dynamic instability of microtubule growth. Nature. 312 (5991), 237-242 (1984).
- Bieling, P., et al. Reconstitution of a microtubule plus-end tracking system in vitro. Nature. 450 (7172), 1100-1105 (2007).
- Bieling, P., Telley, I. A., Surrey, T. A minimal midzone protein module controls formation and length of antiparallel microtubule overlaps. Cell. 142 (3), 420-432 (2010).
- Shimamoto, Y., Forth, S., Kapoor, T. M. Measuring pushing and braking forces generated by ensembles of Kinesin-5 crosslinking two microtubules. Developmental Cell. 34 (6), 669-681 (2015).
- Mani, N., Jiang, S., Neary, A. E., Wijeratne, S. S., Subramanian, R. Differential regulation of single microtubules and bundles by a three-protein module. Nature Chemical Biology. 17 (9), 964-974 (2021).
- Hyman, A. A., Salser, S., Drechsel, D., Unwin, N., Mitchison, T. J. Role of GTP hydrolysis in microtubule dynamics: information from a slowly hydrolyzable analogue, GMPCPP. Molecular Biology of the Cell. 3 (10), 1155-1167 (1992).
- Subramanian, R., et al. Insights into antiparallel microtubule crosslinking by PRC1, a conserved nonmotor microtubule binding protein. Cell. 142 (3), 433-443 (2010).
- Rasnik, I., McKinney, S. A., Ha, T. Nonblinking and long-lasting single-molecule fluorescence imaging. Nature Methods. 3 (11), 891-893 (2006).
- Wijeratne, S., Subramanian, R. Geometry of antiparallel microtubule bundles regulates relative sliding and stalling by PRC1 and Kif4A. eLife. 7, 32595(2018).
- Mani, N., Wijeratne, S. S., Subramanian, R. Micron-scale geometrical features of microtubules as regulators of microtubule organization. eLife. 10, 63880(2021).
- Freal, A., et al. Feedback-driven assembly of the axon initial segment. Neuron. 104 (2), 305-321 (2019).
- Ledbetter, M., Porter, K. A "microtubule" in plant cell fine structure. The Journal of Cell Biology. 19 (1), 239-250 (1963).
- Wijeratne, S. S., Marchan, M. F., Tresback, J. S., Subramanian, R. Atomic force microscopy reveals distinct protofilament-scale structural dynamics in depolymerizing microtubule arrays. Proceedings of the National Academy of Sciences of the United States of America. , 119(2022).
Access restricted. Please log in or start a trial to view this content.
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoExplore Mais Artigos
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados