Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Simultane Visualisierung der Dynamik von vernetzten und einzelnen Mikrotubuli in vitro mittels TIRF-Mikroskopie
* Diese Autoren haben gleichermaßen beigetragen
In diesem Artikel
Zusammenfassung
Hier wird ein TIRF-Mikroskopie-basierter In-vitro-Rekonstitutionsassay vorgestellt, um gleichzeitig die Dynamik zweier Mikrotubulipopulationen zu quantifizieren und zu vergleichen. Es wird ein Verfahren beschrieben, um gleichzeitig die kollektive Aktivität mehrerer Mikrotubuli-assoziierter Proteine auf vernetzten Mikrotubulibündeln und einzelnen Mikrotubuli zu betrachten.
Zusammenfassung
Mikrotubuli sind Polymere von αβ-Tubulin-Heterodimeren, die sich in Zellen zu unterschiedlichen Strukturen organisieren. Mikrotubuli-basierte Architekturen und Netzwerke enthalten häufig Teilmengen von Mikrotubuli-Arrays, die sich in ihren dynamischen Eigenschaften unterscheiden. Zum Beispiel koexistieren in sich teilenden Zellen stabile Bündel vernetzter Mikrotubuli in unmittelbarer Nähe zu dynamischen, nicht vernetzten Mikrotubuli. TIRF-mikroskopiebasierte In-vitro-Rekonstitutionsstudien ermöglichen die gleichzeitige Visualisierung der Dynamik dieser verschiedenen Mikrotubuli-Arrays. Bei diesem Assay wird eine Bildgebungskammer mit oberflächenimmobilisierten Mikrotubuli zusammengesetzt, die entweder als einzelne Filamente vorliegen oder in vernetzten Bündeln organisiert sind. Die Einführung von Tubulin, Nukleotiden und Proteinregulatoren ermöglicht die direkte Visualisierung von assoziierten Proteinen und von dynamischen Eigenschaften einzelner und vernetzter Mikrotubuli. Darüber hinaus können Änderungen, die auftreten, wenn sich dynamische einzelne Mikrotubuli zu Bündeln organisieren, in Echtzeit überwacht werden. Die hier beschriebene Methode ermöglicht eine systematische Bewertung der Aktivität und Lokalisation einzelner Proteine sowie synergistischer Effekte von Proteinregulatoren auf zwei verschiedene Mikrotubuli-Untergruppen unter identischen experimentellen Bedingungen und liefert dadurch mechanistische Erkenntnisse, die mit anderen Methoden nicht zugänglich sind.
Einleitung
Mikrotubuli sind Biopolymere, die strukturelle Gerüste bilden, die für mehrere zelluläre Prozesse unerlässlich sind, vom intrazellulären Transport und der Organellenpositionierung bis hin zur Zellteilung und -dehnung. Um diese vielfältigen Funktionen auszuführen, sind einzelne Mikrotubuli in mikrometergroße Arrays organisiert, wie z.B. mitotische Spindeln, Ziliaraxoneme, neuronale Bündel, Interphasenarrays und pflanzliche kortikale Arrays. Ein allgegenwärtiges architektonisches Motiv, das in diesen Strukturen zu finden ist, ist ein Bündel von Mikrotubuli, die entlang ihrer Länge miteinander verbunden sind1. Ein faszinierendes Merkmal mehrerer Mikrotubuli-basierter Strukturen ist die Koexistenz von gebündelten Mikrotubuli und nicht vernetzten Einzelmikrotubuli in unmittelbarer räumlicher Nähe. Diese Mikrotubuli-Subpopulationen können eine stark unterschiedliche Polymerisationsdynamik aufweisen, die für ihre ordnungsgemäße Funktion erforderlich ist2,3,4,5. Zum Beispiel sind innerhalb der mitotischen Spindel stabile vernetzte Bündel und dynamische einzelne Mikrotubuli innerhalb einer mikrometerskaligen Region im Zellzentrum vorhanden6. Die Untersuchung, wie die dynamischen Eigenschaften koexistierender Mikrotubulipopulationen spezifiziert werden, ist daher von zentraler Bedeutung für das Verständnis des Aufbaus und der Funktion von Mikrotubuli-basierten Strukturen.
Mikrotubuli sind dynamische Polymere, die zwischen Phasen der Polymerisation und Depolymerisation wechseln und zwischen den beiden Phasen in Ereignissen, die als Katastrophe und Rettung bekannt sind7 wechseln. Die Dynamik zellulärer Mikrotubuli wird durch unzählige Mikrotubuli-assoziierte Proteine (MAPs) reguliert, die die Raten der Mikrotubuli-Polymerisation und -Depolymerisation sowie die Häufigkeit von Katastrophen- und Rettungsereignissen modulieren. Es ist eine Herausforderung, die Aktivität von MAPs auf räumlich proximalen Arrays in Zellen zu untersuchen, da die räumliche Auflösung in der Lichtmikroskopie begrenzt ist, insbesondere in Regionen mit hoher Mikrotubulidichte. Darüber hinaus behindert das Vorhandensein mehrerer MAPs in derselben Zellregion die Interpretation zellbiologischer Studien. In-vitro-Rekonstitutionstests, die in Verbindung mit der TIRF-Mikroskopie (Total Internal Reflection Fluorescence) durchgeführt werden, umgehen die Herausforderungen der Untersuchung von Mechanismen, mit denen bestimmte Untergruppen von MAPs die Dynamik proximaler zellulärer Mikrotubuli-Arrays regulieren. Hier wird die Dynamik von in vitro zusammengesetzten Mikrotubuli in Gegenwart eines oder mehrerer rekombinanter MAPs unter kontrollierten Bedingungen untersucht8,9,10. Konventionelle Rekonstitutionsassays werden jedoch typischerweise an einzelnen Mikrotubuli oder an einer Art von Array durchgeführt, was die Visualisierung koexistierender Populationen ausschließt.
Hier stellen wir in vitro Rekonstitutionsassays vor, die die gleichzeitige Visualisierung von zwei Mikrotubulipopulationen unter den gleichen Lösungsbedingungen ermöglichen11. Wir beschreiben eine Methode, um gleichzeitig die kollektive Aktivität mehrerer MAPs auf einzelnen Mikrotubuli und auf Mikrotubulibündeln zu betrachten, die mit dem mitotischen Spindel-assoziierten Protein PRC1 vernetzt sind. Das Protein PRC1 bindet bevorzugt an der Überlappung zwischen antiparallelen Mikrotubuli und vernetzt sie9. Kurz gesagt, dieses Protokoll besteht aus den folgenden Schritten: (i) Herstellung von Stammlösungen und Reagenzien, (ii) Reinigung und Oberflächenbehandlung von Deckgläsern, die zur Herstellung der Bildgebungskammer für Mikroskopieexperimente verwendet werden, (iii) Herstellung stabiler Mikrotubuli-"Samen", von denen während des Experiments eine Polymerisation eingeleitet wird, (iv) Spezifikation von TIRF-Mikroskopeinstellungen zur Visualisierung der Mikrotubuli-Dynamik, (v) Immobilisierung von Mikrotubuli-Samen und Erzeugung von vernetzten Mikrotubuli-Bündeln in der Bildgebungskammer und (vi) Visualisierung der Mikrotubulidynamik in der Bildgebungskammer durch TIRF-Mikroskopie unter Zugabe von löslichem Tubulin, MAPs und Nukleotiden. Diese Assays ermöglichen die qualitative Bewertung und quantitative Untersuchung der MAP-Lokalisierung und ihrer Wirkung auf die Dynamik zweier Mikrotubuli-Populationen. Darüber hinaus erleichtern sie die Bewertung der synergistischen Effekte mehrerer MAPs auf diese Mikrotubulipopulationen unter einer Vielzahl von experimentellen Bedingungen.
Protokoll
1. Reagenzien vorbereiten
- Bereiten Sie Puffer und Reagenzien gemäß Tabelle 1 und Tabelle 2 vor. Bewahren Sie während des Experiments alle Lösungen auf Eis auf, sofern nicht anders angegeben.
| Lösung | Komponenten | Empfohlene Speicherdauer | Notizen | ||
| 5X BRB80 | 400 mM K-ROHRE, 5 mM MgCl2, 5 mM EGTA, pH 6,8 mit KOH, Filter sterilisieren | bis 2 Jahre | Bei 4 °C lagern | ||
| 1X BRB80 | 80 mM K-ROHRE, 1 mM MgCl2, 1 mM EGTA, pH 6,8 | bis 2 Jahre | Bei 4 °C lagern | ||
| BRB80-DVB-T | 1X BRB80, 1 mM DVB-T | bis zu 2 Tage | |||
| Assay-Puffer | 80 mM K-ROHRE, 3 mM MgCl2, 1 mM EGTA, pH 6,8, 5% Saccharose (ODER 1X BRB80, 5% Saccharose, 2 mM MgCl2) | bis zu 1 Jahr | Bei 4 °C lagern | ||
| Master-Puffer (MB) | Assay-Puffer, 5mM TCEP | 1 Woche | Bereiten Sie sich am Tag des Experiments vor; In zwei Röhren trennen: MB-warm bei Raumtemperatur und MB-kalt auf Eis; 1 mM DVB-t enthalten, wenn fluoreszierende Farbstoffe verwendet werden | ||
| Masterpuffer mit Methylcellulose (MBMC) | 1X BRB80, 0,8% Methylcellulose, 5 mM TCEP, 5 mM MgCl2 | 1 Woche | Bereiten Sie sich am Tag des Experiments vor; 1 mM DVB-t enthalten, wenn fluoreszierende Farbstoffe verwendet werden | ||
| Protein-Verdünnungspuffer (DB) | MB, 1 mg/ml Rinderserumalbumin (BSA), 1 μM ATP | 1 Tag, auf Eis | Bereiten Sie sich am Tag des Experiments vor; 1 mM DVB-t enthalten, wenn fluoreszierende Farbstoffe verwendet werden | ||
| Sauerstoff-Fangmischung (OSM) | MB, 389 μg/ml Katalase, 4,44 mg/ml Glucoseoxidase, 15,9 mM 2-Mercaptoethanol (BME) | 1 Tag, auf Eis | Bereiten Sie sich am Tag des Experiments vor | ||
| Sauerstofffänger-Finale (OSF) | MB, 350 μg/ml Katalase, 4mg/ml Glukoseoxidase, 14,3 mM BME, 15 mg/ml Glukose | Nutzung innerhalb von 30 min | Bereiten Sie sich unmittelbar vor Gebrauch vor, indem Sie 1 μL Glukose zu 9 μL OSM hinzufügen | ||
Tabelle 1: Liste der in diesem Protokoll verwendeten Puffer und ihrer Komponenten. In der Spalte "Empfohlene Speicherdauer" finden Sie eine Anleitung, wie weit im Voraus jeder Puffer vorbereitet werden kann.
| Reagenz | Speicherkonzentration | Lösungsmittel für die Lagerung | Lagertemperatur | Arbeitskonzentration | Endkonzentration | Empfohlene Speicherdauer | Notizen | |||||||||
| Neutravidin (NA) | 5 mg/ml | 1X BRB80 | -80°C | 0,2 mg/ml | 0,2 mg/ml | bis zu 1 Jahr | Wird verwendet, um Mikrotubuli über eine Biotin-Neutravidin-Biotin-Bindung zu immobilisieren; Laden Sie in kleinen Aliquots | |||||||||
| Kappa-Casein (KC) | 5 mg/ml | 1X BRB80 | -80°C | 0,5 mg/ml | 0,5 mg/ml | bis 2 Jahre | Wird verwendet, um die Oberfläche der Bildgebungskammer zu blockieren; In kleinen Aliquots lagern; Stellen Sie am Tag des Experiments ein kleines Volumen bei Raumtemperatur beiseite | |||||||||
| Rinderserumalbumin (BSA) | 50 mg/ml | 1X BRB80 | -20°C | 1 mg/ml (in DB) | N/A | bis 2 Jahre | Laden Sie in kleinen Aliquots | |||||||||
| Katalase | 3,5 mg/ml | 1X BRB80 | -80°C | 350 μg/ml (in OSF) | 35 μg/ml | bis 2 Jahre | Bestandteil der Sauerstofffängermischung; Laden Sie in kleinen Aliquots | |||||||||
| Glukoseoxidase | 40 mg/ml | 1X BRB80 | -80°C | 4 mg/ml (in OSF) | 0,4 mg/ml | bis 2 Jahre | Bestandteil der Sauerstofffängermischung; Laden Sie in kleinen Aliquots | |||||||||
| Tubulin | Lyophilisiert | N/A | 4°C | 10 mg/ml | 2,12 mg/ml (in Tubulinmischung) | bis zu 1 Jahr | Sobald Tubulin in Lösung ist, halten Sie es kalt, um eine Polymerisation zu vermeiden. | |||||||||
| Adenosintriphosphat (ATP) | 100 mM | Reinstwasser | -20°C | 10 mM | 1 mM | 6 Monate | Bereiten Sie die Lösung in filtersterilisiertem Wasser vor, stellen Sie den pH-Wert auf ~ 7,0 ein und frieren Sie sie in kleinen Aliquots ein. | |||||||||
| Guanosintriphosphat (GTP) | 100 mM | Reinstwasser | -20°C | 10 mM | 1,29 mM (in Tubulinmischung) | 6 Monate | Bereiten Sie die Lösung in filtersterilisiertem Wasser vor, stellen Sie den pH-Wert auf ~ 7,0 ein und frieren Sie sie in kleinen Aliquots ein. | |||||||||
| Guanosin-5'-[(α,β)-methyleno]-triphosphat (GMPCPP) | 10 mM | Reinstwasser | -20°C | 10 μM | 0,5 μM | 6 Monate | ||||||||||
| Dithiothreitol (DVB-T) | 1 Mio. | steriles Wasser | -20°C | 1 mM | N/A | bis 2 Jahre | ||||||||||
| Tris(2-carboxyethyl)-phosphin (TCEP) | 0,5 Mio. | filtersterilisiertes Wasser | Raumtemperatur | 5 mM | N/A | bis 2 Jahre | ||||||||||
| Methylcellulose | 1% | steriles Wasser | Raumtemperatur | 0,8 % (in MBMC) | 0,21% (in Tubulinmischung) | bis zu 1 Jahr | Lösen Sie Methylcellulose auf, indem Sie sie langsam in fast kochendes Wasser geben. Unter ständigem Rühren abkühlen lassen. | |||||||||
| Beta-Mercaptoethanol (BME) | 143 mM | steriles Wasser | Raumtemperatur | 14,3 mM (in OSF) | 1,43 mM | bis 5 Jahre | 143 mM ist eine 1:100-Verdünnung des Lager-BME | |||||||||
| Traubenzucker | 150 mg/ml | 1X BRB80 | -80°C | 15 mg/ml (in OSF) | 1,5 mg/ml | bis 2 Jahre | Unmittelbar vor der Verwendung zu OSM hinzufügen | |||||||||
| (±)-6-hydroxy-2,5,7,8-tetramethylchroman-2-carbonsäure (Trolox) | 10mM | 1X BRB80 | -80°C | 10 mM | 1 mM | bis zu 1 Jahr | Löst sich nicht vollständig auf. Fügen Sie etwas NaOH hinzu, rühren Sie für ~ 4 Stunden und filtern Sie vor dem Gebrauch sterilisieren | |||||||||
| mPEG-Succinimidylvalerat, MW 5.000 | Pulver | N/A | -20°C | 333 mg/ml (in 0,1 M Natriumbicarbonat) | 324 mg/ml (in 0,1 M Natriumbicarbonat) | 6 Monate | Bereiten Sie ~ 34 mg Aliquots zu und markieren Sie jede Tube mit einem genauen Gewicht des Pulvers. Stickstoffgas über den Feststoff leiten, Rohre mit Parafilm abdichten und bei -20 ° C in einem Behälter mit Trockenmittel lagern. | |||||||||
| Biotin-PEG-SVA, MW 5.000 | Pulver | N/A | -20°C | 111 mg/ml (in 0,1 M Natriumbicarbonat) | 3,24 mg/ml (in 0,1 M Natriumbicarbonat) | 6 Monate | Bereiten Sie ~ 3 mg Aliquots zu und markieren Sie jedes Röhrchen mit einem genauen Gewicht des Pulvers. Stickstoffgas über den Feststoff leiten, Rohre mit Parafilm abdichten und bei -20 ° C in einem Behälter mit Trockenmittel lagern. | |||||||||
Tabelle 2: Liste der in diesem Protokoll verwendeten Reagenzien Eingeschlossen sind die empfohlenen Lagerbedingungen und -konzentrationen, die Arbeitskonzentrationen der während des Experiments verwendeten Stammlösungen und die Endkonzentration in der Bildgebungskammer. Weitere Hinweise finden Sie in der Spalte ganz rechts.
2. Biotin-PEG-Objektträger vorbereiten
HINWEIS: Bereiten Sie die Bildgebungskammern so nah wie möglich am Beginn eines Experiments vor und nicht mehr als 2 Wochen im Voraus.
- Saubere Decklacke
- Positionieren Sie eine gleiche Anzahl von 24 x 60 mm und 18 x 18 mm #1,5 Deckgläsern (Abbildung 1F) in Schieberbeizgläsern bzw. Diawaschgestellen (Abbildung 1A,B). Legen Sie die Diawaschgestelle mit 18 x 18 mm Deckgläsern in ein 100 ml Becherglas.
- Spülen Sie alle Deckgläser 5-6 Mal in Reinstwasser (18,2 MΩ-cm Widerstand) und entfernen Sie überschüssige Flüssigkeit nach jedem Spülen mit einer Pipettenspitze, die an einem Vakuumrohr befestigt ist (Abbildung 1C).
- Bechergläser und Objektträger mit den Deckgläsern mit Reinstwasser füllen, mit Parafilm verschließen und 10 min beschallen.
- Füllen Sie zwei 150 ml Becher mit 200-prozentigem Ethanol. Tauchen Sie mit einer Pinzette jedes Deckglas in ein mit Ethanol gefülltes Becherglas und dann in das andere.
- Mit einer Pinzette Deckgläser auf das Gleitgitter übertragen (Abbildung 1D), unter Stickstoffgasstrom trocknen und bei 37 °C inkubieren, bis sie vollständig trocken sind (~15 min).
- Legen Sie die getrockneten Deckgläser in einer einzigen Schicht in den Plasmareiniger. Bilden Sie die Vakuumdichtung und stellen Sie dann den Hochfrequenzpegel (RF) des Plasmareinigers auf ~ 8 MHz ein.
- Sobald das Plasma erzeugt ist, lassen Sie Deckgläser im Plasmareiniger für 5 min. Schalten Sie den Plasmareiniger aus und lassen Sie das Vakuum langsam ab.
- Sobald die Vakuumdichtung gelöst ist, drehen Sie die Deckgläser um und wiederholen Sie die Plasmareinigung für 5 Minuten für die andere Seite der Deckgläser.
- Alternative zur Plasmareinigung: Anstelle der Schritte 2.1.2-2.1.3 werden Deckgläser in einer warmen Lösung von 2% Reinigungsmittel (in Reinstwasser) für 10 min beschallt. Dann waschen Sie die Deckgläser gründlich mit Reinstwasser und beschallen Sie in Reinstwasser 2-3 mal (jeweils 10 min). Als nächstes in Ethanol waschen und trocknen Sie wie in den Schritten 2.1.4-2.1.5. Überspringen Sie die Schritte 2.1.6-2.1.8.
- Biotin-PEG-Behandlung
- Unmittelbar vor Gebrauch 400 μL 3-Aminopropyltriethoxysilan in 40 ml Aceton auflösen. Bewegen Sie plasmagereinigte Deckgläser mit einer Pinzette in das Schieberwaschgestell und die Schieberfärbgläser. Deckgläser in 3-Aminopropyltriethoxysilan-Lösung tauchen und 5 min12,13 inkubieren.
- Waschen Sie alle Deckgläser 5-6 Mal mit Reinstwasser.
- Deckgläser in den Gleitgitter geben, unter einem Stickstoffgasstrom trocknen und bei 37 °C bis zur vollständigen Trocknung (~20 min) inkubieren.
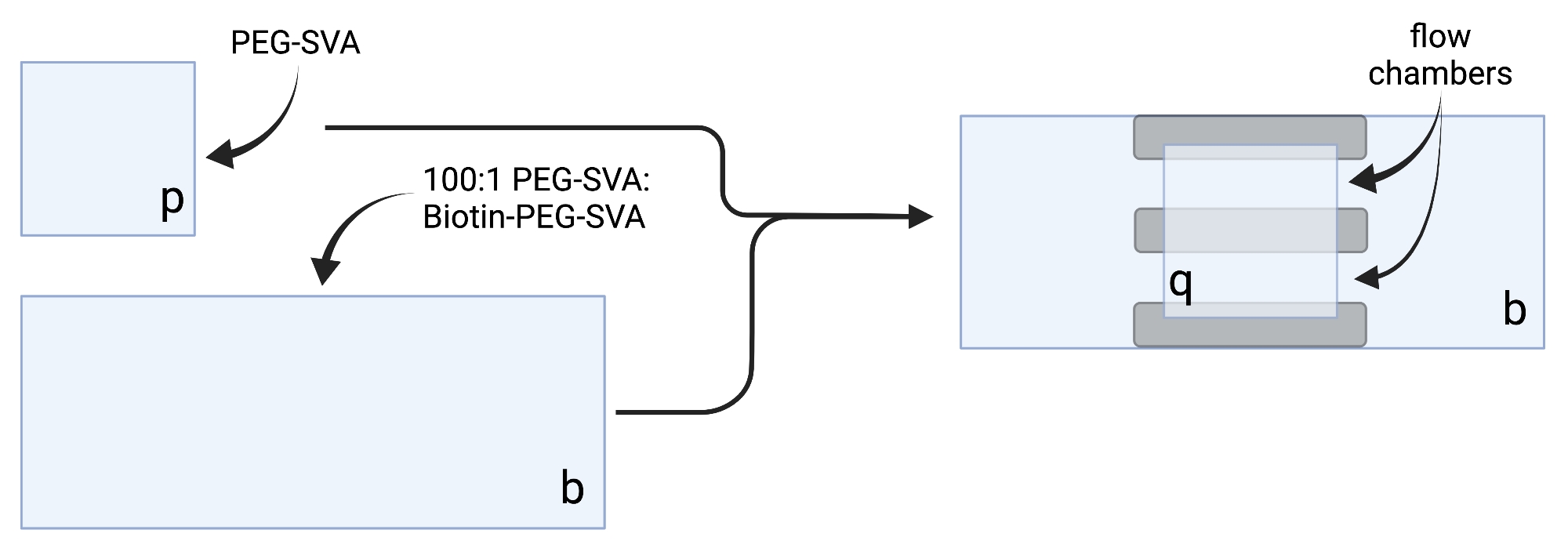
- Legen Sie die getrockneten Deckgläser auf empfindliche Aufgabentücher und beschriften Sie jedes Deckglas an einer Ecke, z. B. "p" auf jedem 18 x 18 mm großen Deckglas und "b" auf jedem 24 x 60 mm großen Deckglas (siehe Abbildung 2).
- Am Tag des Experiments wird eine frische 0,1 M Natriumbicarbonatlösung hergestellt, indem 0,84 mg NaHCO3 in 10 ml Reinstwasser gelöst werden.
- Aliquots von mPEG-Succinimidylvalerat (PEG-SVA) und Biotin-PEG-SVA unmittelbar vor Gebrauch auf Raumtemperatur bringen. Siehe Hinweise zur Aliquot-Zubereitung aus Polyethylenglykol (PEG) in Tabelle 2.
HINWEIS: Arbeiten Sie schnell, da die Hydrolysehalbwertszeit der Succinimidylvalerate (SVA) -Einheit ~ 30 min beträgt. - Fügen Sie 102 μL 0,1 M NaHCO3 zu 34 mg PEG-SVA hinzu, drehen Sie in einer Tischmikrozentrifuge bei 2.656 x g für 20 s und mischen Sie dann durch Pipettieren auf und ab. 3 mg Biotin-PEG-SVA in 27 μL 0,1 m NaHCO3 durch Pipettieren auf und ab auflösen. Passen Sie die Verdünnungsvolumina entsprechend dem genauen Gewicht des auf den Röhrchen notierten PEGs an (siehe Tabelle 2).
- Bereiten Sie ein 100:1 w/w PEG:Biotin-PEG-PEG-Gemisch vor, indem Sie 75 μL PEG-SVA-Lösung und 2,25 μL Biotin-PEG-SVA-Lösung für 20 Deckgläser, 100 μL und 3 μL für 30 Deckgläser oder 125 μL und 3,75 μL für 40 Deckgläser kombinieren.
- Konstruieren Sie eine Trinkkammer, indem Sie nasse Papierhandtücher unter dem Spitzengestell in den Boden einer leeren 10-μL-Spitzenbox legen (Abbildung 1E). Dadurch wird eine Verdunstung der PEG-Lösungen verhindert.
- Pipette 6 μL 100:1 PEG-SVA:biotin-PEG Gemisch auf die Mitte eines 24 x 60 mm Deckglases auf der beschrifteten Seite. Legen Sie ein weiteres 24 x 60 mm großes Deckglas auf das erste Deckglas, so dass das Paar eine X-Form bildet, wobei die mit "b" beschrifteten Seiten einander zugewandt sind. Legen Sie das Paar auf ein leeres Spitzengestell in der Trinkkammer und wiederholen Sie es für die restlichen 24 x 60 mm Deckgläser.
- Pipette 6 μL PEG-SVA auf die Mitte eines 18 x 18 mm Deckglases auf der beschrifteten Seite. Legen Sie ein weiteres 18 x 18 mm großes Deckglas auf das erste Deckglas, wobei die mit "p" beschrifteten Seiten einander zugewandt sind. Legen Sie das Paar auf ein leeres Spitzengestell in der Trinkkammer und wiederholen Sie es für die restlichen 18 x 18 mm Deckgläser.
- Schließen Sie die Trinkkammer und inkubieren Sie für 3 h oder über Nacht.
- Trennen Sie die Paar Deckgläser und spülen Sie sie in Reinstwasser ab.
- Deckgläser mit einem Stickstoffstrahl trocknen und in einen 37 °C Inkubator legen, um sie vollständig zu trocknen.
- Um die Bildgebungskammer zu konstruieren, kleben Sie drei Streifen doppelseitiges Klebeband auf ein 24 x 60 mm großes Deckglas auf der Seite mit der Aufschrift "b". Befestigen Sie auf der anderen Seite der Bandstreifen ein 18 x 18 mm großes Deckglas mit der Seitenbezeichnung "p" gegenüber dem größeren Deckglas. Diese bildet zwei Durchflusskammern für Mikroskopie-Experimente, wobei behandelte Oberflächen einander zugewandt sind (Abbildung 2 und Abbildung 1G).

Abbildung 1: Ausrüstung für die Deckglasbehandlung und die Vorbereitung der Bildgebungskammer . (A) Objektträgerbehältücher für 24 x 60 mm Deckgläser, (B) Diawaschgestelle für 18 x 18 mm Deckgläser, (C) Vakuumaufstellung, (D) Gleittrockner, (E) Trinkkammer, (F) Deckgläser, (G) Bildgebungskammer, (H) Objektträgerhalter. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 2: Schematische Darstellung der Herstellung von Bildgebungskammern mit doppelseitigem Klebeband (grau) und PEG/Biotin-PEG-behandelten Deckgläsern. Erstellt mit BioRender.com. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
3. Mikrotubuli polymerisieren
- Vorbereiten von GMPCPP-Samen
HINWEIS: Bereiten Sie GMPCPP-Samen in einem Kühlraum vor und bewahren Sie alle Reagenzien, Spitzen und Röhrchen bei 4 ° C auf. GMPCPP-Samen können im Voraus vorbereitet und bis zu 1 Jahr bei -80 ° C gelagert werden. Setzen Sie den Ultrazentrifugenrotor mit festem Winkel, der 1 ml Zentrifugenröhrchen enthält, in die Ultrazentrifuge und stellen Sie die Temperatur auf 4 °C ein.- Resuspendiert lyophilisiertes Tubulin (Tabelle 2) auf ~ 10 mg / ml in 1X BRB80, unmittelbar vor der Anwendung.
- Mischen Sie Bestandteile von GMPCPP-Saatgut wie in Tabelle 3 beschrieben.
HINWEIS: Halten Sie alle Tubulinkomponenten so weit wie möglich auf Eis, um die Polymerisation von löslichem Tubulin zu minimieren. - Klären Sie die Mischung in einem Ultrazentrifugenrotor mit festem Winkel bei 352.700 x g für 5 min bei 4 °C.
- Trennen Sie den Überstand in 5 μL Aliquote, schocken Sie sie in flüssigem Stickstoff ein und lagern Sie sie bei -80 ° C.
- Samen am Tag des Experiments polymerisieren
- Warm 1-2 ml BRB80-DTT (Tabelle 1) bis 37 °C.
- 5 μL Aliquot von GMPCPP-Samen (-80 °C) aus Schritt 3.1.4 auf Eis geben und sofort in 20 μL warmem BRB80-DTT auflösen. Bei Raumtemperatur mit 2.000 x g für 5 s drehen und zum Mischen antippen.
HINWEIS: Das anfängliche Verdünnungsvolumen kann zwischen 13 μL und 21 μL variieren und wird empirisch für jede Samencharge bestimmt. Wenn Samen nicht polymerisieren, wird die Fehlerbehebung durchgeführt, indem der anfängliche Verdünnungspuffer (Schritt 3.2.2) mit 0,5 μM GMPCPP ergänzt wird. - Vor Licht schützen und bei 37 °C für 30-45 min inkubieren.
HINWEIS: Die Länge der Mikrotubuli hängt von der Dauer der Inkubation ab. Bei kurzen Mikrotubuli kann die Inkubationszeit bis zu 15 min betragen. Bei langen Mikrotubuli kann die Inkubationszeit bis zu 2 h betragen. Biotinylierte Mikrotubuli benötigen tendenziell längere Inkubationszeiten als nicht-biotinylierte Mikrotubuli. - Setzen Sie den Ultrazentrifugenrotor mit festem Winkel, der 500-μL-Zentrifugenröhrchen enthält, in die Ultrazentrifuge und den Vorwarm auf 30 °C.
- Nach der Inkubation werden 50 μL warmes BRB80-DTT (Schritt 3.2.1) in die polymerisierten GMPCPP-Samen gegeben und die Mischung in ein 500-μL-Zentrifugenröhrchen überführt. Waschen Sie das leere Röhrchen, das die GMPCPP-Samen enthielt, mit weiteren 50 μL warmem BRB80-DTT, pipettieren Sie es auf und ab und fügen Sie diesen Puffer dem 500-μL-Zentrifugenröhrchen hinzu, das die Mischung enthält.
- Markieren Sie vor dem Spinnen den Rand des Zentrifugenröhrchens, um anzugeben, wo sich das Pellet befinden wird (das Pellet ist zu klein, um es zu sehen). Drehen Sie für 10 min bei 244.900 x g bei 30 °C12.
- Pipettieren Sie den Überstand vorsichtig aus und werfen Sie ihn weg. Resuspendiert das Pellet in 100 μL warmem BRB80-DTT. Tippen Sie zum Mixen.
- Drehen Sie für 10 Minuten bei 244.900 x g bei 30 ° C und richten Sie die Markierung mit dem Rotor aus, um an derselben Stelle zu pelletieren.
- Entfernen Sie den Überstand und suspendieren Sie das Pellet in 16 μL warmem BRB80-DTT. Überführen Sie die Mikrotubuli-Lösung in ein sauberes 0,6 ml Mikrozentrifugenröhrchen. Vor Licht schützen und bei oder über Raumtemperatur halten.
HINWEIS: Halten Sie die Mikrotubuli nach der Polymerisation bei oder über Raumtemperatur. Wenn sie kalt werden, depolymerisieren sie. Für zusätzliche Stabilität bei 28 °C inkubieren.
- Mikrotubuli mittels TIRF-Mikroskopie prüfen
- Ein Gemisch aus 4,5 μl BRB80-DTT und 1 μL Mikrotubulilösung (Schritt 3.2.9) auf einen Objektträger pipettieren. Mit einem 18 x 18 mm Deckglas abdecken und die Kanten entweder mit klarem Nagellack oder einem 1:1:1-Gemisch aus Vaseline, Lanolin und Paraffin (Valap-Dichtstoff) verschließen, das bei Raumtemperatur fest und bei 95 °C flüssig ist.
- Positionieren Sie das TIRF-Objektiv unter dem Deckglas (empfohlene Mikroskopeinstellungen siehe Schritt 4) und visualisieren Sie die neu polymerisierten Mikrotubuli bei einer Wellenlänge, die für das fluoreszierend markierte Tubulin in der Bright-Mischung geeignet ist (Tabelle 3), um zu bestimmen, welche Verdünnung von Mikrotubuli in den bevorstehenden Experimenten verwendet werden soll.
| Reagenz | Helle Mischung (μL) | Reihenfolge der Addition | Helle Mischung + Biotin (μL) | Reihenfolge der Addition |
| Fluoreszierendes Tubulin, 10 mg/ml | 2 | 6 | 2 | 7 |
| Biotin-Tubulin, 10 mg/ml | 0 | N/A | 2 | 6 |
| Unmarkiertes Tubulin, 10 mg/ml | 20 | 5 | 18 | 5 |
| GMPCPP, 10 mM | 30 | 4 | 30 | 4 |
| DVB-T, 0,2 m | 0.7 | 3 | 0.7 | 3 |
| 5X BRB80 | 26.4 | 2 | 26.4 | 2 |
| steriles Wasser | 52.9 | 1 | 52.9 | 1 |
| Gesamtvolumen (μL) | 132 | 132 |
Tabelle 3: GMPCPP-Saatgutmischung Bestandteile von GMPCPP-Mikrotubuli-Samen, einschließlich Volumen und Reihenfolge der Zugabe. 5 μL Aliquots vorbereiten und bis zu 1 Jahr bei -80 °C lagern.
4. Mikroskop-Einstellungen
- Temperatur: Stellen Sie die Mikroskoptemperatur auf 28 ° C ein, um dynamische Mikrotubuli anzuzeigen.
- Filter: Verwenden Sie die beste Kombination aus Filterwürfeln und Emissionsfiltern, abhängig von den abzubildenden Fluoreszenzkanälen. Um 488 nm, 560 nm und 647 nm Wellenlängen im selben Experiment zu visualisieren, verwenden Sie ein 405/488/560/647 nm Laser Quad Band Set, gekoppelt mit Emissionsfiltern für die angegebenen Wellenlängen.
- Laser ausrichten: Stellen Sie sicher, dass die im Experiment verwendeten Laserstrahlen ausgerichtet sind. Bestimmen Sie die Laserintensität für das Experiment empirisch, so dass alle fluoreszierenden Proteine mit dem höchstmöglichen Signal-Rausch-Verhältnis abgebildet werden können, aber im Laufe des Experiments keiner signifikanten Photobleiche unterzogen werden.
- Objektiv: Verwenden Sie Linsenpapier, um ein 100-faches Objektiv mit 70% Ethanol zu reinigen. Geben Sie vor der Bildgebung einen Tropfen Mikroskop-Tauchöl in das Objektiv.
- Einrichten einer Bildgebungssequenz
- Für ein Experiment mit 647 nm fluorophormarkierten biotinylierten Mikrotubuli, 560 nm fluorophormarkierten nicht-biotinylierten Mikrotubuli und löslichem Tubulin und GFP-markiertem Protein von Interesse, Bild für 20 min. Stellen Sie sich die 560-nm- und 488-nm-Kanäle alle 10 s und den 647-nm-Kanal alle 30 s vor.
- Um ein Referenzbild von Bündeln vor der Zugabe von löslichem Tubulin und MAPs aufzunehmen, richten Sie eine Sequenz mit je einem Bild in 560 nm und 647 nm Wellenlängen ein.
5. Erzeugen Sie oberflächenimmobilisierte Mikrotubulibündel
HINWEIS: Für die folgenden Schritte lassen Sie alle Lösungen in eine Durchflusskammer ein, indem Sie in eine offene Seite pipettieren, während Sie ein Filterpapier gegen die andere Seite legen. Schützen Sie die Bildgebungskammer vor Licht, um das Photobleichen von fluoreszierend markierten Proteinen zu reduzieren. Kleben Sie die vorbereitete Bildgebungskammer an einen Objektträgerhalter (Abbildung 1G,H). Führen Sie die Schritte in Tabelle 4 aus, die den Protokollsteps 5.2-6.4 entsprechen.
- Lösliche Tubulinmischung gemäß Tabelle 5 zubereiten und auf Eis aufbewahren.
HINWEIS: Lösliches Tubulin muss immer auf Eis gelegt werden, um eine Polymerisation zu verhindern. Bereiten Sie etwa alle 2 Stunden eine frisch lösliche Tubulinmischung vor oder wenn Mikrotubuli nicht mehr polymerisieren. - Um Mikrotubuli über eine Biotin-Neutravidin-Biotin-Bindung zu immobilisieren, fließen Sie zuerst in Neutravidin (NA) -Lösung, bis die Kammer gefüllt ist (~ 7,5 μL) und inkubieren für 5 min.
- Mit 10 μL MB-kalt waschen.
- Fließen Sie in 7,5 μL des blockierenden Proteins κ-Casein (KC) und inkubieren Sie für 2 min.
- Mit 10 μL MB-warm waschen, um die Kammer für die Einführung von Mikrotubuli vorzubereiten.
- Der Bestand an biotinylierten Mikrotubuli (gemäß Beobachtungen in Schritt 3.3.2) wird in 1X BRB80-DTT verdünnt und 1 μL dieser Verdünnung auf 9 μL MB-warm gegeben. Gießen Sie die Mischung in die Kammer und inkubieren Sie für 10 min. Verwenden Sie eine höhere Konzentration von Mikrotubuli für mehr Bündel.
- Nicht immobilisierte Mikrotubuli mit 10 μL MB-warm wegwaschen.
- Gießen Sie 7,5 μL warmes KC in die Kammer und inkubieren Sie es für 2 min.
- Während der Inkubation eine 2 nM Lösung des Vernetzerproteins PRC1 in warmem KC herstellen. Gießen Sie 10 μL dieser Lösung in die Durchflusskammer und inkubieren Sie für 5 min.
HINWEIS: Rekombinantes PRC1 wird wie zuvor beschrieben aus Bakterienzellen exprimiert und gereinigt13. - Um Bündel herzustellen, lassen Sie 10 μL nicht-biotinylierte Mikrotubuli in die Kammer und inkubieren Sie für 10 min. PRC1 vernetzt die nicht biotinylierten und immobilisierten biotinylierten Mikrotubuli15,16 (Abbildung 3).
HINWEIS: Siehe Schritt 6.1 für die Vorbereitung des Assay-Mixes während der Inkubationszeit. - Waschen Sie die Kammer zweimal mit 10 μL MB-warm. Die angebrachten Mikrotubuli sind von diesem Punkt an etwa 20 min stabil.
| Schritt | Reagenz | Volumen (μL) | Inkubationszeit (Minuten) |
| 1 | Neutravidin | 7.5 | 5 |
| 2 | MB-kalt | 10 | - |
| 3 | κ-Kasein | 7.5 | 2 |
| 4 | MB-warm | 10 | - |
| 5 | Biotinylierter Mikrotubuli (verdünnt in MB-warm) | 10 | 10 |
| 6 | MB-warm | 10 | - |
| 7 | Warmes κ-Kasein | 7.5 | 2 |
| 8 | 2 nM PRC1 verdünnt in κ-Casein | 10 | 5 |
| 9 | Nicht-biotinylierter Mikrotubuli | 10 | 10 |
| 10 | MB-warm x 2 | 10 | - |
| 11 | Assay-Mix | 10 | - |
| Angehängte Samen sind zu diesem Zeitpunkt etwa 20 Minuten stabil | |||
Tabelle 4: Assay-Schritte Liste der Reagenzien, die der Bildgebungskammer zugesetzt werden, mit Angabe der Wasch- (-) oder Inkubationszeit.
| Reagenz | Volumen (μL) |
| Recyceltes Tubulin, 10 mg/ml | 10 |
| MB-Kalt | 10.3 |
| MBMC | 13.7 |
| BRB80-DVB-T | 3.4 |
| GTP, 10 mM | 6.7 |
| ATP, 10 mM (Bei Verwendung von Kinesinen) | 6.7 |
| Fluoreszierend markiertes Tubulin, 10 mg/ml | 1 (Resuspendiert lyophilisiertes markiertes Tubulin in kaltem BRB80-DTT) |
Tabelle 5: Lösliche Tubulinmischungskomponenten. Zu Beginn des Experiments mischen und auf Eis halten.

Abbildung 3: Schematische Darstellung der Zugabe von Assay-Komponenten zur Herstellung und Abbildung fluoreszierend markierter Bündel und einzelner Mikrotubuli. Biotinylierte Samen sind in blauen, nicht biotinylierten Samen und löslichem Tubulin in Rot, PRC1 in Schwarz und Protein von Interesse in Cyan dargestellt. Die Schrittzahlen in der Abbildung entsprechen denen in Tabelle 4. Das Feld, das Schritt 9 entspricht, zeigt ein vorgeformtes Bündel (unten links); Schritt 11 zeigt ein neu gebildetes Bündel (oben links). Erstellt mit BioRender.com. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
6. Bildmikrotubuli-Dynamik
- Während der 10-minütigen Inkubationszeit in Schritt 5.10 werden 10 μL Assay-Mix mit interessanten Proteinen, löslichem Tubulin, Nukleotiden, Sauerstofffängern14 und Antioxidantien gemäß Tabelle 6 hergestellt. Halten Sie die Mischung auf Eis.
- Laden Sie die vorbereitete Bildgebungskammer, die auf den Objektträgerhalter geklebt ist, auf das 100-fache TIRF-Objektiv. Verwenden Sie die Kanäle 560 nm und 647 nm, um ein Sichtfeld zu finden, das eine optimale Anzahl und Dichte einzelner Mikrotubuli und Bündel enthält.
HINWEIS: Wenn sowohl biotinylierte als auch nicht-biotinylierte Mikrotubuli mit demselben Fluorophor markiert sind, können Zeilenscan-Analysen der Fluoreszenzintensitäten zwischen einzelnen Mikrotubuli und Bündeln unterscheiden. - Sobald ein Sichtfeld identifiziert ist, nehmen Sie ein Referenzbild auf.
- Fließen Sie vorsichtig in die Assay-Mischung, ohne die Bildgebungskammer zu stören.
- Versiegeln Sie die offenen Enden der Kammer mit Valap-Dichtmittel.
- Starten Sie die Bildgebungssequenz wie in Schritt 4.5.1 beschrieben.
| Reagenz | Volumen (μL) |
| Lösliche Tubulinmischung | 4 |
| OSF | 1 |
| Trolox (bei Verwendung von Mikrotubuli, die mit einem leicht photobleichenden Fluorophor gekennzeichnet sind) | 1 |
| ATP, 10 mM (Bei Verwendung von Kinesinen) | 1 |
| PRC1 (oder Vernetzer Ihrer Wahl) | 1 |
| Proteine von Interesse | X |
| MB-kalt | 2-fach |
Tabelle 6: Assay-Mix-Komponenten. Mischen Sie, fließen Sie in die Bildgebungskammer und bilden Sie die Mikrotubulidynamik innerhalb von 30 Minuten ab.
Ergebnisse
Das oben beschriebene Experiment wurde unter Verwendung von 647 nm fluorophormarkierten biotinylierten Mikrotubuli, 560 nm fluorophormarkierten nicht biotinylierten Mikrotubuli und 560 nm fluorophormarkierten löslichen Tubulinmischungen durchgeführt. Mikrotubuli wurden durch das Vernetzerprotein PRC1 (GFP-markiert) vernetzt. Nachdem oberflächenimmobilisierte Bündel und einzelne Mikrotubuli erzeugt wurden (Schritt 5.11), wurde die Bildgebungskammer auf einem TIRF 100X 1.49 NA Ölobjektiv montiert und in den 560 nm und...
Diskussion
Das hier beschriebene Experiment erweitert den Umfang und die Komplexität herkömmlicher Mikrotubuli-Rekonstitutionsassays, die traditionell an einzelnen Mikrotubuli oder an einem Array-Typ durchgeführt werden, erheblich. Der aktuelle Assay bietet eine Methode zur gleichzeitigen Quantifizierung und zum Vergleich der regulatorischen MAP-Aktivität an zwei Populationen, nämlich einzelnen Mikrotubuli und vernetzten Bündeln. Darüber hinaus ermöglicht dieser Assay die Untersuchung von zwei Arten von Bündeln: solchen, d...
Offenlegungen
Die Autoren erklären keine konkurrierenden Interessen.
Danksagungen
Diese Arbeit wurde durch einen Zuschuss des NIH (Nr. 1DP2GM126894-01) und durch Mittel der Pew Charitable Trusts und der Smith Family Foundation an R.S. unterstützt. Die Autoren danken Dr. Shuo Jiang für seinen Beitrag zur Entwicklung und Optimierung der Protokolle.
Materialien
| Name | Company | Catalog Number | Comments |
| (±)-6-Hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (Trolox) | Sigma Aldrich | 238813 | |
| 1,4-piperazinediethanesulfonic acid (PIPES) | Sigma Aldrich | P6757 | |
| 18x18 mm #1.5 coverslips | Electron Microscopy Sciences | 63787 | |
| 2-Mercaptoethanol (BME) | Sigma Aldrich | M-6250 | |
| 24x60 mm #1.5 coverslips | Electron Microscopy Sciences | 63793 | |
| 405/488/560/647 nm Laser Quad Band | Chroma | TRF89901-NK | |
| Acetone | Sigma Aldrich | 320110 | |
| Adenosine 5'-triphosphate disodium salt hydrate (ATP) | Sigma Aldrich | A7699-5G | |
| Avidin, NeutrAvidin® Biotin-binding Protein (Molecular Probes®) | Thermo Fischer Scientific | A2666 | |
| Bath sonicator: Branson 2800 Cleaner | Branson | CPX2800H | |
| Beckman Coulter Polycarbonate Thickwall Tubes, 11 x 34 mm | Beckman-Coulter | 343778 | |
| Beckman Coulter Polycarbonate Thickwall Tubes, 8 x 34 mm | Beckman-Coulter | 343776 | |
| Biotin-PEG-SVA, MW 5,000 | Laysan Bio | #Biotin-PEG-SVA-5000 | |
| Bovine Serum Albumin (BSA) | Sigma Aldrich | 2905 | |
| Catalase | Sigma Aldrich | C40 | |
| Corning LSE Mini Microcentrifuge, AC100-240V | Corning | 6670 | |
| Delicate Task Wipes | Kimtech | 34120 | |
| Dithiothreitol (DTT) | GoldBio | DTT10 | |
| Emission filter | Chroma | ET610/75m | |
| Ethanol (200-proof) | Decon Labs | 2705 | |
| Ethylene glycol tetraacetic acid (EGTA) | Sigma Aldrich | 3777 | |
| Glucose Oxidase | Sigma Aldrich | G2133 | |
| GMPCPP | Jena Bioscience | NU-405 | |
| Guanosine 5'-triphosphate sodium salt hydrate (GTP) | Sigma Aldrich | G8877 | |
| Hellmanex III detergent | Sigma Aldrich | Z805939 | |
| Immersion oil, Type A | Fisher Scientific | 77010 | |
| Kappa-casein | Sigma Aldrich | C0406 | |
| Lanolin | Fisher Scientific | S25376 | |
| Lens Cleaning Tissue | ThorLabs | MC-5 | |
| Magnesium Chloride (MgCl2) | Sigma Aldrich | M9272 | |
| Methylcellulose | Sigma Aldrich | M0512 | |
| Microfuge 16 Benchtop Centrifuge | Beckman-Coulter | A46474 | |
| Microscope Slides, Diamond White Glass, 25 x 75mm, 90° Ground Edges, WHITE Frosted | Globe Scientific | 1380-50W | |
| mPEG-Succinimidyl Valerate, MW 5,000 | Laysan Bio | #NH2-PEG-VA-5K | |
| Optima™ Max-XP Tabletop Ultracentrifuge | Beckman-Coulter | 393315 | |
| Paraffin | Fisher Scientific | P31-500 | |
| PELCO Reverse (self-closing), Fine Tweezers | Ted Pella | 5377-NM | |
| Petrolatum, White | Fisher Scientific | 18-605-050 | |
| Plasma Cleaner, 115V | Harrick Plasma | PDC-001 | |
| Potassium Hydroxide (KOH) | Sigma Aldrich | 221473 | |
| Sodium bicarbonate | Sigma Aldrich | S6014 | |
| Sucrose | Sigma Aldrich | S7903 | |
| Thermal-Lok 1-Position Dry Heat Bath | USA Scientific | 2510-1101 | |
| Thermal-Lok Block for 1.5 and 2.0 mL Tubes | USA Scientific | 2520-0000 | |
| Thermo Scientific™ Pierce™ Bond-Breaker™ TCEP Solution, Neutral pH; 500mM | Thermo Fischer Scientific | PI-77720 | |
| TIRF 100X NA 1.49 Oil Objective | Nikon | CFI Apochromat TIRF 100XC Oil | |
| TIRF microscope | Nikon | Eclipse Ti | |
| TLA 120.1 rotor | Beckman-Coulter | 362224 | |
| TLA 120.2 rotor | Beckman-Coulter | 357656 | |
| Tubulin protein (>99% pure): porcine brain | Cytoskeleton | T240 | |
| Tubulin Protein (Biotin): Porcine Brain | Cytoskeleton | T333P | |
| Tubulin protein (fluorescent HiLyte 647): porcine brain | Cytoskeleton | TL670M | |
| Tubulin protein (X-rhodamine): bovine brain | Cytoskeleton | TL620M | |
| VECTABOND® Reagent, Tissue Section Adhesion | Vector Biolabs | SP-1800-7 | |
| VWR® Personal-Sized Incubator, 120V, 50/60Hz, 0.6A | VWR | 97025-630 | |
Referenzen
- Subramanian, R., Kapoor, T. M. Building complexity: insights into self-organized assembly of microtubule-based architectures. Developmental Cell. 23 (5), 874-885 (2012).
- Baas, P. W., Rao, A. N., Matamoros, A. J., Leo, L. Stability properties of neuronal microtubules. Cytoskeleton (Hoboken). 73 (9), 442-460 (2016).
- Bitan, A., Rosenbaum, I., Abdu, U. Stable and dynamic microtubules coordinately determine and maintain Drosophila bristle shape. Development. 139 (11), 1987-1996 (2012).
- Foe, V. E., von Dassow, G. Stable and dynamic microtubules coordinately shape the myosin activation zone during cytokinetic furrow formation. The Journal of Cell Biology. 183 (3), 457-470 (2008).
- Pous, C., et al. Functional specialization of stable and dynamic microtubules in protein traffic in WIF-B cells. The Journal of Cell Biology. 142 (1), 153-165 (1998).
- Uehara, R., Goshima, G. Functional central spindle assembly requires de novo microtubule generation in the interchromosomal region during anaphase. The Journal of Cell Biology. 191 (2), 259-267 (2010).
- Mitchison, T., Kirschner, M. Dynamic instability of microtubule growth. Nature. 312 (5991), 237-242 (1984).
- Bieling, P., et al. Reconstitution of a microtubule plus-end tracking system in vitro. Nature. 450 (7172), 1100-1105 (2007).
- Bieling, P., Telley, I. A., Surrey, T. A minimal midzone protein module controls formation and length of antiparallel microtubule overlaps. Cell. 142 (3), 420-432 (2010).
- Shimamoto, Y., Forth, S., Kapoor, T. M. Measuring pushing and braking forces generated by ensembles of Kinesin-5 crosslinking two microtubules. Developmental Cell. 34 (6), 669-681 (2015).
- Mani, N., Jiang, S., Neary, A. E., Wijeratne, S. S., Subramanian, R. Differential regulation of single microtubules and bundles by a three-protein module. Nature Chemical Biology. 17 (9), 964-974 (2021).
- Hyman, A. A., Salser, S., Drechsel, D., Unwin, N., Mitchison, T. J. Role of GTP hydrolysis in microtubule dynamics: information from a slowly hydrolyzable analogue, GMPCPP. Molecular Biology of the Cell. 3 (10), 1155-1167 (1992).
- Subramanian, R., et al. Insights into antiparallel microtubule crosslinking by PRC1, a conserved nonmotor microtubule binding protein. Cell. 142 (3), 433-443 (2010).
- Rasnik, I., McKinney, S. A., Ha, T. Nonblinking and long-lasting single-molecule fluorescence imaging. Nature Methods. 3 (11), 891-893 (2006).
- Wijeratne, S., Subramanian, R. Geometry of antiparallel microtubule bundles regulates relative sliding and stalling by PRC1 and Kif4A. eLife. 7, 32595 (2018).
- Mani, N., Wijeratne, S. S., Subramanian, R. Micron-scale geometrical features of microtubules as regulators of microtubule organization. eLife. 10, 63880 (2021).
- Freal, A., et al. Feedback-driven assembly of the axon initial segment. Neuron. 104 (2), 305-321 (2019).
- Ledbetter, M., Porter, K. A "microtubule" in plant cell fine structure. The Journal of Cell Biology. 19 (1), 239-250 (1963).
- Wijeratne, S. S., Marchan, M. F., Tresback, J. S., Subramanian, R. Atomic force microscopy reveals distinct protofilament-scale structural dynamics in depolymerizing microtubule arrays. Proceedings of the National Academy of Sciences of the United States of America. , 119 (2022).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten