
Для просмотра этого контента требуется подписка на Jove Войдите в систему или начните бесплатную пробную версию.
Method Article
Одновременная визуализация динамики сшитых и одиночных микротрубочек in vitro методом TIRF-микроскопии
В этой статье
Резюме
Здесь представлен анализ восстановления in vitro на основе микроскопии TIRF для одновременной количественной оценки и сравнения динамики двух популяций микротрубочек. Описан способ одновременного просмотра коллективной активности нескольких микротрубочек-ассоциированных белков на сшитых пучках микротрубочек и одиночных микротрубочек.
Аннотация
Микротрубочки представляют собой полимеры гетеродимеров αβ-тубулина, которые организуются в различные структуры в клетках. Архитектуры и сети на основе микротрубочек часто содержат подмножества массивов микротрубочек, которые различаются по своим динамическим свойствам. Например, в делящихся клетках стабильные пучки сшитых микротрубочек сосуществуют в непосредственной близости от динамических несшитых микротрубочек. Исследования восстановления in vitro на основе TIRF-микроскопии позволяют одновременно визуализировать динамику этих различных массивов микротрубочек. В этом анализе камера визуализации собирается с поверхностно-иммобилизованными микротрубочками, которые либо присутствуют в виде одиночных нитей, либо организованы в сшитые пучки. Введение тубулина, нуклеотидов и белковых регуляторов позволяет непосредственно визуализировать ассоциированные белки и динамические свойства одиночных и сшитых микротрубочек. Кроме того, изменения, которые происходят, когда динамические одиночные микротрубочки организуются в пучки, могут отслеживаться в режиме реального времени. Описанный здесь способ позволяет проводить систематическую оценку активности и локализации отдельных белков, а также синергетических эффектов белковых регуляторов на два разных подмножества микротрубочек в идентичных экспериментальных условиях, тем самым обеспечивая механистические идеи, недоступные другим методам.
Введение
Микротрубочки представляют собой биополимеры, которые образуют структурные каркасы, необходимые для нескольких клеточных процессов, начиная от внутриклеточного транспорта и позиционирования органелл до деления и удлинения клеток. Для выполнения этих разнообразных функций отдельные микротрубочки организованы в массивы размером с микрон, такие как митотические веретена, цилиарные аксонемы, пучки нейронов, межфазные массивы и растительные корковые массивы. Вездесущим архитектурным мотивом, найденным в этих структурах, является пучок микротрубочек, сшитых по их длине1. Интригующей особенностью нескольких структур на основе микротрубочек является сосуществование пучковых микротрубочек и несшитых одиночных микротрубочек в непосредственной пространственной близости. Эти субпопуляции микротрубочек могут демонстрировать резко отличающуюся динамику полимеризации друг от друга, как это необходимо для их правильного функционирования2,3,4,5. Например, внутри митотического шпинделя стабильные сшитые пучки и динамические одиночные микротрубочки присутствуют в микронной области в центре ячейки6. Таким образом, изучение того, как определяются динамические свойства сосуществующих популяций микротрубочек, имеет решающее значение для понимания сборки и функции структур на основе микротрубочек.
Микротрубочки представляют собой динамические полимеры, которые циклически переключаются между фазами полимеризации и деполимеризации, переключаясь между двумя фазами в событиях, известных как катастрофа и спасение7. Динамика клеточных микротрубочек регулируется мириадами микротрубочек,ассоциированных белков (MAP), которые модулируют скорость полимеризации и деполимеризации микротрубочек и частоты катастроф и спасательных событий. Исследовать активность MAP на пространственно проксимальных массивах в клетках сложно из-за ограничений пространственного разрешения в световой микроскопии, особенно в областях с высокой плотностью микротрубочек. Более того, наличие нескольких MAP в одной и той же клеточной области затрудняет интерпретацию клеточных биологических исследований. Анализы восстановления in vitro, выполняемые в сочетании с микроскопией полной флуоресценции внутреннего отражения (TIRF), обходят проблемы изучения механизмов, с помощью которых конкретные подмножества MAP регулируют динамику проксимальных клеточных микротрубочек. Здесь исследуется динамика собранных in vitro микротрубочек в присутствии одного или нескольких рекомбинантных MAP в контролируемых условиях8,9,10. Однако обычные анализы восстановления обычно выполняются на одиночных микротрубочках или на одном типе массива, что исключает визуализацию сосуществующих популяций.
Здесь мы представляем анализы восстановления in vitro , которые позволяют одновременно визуализировать две популяции микротрубочек в одних и тех же условиях решения11. Описан способ одновременного просмотра коллективной активности нескольких MAP на одиночных микротрубочках и на пучках микротрубочек, сшитых митотическим веретенно-ассоциированным белком PRC1. Белок PRC1 преимущественно связывается при перекрытии между антипараллельными микротрубочками, сшивая их9. Вкратце, этот протокол состоит из следующих этапов: (i) подготовка исходных растворов и реагентов, (ii) очистка и обработка поверхности покровных пластин, используемых для создания камеры визуализации для микроскопических экспериментов, (iii) подготовка стабильных «семян» микротрубочек, из которых полимеризация инициируется в ходе эксперимента, (iv) спецификация настроек микроскопа TIRF для визуализации динамики микротрубочек, (v) иммобилизация семян микротрубочек и генерация сшитых пучков микротрубочек в камере визуализации и (vi) визуализация динамики микротрубочек в камере визуализации с помощью микроскопии TIRF при добавлении растворимого тубулина, MAP и нуклеотидов. Эти анализы позволяют качественно оценить и количественно изучить локализацию MAP и их влияние на динамику двух популяций микротрубочек. Кроме того, они облегчают оценку синергетического воздействия нескольких MAP на эти популяции микротрубочек в широком диапазоне экспериментальных условий.
Access restricted. Please log in or start a trial to view this content.
протокол
1. Подготовьте реагенты
- Подготовьте буферы и реагенты, как показано в таблице 1 и таблице 2. Во время эксперимента держите все растворы на льду, если не указано иное.
| Решение | Компоненты | Рекомендуемая продолжительность хранения | Примечания | ||
| 5X BRB80 | 400 мМ K-PIPES, 5 мМ MgCl2, 5 мМ EGTA, рН 6,8 с KOH, фильтр стерилизуется | до 2 лет | Хранить при температуре 4 °C | ||
| 1X BRB80 | 80 мМ K-PIPES, 1 мМ MgCl2, 1 мМ EGTA, pH 6,8 | до 2 лет | Хранить при температуре 4 °C | ||
| БРБ80-ДТТ | 1X BRB80, 1 мМ DTT | до 2 дней | |||
| Буфер анализа | 80 мМ K-PIPES, 3 мМ MgCl2, 1 мМ EGTA, рН 6,8, 5% сахарозы (OR 1X BRB80, 5% сахарозы, 2 мМ MgCl2) | до 1 года | Хранить при температуре 4 °C | ||
| Главный буфер (МБ) | Буфер анализа, 5 мМ TCEP | 1 неделя | Подготовьтесь в день эксперимента; Разделите на две трубки: MB-теплый при комнатной температуре и MB-холодный на льду; включают 1 мМ DTT при использовании флуоресцентных красителей | ||
| Главный буфер с метилцеллюлозой (MBMC) | 1X BRB80, 0,8% метилцеллюлоза, 5 мМ TCEP, 5 мМ MgCl2 | 1 неделя | Подготовьтесь в день эксперимента; включают 1 мМ DTT при использовании флуоресцентных красителей | ||
| Буфер разбавления белка (DB) | MB, 1 мг/мл бычьего сывороточного альбумина (BSA), 1 мкМ АТФ | 1 день, на льду | Подготовьтесь в день эксперимента; включают 1 мМ DTT при использовании флуоресцентных красителей | ||
| Кислородопоглощающая смесь (OSM) | MB, 389 мкг/мл каталазы, 4,44 мг/мл глюкозооксидазы, 15,9 мМ 2-меркаптоэтанола (BME) | 1 день, на льду | Подготовьтесь в день эксперимента | ||
| Финал очистки кислорода (OSF) | MB, 350 мкг/мл каталазы, 4 мг/мл глюкозооксидазы, 14,3 мМ BME, 15 мг/мл глюкозы | использовать в течение 30 мин | Подготовьте непосредственно перед использованием, добавив 1 мкл глюкозы к 9 мкл OSM | ||
Таблица 1: Список буферов, используемых в этом протоколе, и их компонентов. В столбце «Рекомендуемая продолжительность хранения» приведены рекомендации о том, насколько заблаговременно можно подготовить каждый буфер.
| Реагент | Концентрация при хранении | Растворитель для хранения | Температура хранения | Рабочая концентрация | Конечная концентрация | Рекомендуемая продолжительность хранения | Примечания | |||||||||
| Нейтравидин (NA) | 5 мг/мл | 1X BRB80 | -80°С | 0,2 мг/мл | 0,2 мг/мл | до 1 года | Используется для иммобилизации микротрубочек через связь биотин-нейтравидин-биотин; хранить в небольших аликвотах | |||||||||
| Каппа-казеин (KC) | 5 мг/мл | 1X BRB80 | -80°С | 0,5 мг/мл | 0,5 мг/мл | до 2 лет | Используется для блокировки поверхности камеры визуализации; Хранить в небольших аликвотах; В день эксперимента отложите небольшой объем в сторону при комнатной температуре | |||||||||
| Бычий сывороточный альбумин (BSA) | 50 мг/мл | 1X BRB80 | -20°С | 1 мг/мл (в БД) | Н/Д | до 2 лет | хранить в небольших аликвотах | |||||||||
| Каталазы | 3,5 мг/мл | 1X BRB80 | -80°С | 350 мкг/мл (в OSF) | 35 мкг/мл | до 2 лет | компонент кислородопоглощающей смеси; хранить в небольших аликвотах | |||||||||
| Глюкозооксидаза | 40 мг/мл | 1X BRB80 | -80°С | 4 мг/мл (в OSF) | 0,4 мг/мл | до 2 лет | компонент кислородопоглощающей смеси; хранить в небольших аликвотах | |||||||||
| Тубулин | Лиофилизированный | Н/Д | 4°С | 10 мг/мл | 2,12 мг/мл (в смеси тубулина) | до 1 года | Как только тубулин окажется в растворе, держите его холодным, чтобы избежать полимеризации. | |||||||||
| Аденозинтрифосфат (АТФ) | 100 мМ | сверхчистая вода | -20°С | 10 мМ | 1 мМ | 6 месяцев | Приготовьте раствор в фильтрованной стерилизованной воде, отрегулируйте рН до ~7,0 и заморозьте небольшими аликвотами. | |||||||||
| Гуанозинтрифосфат (ГТФ) | 100 мМ | сверхчистая вода | -20°С | 10 мМ | 1,29 мМ (в тубулиновой смеси) | 6 месяцев | Приготовьте раствор в фильтрованной стерилизованной воде, отрегулируйте рН до ~7,0 и заморозьте небольшими аликвотами. | |||||||||
| Гуанозин-5'-[(α,β)-метилено]трифосфат (GMPCPP) | 10 мМ | сверхчистая вода | -20°С | 10 мкМ | 0.5 мкМ | 6 месяцев | ||||||||||
| Дитиотрейтол (DTT) | 1 М | стерильная вода | -20°С | 1 мМ | Н/Д | до 2 лет | ||||||||||
| Трис(2-карбоксиэтил)фосфин (TCEP) | 0.5 млн | фильтрованно-стерилизованная вода | Комнатная температура | 5 мМ | Н/Д | до 2 лет | ||||||||||
| Метилцеллюлоза | 1% | стерильная вода | Комнатная температура | 0,8% (в МБМК) | 0,21% (в смеси тубулина) | до 1 года | Растворите метилцеллюлозу, медленно добавляя ее в почти кипящую воду. Дайте остыть, постоянно помешивая. | |||||||||
| Бета-меркаптоэтанол (BME) | 143 мМ | стерильная вода | Комнатная температура | 14,3 мМ (в OSF) | 1,43 мМ | до 5 лет | 143 мМ - разбавление запаса BME 1:100 | |||||||||
| Глюкоза | 150 мг/мл | 1X BRB80 | -80°С | 15 мг/мл (в OSF) | 1,5 мг/мл | до 2 лет | Добавление в OSM непосредственно перед использованием | |||||||||
| (±)-6-гидрокси-2,5,7,8-тетраметилхроман-2-карбоновая кислота (Тролокс) | 10мМ | 1X BRB80 | -80°С | 10 мМ | 1 мМ | до 1 года | Не растворяется полностью. Добавьте немного NaOH, перемешайте в течение ~ 4 часов и фильтруйте стерилизовать перед использованием | |||||||||
| мПЕГ-Сукцинимидил валерат, МВт 5,000 | порошок | Н/Д | -20°С | 333 мг/мл (в 0,1 М бикарбоната натрия) | 324 мг/мл (в 0,1 М бикарбоната натрия) | 6 месяцев | Приготовьте ~34 мг аликвоты, пометив каждый тюбик точным весом порошка. Пропустите газообразный азот над твердым веществом, запечатайте трубки с парапленкой и храните при -20°C в контейнере с осушителем. | |||||||||
| Биотин-ПЭГ-СВА, МВт 5,000 | порошок | Н/Д | -20°С | 111 мг/мл (в 0,1 М бикарбоната натрия) | 3,24 мг/мл (в 0,1 М бикарбоната натрия) | 6 месяцев | Приготовьте ~3 мг аликвот, пометив каждый тюбик точным весом порошка. Пропустите газообразный азот над твердым веществом, запечатайте трубки с парапленкой и храните при -20°C в контейнере с осушителем. | |||||||||
Таблица 2: Список реагентов, используемых в данном протоколе. Включены рекомендуемые условия хранения и концентрации, рабочие концентрации исходных растворов, используемых во время эксперимента, и конечная концентрация в камере визуализации. Дополнительные примечания приведены в крайнем правом столбце.
2. Подготовьте слайды Biotin-PEG
ПРИМЕЧАНИЕ: Подготовьте камеры визуализации как можно ближе к началу эксперимента и не более чем за 2 недели.
- Чистые крышки
- Расположите равное количество крышек размером 24 x 60 мм и 18 x 18 мм #1,5 (рисунок 1F) в банках для окрашивания слайдов и стеллажах для мойки слайдов соответственно (рисунок 1A, B). Поместите стеллажи для стирки для скольжения, содержащие крышки размером 18 x 18 мм, в стакан емкостью 100 мл.
- Промывайте все крышки 5-6 раз в сверхчистой воде (удельное сопротивление 18,2 МОм-см) и удаляйте лишнюю жидкость после каждого ополаскивания с помощью наконечника пипетки, прикрепленного к вакуумной трубке (рисунок 1C).
- Наполните стаканы и банки для окрашивания слайдов, содержащие крышки, сверхчистой водой, запечатайте парапленкой и обработайте ультразвуком в течение 10 минут.
- Наполните два стакана по 150 мл 200-стойким этанолом. Используя пинцет, окуните каждую крышку в один стакан, наполненный этанолом, а затем в другой.
- С помощью пинцета переложите крышки в скользящую сушильную стойку (рисунок 1D), высушите их под потоком газообразного азота и инкубируйте при 37 °C до полного высыхания (~15 мин).
- Поместите высушенные крышки в один слой внутри плазменного очистителя. Сформируйте вакуумное уплотнение, а затем установите радиочастотный (РЧ) уровень плазменного очистителя на ~8 МГц.
- Как только плазма сгенерирована, оставьте крышки в плазмоочистителе на 5 минут. Выключите плазмоочиститель и медленно отпустите вакуум.
- Как только вакуумное уплотнение будет освобождено, переверните крышки и повторите плазменную очистку в течение 5 минут для другой стороны обшивки.
- Альтернатива плазменной очистке: Вместо шагов 2.1.2-2.1.3, обтекайте ультразвуком обтекайте крышки в теплом растворе 2% моющего средства (в сверхчистой воде) в течение 10 мин. Затем тщательно промыть крышки сверхчистой водой и обработать ультразвуком в сверхчистой воде 2-3 раза (по 10 мин каждая). Далее промыть в этаноле и высушить, как на этапах 2.1.4-2.1.5. Пропустите шаги 2.1.6-2.1.8.
- Лечение биотин-ПЭГ
- Непосредственно перед применением растворяют 400 мкл 3-аминопропилтриэтоксисилана в 40 мл ацетона. С помощью пинцета переместите очищенные плазмой крышки в стойку для стирки слайдов и банки для окрашивания слайдов. Погружайте покровы в 3-аминопропилтриэтоксисилановый раствор и инкубируйте в течение 5 мин12,13.
- Вымойте все крышки 5-6 раз сверхчистой водой.
- Переложите крышки в сушильную стойку, высушите их под потоком газообразного азота и высиживаете при 37 °C до полного высыхания (~20 мин).
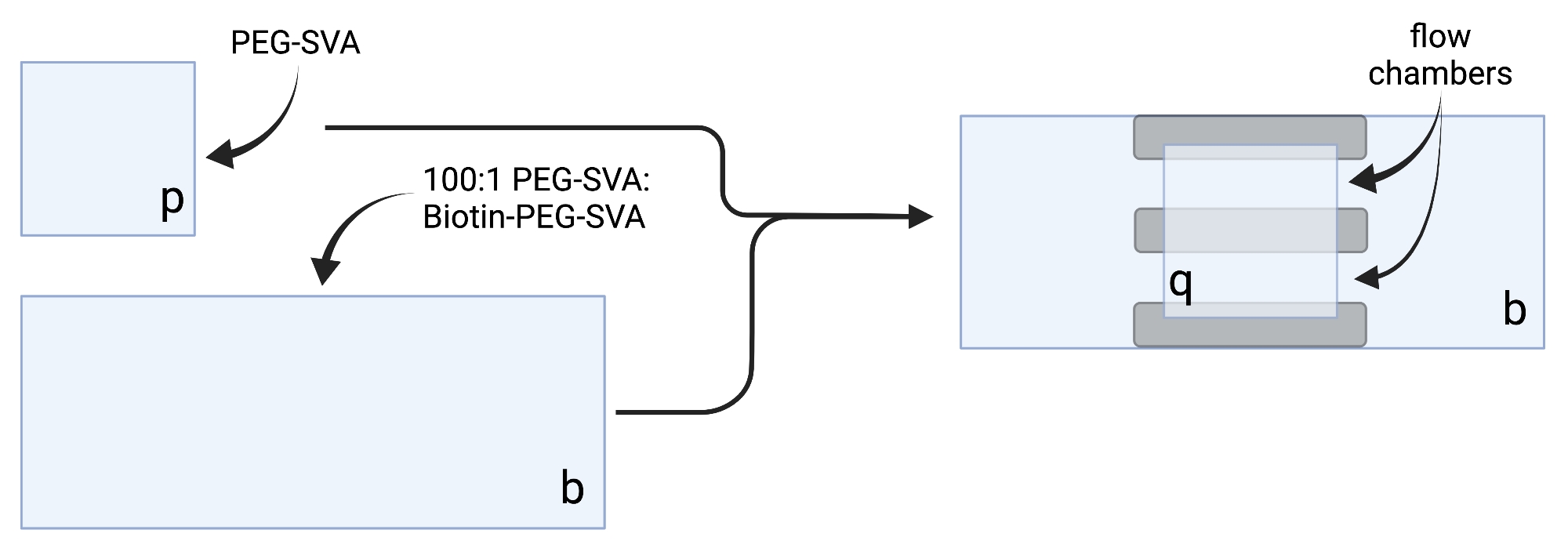
- Положите высушенные крышки на деликатные салфетки и пометьте каждый обложек на одном углу, например, 'p' на каждом чехле 18 x 18 мм и 'b' на каждом чехле 24 x 60 мм (см. Рисунок 2).
- В день эксперимента готовят свежий 0,1 М раствор бикарбоната натрия, растворяя 0,84 мг NaHCO3 в 10 мл сверхчистой воды.
- Доведите аликвоты mPEG-Сукцинимидил валерата (PEG-SVA) и Биотин-PEG-SVA до комнатной температуры непосредственно перед использованием. См. примечания по приготовлению аликвоты полиэтиленгликоля (ПЭГ) в таблице 2.
ПРИМЕЧАНИЕ: Работайте быстро, потому что период полураспада гидролиза фрагмента сукцинимидила валерата (SVA) составляет ~ 30 мин. - Добавьте 102 мкл 0,1 М NaHCO3 к 34 мг ПЭГ-СВА, вращайте в настольной микроцентрифуге при 2,656 х г в течение 20 с, а затем перемешайте путем пипетки вверх и вниз. Растворить 3 мг биотина-ПЭГ-СВА в 27 мкл 0,1 М NaHCO3 путем пипетирования вверх и вниз. Отрегулируйте объемы разбавления в соответствии с точным весом ПЭГ, указанным на трубах (см. таблицу 2).
- Приготовьте смесь 100:1 п/б ПЭГ:биотин-ПЭГ, объединив 75 мкл раствора ПЭГ-СВА и 2,25 мкл раствора Биотин-ПЭГ-СВА для 20 облицовок, 100 мкл и 3 мкл для 30 обшивок или 125 мкл и 3,75 мкл для 40 обложек.
- Постройте камеру гидратации, поместив влажные бумажные полотенца под стойку наконечника в нижней части пустого наконечника объемом 10 мкл (рисунок 1E). Это предотвратит испарение растворов ПЭГ.
- Пипетка 6 мкл смеси PEG-SVA:biotin-PEG 100:1 на центр одного чехла размером 24 x 60 мм на маркированной стороне. Поместите еще один обшивочный лист размером 24 x 60 мм поверх первого чехлового листа таким образом, чтобы пара образовывала X-образную форму, а стороны с надписью «b» были обращены друг к другу. Поместите пару на пустую стойку наконечника в увлажняющей камере и повторите для оставшихся 24 x 60 мм обшивочных прорезей.
- Пипетка 6 мкл PEG-SVA по центру одного чехла размером 18 x 18 мм на маркированной стороне. Поместите еще одну крышку размером 18 x 18 мм поверх первой крышки, с боковыми сторонами с надписью «p» лицом друг к другу. Поместите пару на пустую стойку наконечника в гидратационной камере и повторите для оставшихся 18 x 18 мм крышек.
- Закройте гидратационную камеру и инкубируйте в течение 3 ч или на ночь.
- Отделите пары чехлов и промойте в сверхчистой воде.
- Высушите покровы струей азота и поместите их в инкубатор при температуре 37 °C для полного высыхания.
- Чтобы построить камеру визуализации, наклейте три полосы двусторонней ленты на крышку размером 24 x 60 мм с боковой маркировкой «b». К другой стороне ленточных полос прикрепите крышку размером 18 x 18 мм с боковой пометкой «p», обращенную к более крупному чехловому листу. Это образует две проточные камеры для микроскопических экспериментов, с обработанными поверхностями, обращенными друг к другу (рисунок 2 и рисунок 1G).

Рисунок 1: Оборудование для обработки крышки и подготовки камеры визуализации. (A) банки для окрашивания слайдов для 24 x 60 мм обшивки, (B) стойки для стирки для скольжения для 18 x 18 мм чехлов, (C) вакуумная установка, (D) стеллаж для сушки слайдов, (E) гидратационная камера, (F) крышки, (G) камера визуализации, (H) держатель слайдов. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}

Рисунок 2: Схема подготовки камер визуализации с использованием двусторонней ленты (серого цвета) и покрытых крышек, обработанных ПЭГ/Биотин-ПЭГ. Создано с помощью BioRender.com. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
3. Полимеризуйте микротрубочки
- Подготовка семян ГМПЦПП
ПРИМЕЧАНИЕ: Подготовьте семена GMPCPP в холодном помещении, сохраняя все реагенты, наконечники и трубки при 4 °C. Семена GMPCPP могут быть подготовлены заранее и храниться при -80 °C в течение 1 года. Поместите в ультрацентрифугу ротор ультрацентрифуги с фиксированным углом наклона, содержащий трубки центрифуги объемом 1 мл, и установите температуру до 4 °C.- Повторное суспендирование лиофилизированного тубулина (таблица 2) до ~10 мг/мл в 1X BRB80 непосредственно перед применением.
- Смешайте компоненты семян GMPCPP, как описано в таблице 3.
ПРИМЕЧАНИЕ: Держите все компоненты тубулина на льду как можно больше, чтобы свести к минимуму полимеризацию растворимого тубулина. - Осветлить смесь в ультрацентрифужном роторе с фиксированным углом при 352 700 х г в течение 5 мин при 4 °C.
- Разделите супернатант на 5 мкл аликвот, заморозьте их в жидком азоте и храните при -80 °C.
- Полимеризуйте семена в день эксперимента
- Теплый 1-2 мл BRB80-DTT (таблица 1) до 37 °C.
- Поместите 5 мкл аликвоты семян GMPCPP (-80 °C) со стадии 3.1.4 на лед и немедленно растворите в 20 мкл теплого BRB80-DTT. Открутите при 2 000 х г в течение 5 с при комнатной температуре и постучите для смешивания.
ПРИМЕЧАНИЕ: Первоначальный объем разбавления может варьироваться от 13 мкл до 21 мкл и определяется эмпирически для каждой партии семян. Если семена не полимеризуются, устраните неполадки, дополнив первоначальный буфер разбавления (этап 3.2.2) 0,5 мкМ GMPCPP. - Беречь от света и инкубировать при 37 °C в течение 30-45 мин.
ПРИМЕЧАНИЕ: Длина микротрубочек зависит от продолжительности инкубации. Для коротких микротрубочек время инкубации может составлять всего 15 минут. Для длинных микротрубочек время инкубации может составлять до 2 ч. Биотинилированные микротрубочки, как правило, требуют более длительного времени инкубации, чем небиотинилированные микротрубочки. - Поместите ультрацентрифужный ротор с фиксированным углом наклона, содержащий 500 мкл центрифужных трубок, в ультрацентрифугу и предварительно прогрет до 30 °C.
- После инкубации добавляют 50 мкл теплого BRB80-DTT (стадия 3.2.1) к полимеризованным семенам GMPCPP и переносят смесь в центрифужную трубку объемом 500 мкл. Промыть пустую трубку, содержащую семена GMPCPP, еще 50 мкл теплого BRB80-DTT, пипеткой вверх и вниз, и добавить этот буфер в 500-мкл центрифужную трубку, содержащую смесь.
- Перед вращением отметьте ободок трубки центрифуги, чтобы указать, где будет находиться гранула (гранула будет слишком мала, чтобы ее можно было увидеть). Отжим в течение 10 мин при 244 900 х г при 30 °C12.
- Осторожно выложите супернатант и выбросьте. Повторно суспендировать гранулу в 100 мкл теплого BRB80-DTT. Нажмите, чтобы смешать.
- Отжим в течение 10 мин при 244 900 х г при 30 °C, выравнивая маркировку с ротором на гранулу в том же месте.
- Удалите супернатант и повторно суспендируйте гранулу в 16 мкл теплого BRB80-DTT. Перенесите раствор микротрубочек в чистую микроцентрифужную трубку объемом 0,6 мл. Защитите от света и храните при комнатной температуре или выше.
ПРИМЕЧАНИЕ: После полимеризации держите микротрубочки при комнатной температуре или выше. Если они замерзнут, они деполимеризуются. Инкубировать при температуре 28 °C для дополнительной стабильности.
- Проверка микротрубочек с помощью TIRF-микроскопии
- Пипетка смесью 4,5 мкл BRB80-DTT и 1 мкл раствора микротрубочек (шаг 3.2.9) на предметное стекло микроскопа. Накройте крышкой размером 18 x 18 мм и запечатайте края либо прозрачным лаком для ногтей, либо смесью вазелина, ланолина и парафина (валапный герметик) 1:1: 1, которая является твердой при комнатной температуре и жидкой при 95 ° C.
- Поместите объектив TIRF под крышкой (рекомендуемые настройки микроскопа см. на этапе 4) и визуализируйте вновь полимеризованные микротрубочки на длине волны, соответствующей флуоресцентно меченому тубулину в смеси Bright (таблица 3), чтобы определить, какое разбавление микротрубочек использовать в предстоящих экспериментах.
| Реагент | Яркая смесь (мкл) | Порядок добавления | Яркая смесь + биотин (мкл) | Порядок добавления |
| Флуоресцентный тубулин, 10 мг/мл | 2 | 6 | 2 | 7 |
| Биотин-тубулин, 10 мг/мл | 0 | Н/Д | 2 | 6 |
| Немаркированный тубулин, 10 мг/мл | 20 | 5 | 18 | 5 |
| ГМПКЭС, 10 мМ | 30 | 4 | 30 | 4 |
| Диафрагма, 0,2 М | 0.7 | 3 | 0.7 | 3 |
| 5X BRB80 | 26.4 | 2 | 26.4 | 2 |
| стерильная вода | 52.9 | 1 | 52.9 | 1 |
| Общий объем (мкл) | 132 | 132 |
Таблица 3: Смесь семян ГМПЦПП. Компоненты семян микротрубочек GMPCPP, включая объем и порядок добавления. Приготовьте 5 мкл аликвот и храните до 1 года при -80 °C.
4. Настройки микроскопа
- Температура: Установите температуру микроскопа на 28 °C для просмотра динамических микротрубочек.
- Фильтры: Используйте наилучшую комбинацию кубиков фильтров и эмиссионных фильтров, в зависимости от флуоресцентных каналов, которые будут визуализированы. Чтобы визуализировать длины волн 488 нм, 560 нм и 647 нм в том же эксперименте, используйте лазерный четырехдиапазонный набор 405/488/560/647 нм в сочетании с эмиссионными фильтрами для обозначенных длин волн.
- Выровнять лазеры: убедитесь, что лазерные лучи, используемые в эксперименте, выровнены. Определите интенсивность лазера для эксперимента эмпирически, чтобы все флуоресцентные белки могли быть визуализированы с максимально возможным отношением сигнал/шум, но не подвергались значительному фотоотбеливанию в течение времени эксперимента.
- Цель: Используйте бумагу для линз для очистки 100-кратного объектива с 70% этанолом. Перед визуализацией добавьте каплю погружного масла микроскопа к объективу.
- Настройка последовательности изображений
- Для эксперимента с 647 нм флуорофор-мечеными биотинилированными микротрубочками, 560 нм флуорофор-мечеными небиотинилированными микротрубочками и растворимым тубулином, и интересующим белком, меченым GFP, изображение в течение 20 мин. Снимайте каналы 560 нм и 488 нм каждые 10 с, а канал 647 нм каждые 30 с.
- Чтобы захватить эталонное изображение пучков перед добавлением растворимого тубулина и MAP, настройте последовательность с одним изображением каждый в длинах волн 560 нм и 647 нм.
5. Генерация поверхностно-иммобилизованных пучков микротрубочек
ПРИМЕЧАНИЕ: Для следующих этапов перетейте все растворы в проточную камеру путем пипетки в одну открытую сторону, поместив фильтровальную бумагу на другую сторону. Защитите камеру визуализации от света, чтобы уменьшить фотоотбеливание флуоресцентно меченых белков. Прикрепите подготовленную камеру визуализации к держателю слайдов (рисунок 1G,H). Выполните действия, описанные в таблице 4, которые соответствуют шагам протокола 5.2-6.4.
- Приготовить растворимую тубулиновую смесь по таблице 5 и держать на льду.
ПРИМЕЧАНИЕ: Растворимый тубулин всегда должен быть помещен на лед, чтобы предотвратить полимеризацию. Готовьте свежую растворимую смесь тубулина примерно каждые 2 ч, или когда микротрубочки больше не полимеризуются. - Чтобы обездвижить микротрубочки через связь биотин-нейтравидин-биотин, сначала текут в растворе нейтравидина (NA) до тех пор, пока камера не будет заполнена (~ 7,5 мкл) и инкубирует в течение 5 мин.
- Промыть 10 мкл МБ-холода.
- Протойте в 7,5 мкл блокирующего белка κ-казеина (KC) и инкубируйте в течение 2 мин.
- Промыть 10 мкл МБ-тепла для подготовки камеры к введению микротрубочек.
- Разбавляют запас биотинилированных микротрубочек (по наблюдениям на стадии 3.3.2) в 1X BRB80-DTT и добавляют 1 мкл этого разведения к 9 мкл MB-теплого. Протейте смесь в камеру и инкубируйте в течение 10 мин. Используйте более высокую концентрацию микротрубочек для большего количества пучков.
- Смойте неиммобилизованные микротрубочки 10 мкл MB-тепла.
- Влить в камеру 7,5 мкл теплого KC и инкубировать в течение 2 мин.
- Во время инкубации готовят 2 нМ раствора сшивающего белка PRC1 в теплом KC. Перетекают 10 мкл этого раствора в проточную камеру и инкубируют в течение 5 мин.
ПРИМЕЧАНИЕ: Рекомбинантный PRC1 экспрессируется и очищается из бактериальных клеток, как описано ранее13. - Чтобы сделать пучки, перетекайте 10 мкл небиотинилированных микротрубочек в камеру и инкубируйте в течение 10 мин. PRC1 будет сшивать небиотинилированные и иммобилизованные биотинилированные микротрубочки15,16 (рисунок 3).
ПРИМЕЧАНИЕ: См. этап 6.1 для приготовления пробирной смеси во время инкубации. - Дважды промыть камеру 10 мкл в тепле. Прикрепленные микротрубочки стабильны в течение примерно 20 минут с этой точки.
| Шаг | Реагент | Объем (мкл) | Время инкубации (минуты) |
| 1 | Нейтравидин | 7.5 | 5 |
| 2 | МБ-холодный | 10 | - |
| 3 | κ-казеин | 7.5 | 2 |
| 4 | МБ-теплый | 10 | - |
| 5 | Биотинилированная микротрубочка (разведенная в MB-теплой) | 10 | 10 |
| 6 | МБ-теплый | 10 | - |
| 7 | Теплый κ-казеин | 7.5 | 2 |
| 8 | 2 нМ PRC1, разбавленный в κ-казеине | 10 | 5 |
| 9 | Небиотинилированная микротрубочка | 10 | 10 |
| 10 | МБ-теплый x 2 | 10 | - |
| 11 | Пробирная смесь | 10 | - |
| Прикрепленные семена стабильны в течение примерно 20 минут в этот момент. | |||
Таблица 4: Этапы анализа. Перечень реагентов, добавляемых в камеру визуализации, с указанием времени промывки (-) или инкубации.
| Реагент | Объем (мкл) |
| Переработанный тубулин, 10 мг/мл | 10 |
| МБ-Холодный | 10.3 |
| МБМК | 13.7 |
| БРБ80-ДТТ | 3.4 |
| ГТП, 10 мМ | 6.7 |
| СПС, 10 мМ (При использовании кинезинов) | 6.7 |
| Флуоресцентно меченый тубулин, 10 мг/мл | 1 (Ресуспенд лиофилизированного меченого тубулина в холодном BRB80-DTT) |
Таблица 5: Компоненты смеси растворимых тубулинов. Перемешайте в начале эксперимента и держите на льду.

Рисунок 3: Схема добавления компонентов анализа для изготовления и изображения флуоресцентно меченых пучков и одиночных микротрубочек. Биотинилированные семена показаны в синих, небиотинилированных семенах и растворимый тубулин в красном, PRC1 в черном, а белок, представляющий интерес, в голубом. Номера шагов на рисунке соответствуют номерам, приведенным в таблице 4. Панель, соответствующая шагу 9, показывает предварительно сформированный пучок (внизу слева); на шаге 11 показан вновь образованный пучок (вверху слева). Создано с помощью BioRender.com. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
6. Динамика микротрубочек изображения
- В течение 10 мин инкубационного времени на стадии 5.10 готовят 10 мкл пробирной смеси, содержащей интересующие белки, растворимый тубулин, нуклеотиды, поглотители кислорода14 и антиоксиданты согласно таблице 6. Держите смесь на льду.
- Загрузите подготовленную камеру визуализации, приклеенную к держателю слайда, на объектив 100x TIRF. Используйте каналы 560 нм и 647 нм, чтобы найти поле зрения, содержащее оптимальное количество и плотность одиночных микротрубочек и пучков.
ПРИМЕЧАНИЕ: Если как биотинилированные, так и небиотинилированные микротрубочки помечены одним и тем же флуорофором, анализ интенсивности флуоресценции при линейном сканировании может различать отдельные микротрубочки и пучки. - После определения поля зрения сделайте эталонное изображение.
- Осторожно течь в пробирную смесь, не нарушая камеру визуализации.
- Запечатайте открытые концы камеры валапным герметиком.
- Запустите последовательность изображений, как описано в шаге 4.5.1.
| Реагент | Объем (мкл) |
| Растворимая тубулиновая смесь | 4 |
| ОСФ | 1 |
| Тролокс (при использовании микротрубочек, помеченных легкоотбеливающим флуорофором) | 1 |
| СПС, 10 мМ (При использовании кинезинов) | 1 |
| PRC1 (или поперечный по выбору) | 1 |
| Интересующие белки | X |
| МБ-холодный | 2-Х |
Таблица 6: Компоненты пробирной смеси. Смешайте, перетейте в камеру визуализации и изобразите динамику микротрубочек в течение 30 минут.
Access restricted. Please log in or start a trial to view this content.
Результаты
Эксперимент, описанный выше, проводили с использованием 647 нм флуорофор-меченых биотинилированных микротрубочек, 560 нм флуорофор-меченых небиотинилированных микротрубочек и 560 нм флуорофор-меченой растворимой тубулиновой смеси. Микротрубочки были сшиты сшиваемым белком PRC1 (меченым GFP...
Access restricted. Please log in or start a trial to view this content.
Обсуждение
Описанный здесь эксперимент значительно расширяет сферу применения и сложность обычных анализов восстановления микротрубочек, которые традиционно выполняются на одиночных микротрубочках или на одном типе массива. Текущий анализ обеспечивает метод одновременной количественной оце...
Access restricted. Please log in or start a trial to view this content.
Раскрытие информации
Авторы заявляют об отсутствии конкурирующих интересов.
Благодарности
Эта работа была поддержана грантом NIH (No 1DP2GM126894-01), а также средствами Pew Charitable Trusts и Smith Family Foundation для R.S. Авторы благодарят доктора Шуо Цзяна за его вклад в разработку и оптимизацию протоколов.
Access restricted. Please log in or start a trial to view this content.
Материалы
| Name | Company | Catalog Number | Comments |
| (±)-6-Hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (Trolox) | Sigma Aldrich | 238813 | |
| 1,4-piperazinediethanesulfonic acid (PIPES) | Sigma Aldrich | P6757 | |
| 18x18 mm #1.5 coverslips | Electron Microscopy Sciences | 63787 | |
| 2-Mercaptoethanol (BME) | Sigma Aldrich | M-6250 | |
| 24x60 mm #1.5 coverslips | Electron Microscopy Sciences | 63793 | |
| 405/488/560/647 nm Laser Quad Band | Chroma | TRF89901-NK | |
| Acetone | Sigma Aldrich | 320110 | |
| Adenosine 5'-triphosphate disodium salt hydrate (ATP) | Sigma Aldrich | A7699-5G | |
| Avidin, NeutrAvidin® Biotin-binding Protein (Molecular Probes®) | Thermo Fischer Scientific | A2666 | |
| Bath sonicator: Branson 2800 Cleaner | Branson | CPX2800H | |
| Beckman Coulter Polycarbonate Thickwall Tubes, 11 x 34 mm | Beckman-Coulter | 343778 | |
| Beckman Coulter Polycarbonate Thickwall Tubes, 8 x 34 mm | Beckman-Coulter | 343776 | |
| Biotin-PEG-SVA, MW 5,000 | Laysan Bio | #Biotin-PEG-SVA-5000 | |
| Bovine Serum Albumin (BSA) | Sigma Aldrich | 2905 | |
| Catalase | Sigma Aldrich | C40 | |
| Corning LSE Mini Microcentrifuge, AC100-240V | Corning | 6670 | |
| Delicate Task Wipes | Kimtech | 34120 | |
| Dithiothreitol (DTT) | GoldBio | DTT10 | |
| Emission filter | Chroma | ET610/75m | |
| Ethanol (200-proof) | Decon Labs | 2705 | |
| Ethylene glycol tetraacetic acid (EGTA) | Sigma Aldrich | 3777 | |
| Glucose Oxidase | Sigma Aldrich | G2133 | |
| GMPCPP | Jena Bioscience | NU-405 | |
| Guanosine 5'-triphosphate sodium salt hydrate (GTP) | Sigma Aldrich | G8877 | |
| Hellmanex III detergent | Sigma Aldrich | Z805939 | |
| Immersion oil, Type A | Fisher Scientific | 77010 | |
| Kappa-casein | Sigma Aldrich | C0406 | |
| Lanolin | Fisher Scientific | S25376 | |
| Lens Cleaning Tissue | ThorLabs | MC-5 | |
| Magnesium Chloride (MgCl2) | Sigma Aldrich | M9272 | |
| Methylcellulose | Sigma Aldrich | M0512 | |
| Microfuge 16 Benchtop Centrifuge | Beckman-Coulter | A46474 | |
| Microscope Slides, Diamond White Glass, 25 x 75mm, 90° Ground Edges, WHITE Frosted | Globe Scientific | 1380-50W | |
| mPEG-Succinimidyl Valerate, MW 5,000 | Laysan Bio | #NH2-PEG-VA-5K | |
| Optima™ Max-XP Tabletop Ultracentrifuge | Beckman-Coulter | 393315 | |
| Paraffin | Fisher Scientific | P31-500 | |
| PELCO Reverse (self-closing), Fine Tweezers | Ted Pella | 5377-NM | |
| Petrolatum, White | Fisher Scientific | 18-605-050 | |
| Plasma Cleaner, 115V | Harrick Plasma | PDC-001 | |
| Potassium Hydroxide (KOH) | Sigma Aldrich | 221473 | |
| Sodium bicarbonate | Sigma Aldrich | S6014 | |
| Sucrose | Sigma Aldrich | S7903 | |
| Thermal-Lok 1-Position Dry Heat Bath | USA Scientific | 2510-1101 | |
| Thermal-Lok Block for 1.5 and 2.0 mL Tubes | USA Scientific | 2520-0000 | |
| Thermo Scientific™ Pierce™ Bond-Breaker™ TCEP Solution, Neutral pH; 500mM | Thermo Fischer Scientific | PI-77720 | |
| TIRF 100X NA 1.49 Oil Objective | Nikon | CFI Apochromat TIRF 100XC Oil | |
| TIRF microscope | Nikon | Eclipse Ti | |
| TLA 120.1 rotor | Beckman-Coulter | 362224 | |
| TLA 120.2 rotor | Beckman-Coulter | 357656 | |
| Tubulin protein (>99% pure): porcine brain | Cytoskeleton | T240 | |
| Tubulin Protein (Biotin): Porcine Brain | Cytoskeleton | T333P | |
| Tubulin protein (fluorescent HiLyte 647): porcine brain | Cytoskeleton | TL670M | |
| Tubulin protein (X-rhodamine): bovine brain | Cytoskeleton | TL620M | |
| VECTABOND® Reagent, Tissue Section Adhesion | Vector Biolabs | SP-1800-7 | |
| VWR® Personal-Sized Incubator, 120V, 50/60Hz, 0.6A | VWR | 97025-630 |
Ссылки
- Subramanian, R., Kapoor, T. M. Building complexity: insights into self-organized assembly of microtubule-based architectures. Developmental Cell. 23 (5), 874-885 (2012).
- Baas, P. W., Rao, A. N., Matamoros, A. J., Leo, L. Stability properties of neuronal microtubules. Cytoskeleton (Hoboken). 73 (9), 442-460 (2016).
- Bitan, A., Rosenbaum, I., Abdu, U. Stable and dynamic microtubules coordinately determine and maintain Drosophila bristle shape. Development. 139 (11), 1987-1996 (2012).
- Foe, V. E., von Dassow, G. Stable and dynamic microtubules coordinately shape the myosin activation zone during cytokinetic furrow formation. The Journal of Cell Biology. 183 (3), 457-470 (2008).
- Pous, C., et al. Functional specialization of stable and dynamic microtubules in protein traffic in WIF-B cells. The Journal of Cell Biology. 142 (1), 153-165 (1998).
- Uehara, R., Goshima, G. Functional central spindle assembly requires de novo microtubule generation in the interchromosomal region during anaphase. The Journal of Cell Biology. 191 (2), 259-267 (2010).
- Mitchison, T., Kirschner, M. Dynamic instability of microtubule growth. Nature. 312 (5991), 237-242 (1984).
- Bieling, P., et al. Reconstitution of a microtubule plus-end tracking system in vitro. Nature. 450 (7172), 1100-1105 (2007).
- Bieling, P., Telley, I. A., Surrey, T. A minimal midzone protein module controls formation and length of antiparallel microtubule overlaps. Cell. 142 (3), 420-432 (2010).
- Shimamoto, Y., Forth, S., Kapoor, T. M. Measuring pushing and braking forces generated by ensembles of Kinesin-5 crosslinking two microtubules. Developmental Cell. 34 (6), 669-681 (2015).
- Mani, N., Jiang, S., Neary, A. E., Wijeratne, S. S., Subramanian, R. Differential regulation of single microtubules and bundles by a three-protein module. Nature Chemical Biology. 17 (9), 964-974 (2021).
- Hyman, A. A., Salser, S., Drechsel, D., Unwin, N., Mitchison, T. J. Role of GTP hydrolysis in microtubule dynamics: information from a slowly hydrolyzable analogue, GMPCPP. Molecular Biology of the Cell. 3 (10), 1155-1167 (1992).
- Subramanian, R., et al. Insights into antiparallel microtubule crosslinking by PRC1, a conserved nonmotor microtubule binding protein. Cell. 142 (3), 433-443 (2010).
- Rasnik, I., McKinney, S. A., Ha, T. Nonblinking and long-lasting single-molecule fluorescence imaging. Nature Methods. 3 (11), 891-893 (2006).
- Wijeratne, S., Subramanian, R. Geometry of antiparallel microtubule bundles regulates relative sliding and stalling by PRC1 and Kif4A. eLife. 7, 32595(2018).
- Mani, N., Wijeratne, S. S., Subramanian, R. Micron-scale geometrical features of microtubules as regulators of microtubule organization. eLife. 10, 63880(2021).
- Freal, A., et al. Feedback-driven assembly of the axon initial segment. Neuron. 104 (2), 305-321 (2019).
- Ledbetter, M., Porter, K. A "microtubule" in plant cell fine structure. The Journal of Cell Biology. 19 (1), 239-250 (1963).
- Wijeratne, S. S., Marchan, M. F., Tresback, J. S., Subramanian, R. Atomic force microscopy reveals distinct protofilament-scale structural dynamics in depolymerizing microtubule arrays. Proceedings of the National Academy of Sciences of the United States of America. , 119(2022).
Access restricted. Please log in or start a trial to view this content.
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеСмотреть дополнительные статьи
This article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены