
Method Article
Descifrando la organización de la cromatina 3D de alta resolución a través de Capture Hi-C
En este artículo
Resumen
Este protocolo describe el método Capture Hi-C utilizado para caracterizar la organización 3D de regiones genómicas específicas de tamaño megabasado en alta resolución, incluidos los límites de los dominios de asociación topológica (TAD) y las interacciones de cromatina de largo alcance entre los elementos reguladores y otros elementos de secuencia de ADN.
Resumen
La organización espacial del genoma contribuye a su función y regulación en muchos contextos, incluyendo la transcripción, replicación, recombinación y reparación. Por lo tanto, comprender la causalidad exacta entre la topología y la función del genoma es crucial y cada vez más objeto de una investigación intensiva. Las tecnologías de captura de conformación cromosómica (3C) permiten inferir la estructura 3D de la cromatina midiendo la frecuencia de interacciones entre cualquier región del genoma. Aquí describimos un protocolo rápido y simple para realizar Capture Hi-C, un método de enriquecimiento de objetivos basado en 3C que caracteriza la organización 3D específica de alelos de objetivos genómicos de tamaño megabasado en alta resolución. En Capture Hi-C, las regiones objetivo son capturadas por una serie de sondas biotiniladas antes de la secuenciación de alto rendimiento aguas abajo. Por lo tanto, se logra una mayor resolución y alelo especificidad al tiempo que se mejora la efectividad del tiempo y la asequibilidad de la tecnología. Para demostrar sus fortalezas, se aplicó el protocolo Capture Hi-C al centro de inactivación X ( Xic) del ratón, el locus regulador maestro de la inactivación del cromosoma X (XCI).
Introducción
El genoma lineal contiene toda la información necesaria para que un organismo experimente el desarrollo embrionario y sobreviva hasta la edad adulta. Sin embargo, instruir a células genéticamente idénticas para que realicen diferentes funciones es fundamental para controlar con precisión qué información se utiliza en contextos específicos, incluidos diferentes tejidos y / o etapas de desarrollo. Se cree que la organización tridimensional del genoma participa en esta regulación espacio-temporal precisa de la actividad génica al facilitar o prevenir la interacción física entre elementos reguladores que pueden separarse por varios cientos de kilobases en el genoma lineal (para revisiones 1,2,3). En los últimos 20 años, nuestra comprensión de la interacción entre el plegamiento del genoma y la actividad ha aumentado rápidamente, en gran parte debido al desarrollo de tecnologías de captura de conformación cromosómica (3C) (para revisión 4,5,6,7). Estos métodos miden la frecuencia de las interacciones entre cualquier región del genoma y se basan en la ligadura de secuencias de ADN que están en estrecha proximidad 3D dentro del núcleo. Los protocolos 3C más comunes comienzan con la fijación de poblaciones celulares con un agente de reticulación como el formaldehído. La cromatina reticulada se digiere entonces con una enzima de restricción, aunque también se ha utilizado la digestión de la MNasa 8,9. Después de la digestión, el ADN libre termina en estrecha proximidad espacial y se vuelve a ligar y se invierte la reticulación. Este paso da lugar a la 'biblioteca' o 'plantilla' 3C, un grupo mixto de fragmentos híbridos en el que las secuencias que estaban en 3D cerca del núcleo tienen mayores posibilidades de ligarse en el mismo fragmento de ADN. La cuantificación posterior de estos fragmentos híbridos permite inferir la conformación 3D de regiones genómicas que se encuentran a miles de pares de bases separadas en el genoma lineal, pero que podrían interactuar en el espacio 3D.
Se han desarrollado muchos enfoques diferentes para caracterizar la biblioteca 3C, difiriendo tanto en términos de qué subconjuntos de fragmentos de ligadura se analizan como de qué tecnología se utiliza para su cuantificación posterior. El protocolo 3C original se basó en la selección de dos regiones de interés y la cuantificación de su frecuencia de interacción "uno contra uno" mediante PCR10,11. El enfoque 4C (captura de conformación cromosómica circular) mide las interacciones entre un único locus de interés (es decir, el 'punto de vista') y el resto del genoma ('uno contra todos')12,13,14. En 4C, la biblioteca 3C se somete a una segunda ronda de digestión y religación para generar pequeñas moléculas circulares de ADN que son amplificadas por PCR por cebadores específicos de punto de vista15. 5C (copia de carbono de captura de conformación cromosómica) permite la caracterización de interacciones 3D en regiones de interés más grandes, proporcionando información sobre el plegamiento de cromatina de orden superior dentro de esa región ('muchos versus muchos')16. En 5C, la biblioteca 3C se hibrida a un conjunto de oligonucleótidos que se superponen a sitios de restricción que pueden ser amplificados posteriormente por PCR múltiplex con cebadores universales15. Tanto en 4C como en 5C, los fragmentos de ADN informativos fueron cuantificados inicialmente por microarrays y posteriormente por secuenciación de próxima generación (NGS)17,18,19. Estas estrategias caracterizan las regiones específicas de interés, pero no se pueden aplicar para mapear las interacciones de todo el genoma. Este último objetivo se logra con Hi-C, una estrategia de alto rendimiento basada en 3C en la que la secuenciación masivamente paralela de la plantilla 3C permite la caracterización imparcial del plegamiento de la cromatina a nivel de todo el genoma ('todos contra todos')20. El protocolo Hi-C incluye la incorporación de un residuo biotinilado en los extremos de los fragmentos digeridos, seguido por la extracción de fragmentos de ligadura con perlas de estreptavidina para aumentar la recuperación de fragmentos ligados20.
Hi-C reveló que los genomas de mamíferos están organizados estructuralmente a múltiples escalas en el núcleo 3D. A escala de megabase, el genoma se divide en regiones de cromatina activa e inactiva, los compartimentos A y B, respectivamente20,21. La existencia de subcompartimentos adicionales representados por diferentes estados de cromatina y actividad también se mostró posteriormente22. A mayor resolución, el genoma se divide aún más en dominios de autointeracción sub-megabase llamados dominios de asociación topológica (TADs), revelados por primera vez por el análisis Hi-C y 5C de los genomas humano y de ratón23,24. A diferencia de los compartimentos que varían de manera específica del tejido, los TAD tienden a ser constantes (aunque hay muchas excepciones). Es importante destacar que los límites de TAD se conservan en todas las especies25. En las células de mamíferos, los TAD con frecuencia abarcan genes que comparten el mismo panorama regulatorio y se ha demostrado que representan un marco estructural que facilita la corregulación génica al tiempo que limita las interacciones con los dominios reguladores vecinos (para revisión 3,26,27,28). Además, dentro de los TAD, las interacciones debidas a los sitios CTCF en la base de los bucles extruidos de cohesina pueden aumentar la probabilidad de interacciones promotor-potenciador o potenciador-potenciador (para la revisión29).
En Hi-C, los compartimentos y TAD se pueden detectar a una resolución de 1 Mb a 40 kb, pero se puede lograr una resolución más alta para caracterizar contactos de menor escala, como interacciones de bucle entre elementos distales a una escala de 5-10 kb. Sin embargo, aumentar la resolución para poder detectar dichos bucles de manera eficiente mediante HiC requiere un aumento significativo en la profundidad de secuenciación y, por lo tanto, en los costos de secuenciación. Esto se agrava si el análisis debe ser alelo específico. De hecho, un aumento de X veces en la resolución requiere un aumento X2 en la profundidad de secuenciación, lo que significa que los enfoques de alta resolución y alelos específicos de todo el genoma pueden ser prohibitivamente costosos30.
Para mejorar la rentabilidad y la asequibilidad mientras se mantiene una alta resolución, las regiones objetivo de interés se pueden extraer físicamente de las bibliotecas 3C o Hi-C de todo el genoma después de su hibridación con sondas de oligonucleótidos marcadas con biotina complementarias antes de la secuenciación posterior. Estas estrategias de enriquecimiento de objetivos se conocen como métodos de captura-C y permiten interrogar las interacciones de cientos de loci objetivo dispersos por todo el genoma (es decir, Promoter Capture (PC) Hi-C; Captura de próxima generación (NG) C; Captura de entrada baja (LI)-C; Captura de Título Nuclear (NuTi)-C; Tri-C)31,32,33,34,35,36,37,38,39,40, o a través de regiones que abarcan hasta varias megabases (es decir, Capture HiC; HYbrid Capture Hi-C (Hi-C2); Azulejo-C)41,42,43. Dos aspectos pueden variar en los métodos basados en la captura: (1) la naturaleza y el diseño de oligonucleótidos biotinilados (es decir, ARN o ADN, oligos individuales que capturan objetivos genómicos dispersos o múltiples oligos que teselan una región de interés); y (2) la plantilla que se utiliza para derribar objetivos que pueden ser la biblioteca 3C o Hi-C, esta última consiste en fragmentos de restricción biotinilados extraídos de la biblioteca 3C.
Aquí se describe un protocolo Capture Hi-C basado en el enriquecimiento de los contactos de destino de la biblioteca 3C. El protocolo se basa en el diseño de una matriz de mosaico personalizada de sondas de ARN biotiniladas y se puede realizar en 1 semana desde la preparación de la biblioteca 3C hasta la secuenciación NGS. El protocolo es rápido, simple y permite caracterizar la organización 3D de orden superior de las regiones de interés de tamaño megabase con una resolución de 5 kb, al tiempo que mejora la efectividad del tiempo y la asequibilidad en comparación con otros métodos 3C. El protocolo Capture Hi-C se aplicó al locus regulador maestro de la inactivación del cromosoma X (XCI), el centro de inactivación X (Xic), que alberga el ARN no codificante Xist. El Xic ha sido previamente objeto de extensos análisis estructurales y funcionales (para revisión44,45). En los mamíferos, XCI compensa la dosis de genes ligados al cromosoma X entre hembras (XX) y machos (XY) e implica el silenciamiento transcripcional de casi la totalidad de uno de los dos cromosomas X en las células femeninas. El Xic ha representado un poderoso locus estándar de oro para estudios en topología genómica 3D y la interacción con la regulación génica44. El análisis de 5C del Xic en células madre embrionarias de ratón (mESCs) condujo al descubrimiento y denominación de TADs, proporcionando los primeros conocimientos sobre la relevancia funcional de la partición topológica y la corregulación génica24. Posteriormente, se demostró que la organización topológica del Xic estaba críticamente involucrada en el momento apropiado del desarrollo de la regulación positiva del Xist y XCI 46, y también se descubrieron recientemente elementos reguladores cis insospechados que pueden influir en la actividad génica dentro y entre los TAD dentro del Xic47,48,49. La aplicación de Capture Hi-C a 3 Mb del cromosoma X del ratón que abarca el Xic demuestra el poder de este enfoque para diseccionar el plegamiento de cromatina a gran escala a alta resolución. Se proporciona un protocolo detallado y fácil de seguir, desde el diseño de la matriz de sondas biotiniladas en cada sitio de restricción de DpnII dentro de la región de interés hasta la generación de la biblioteca 3C de todo el genoma, la hibridación y captura de contactos objetivo y el análisis de datos posteriores. También se incluye una visión general de los controles de calidad apropiados y los resultados esperados, y tanto las fortalezas como las limitaciones del enfoque se discuten a la luz de métodos similares existentes.
Protocolo
Las células madre embrionarias de ratón (mESCs) utilizadas en este estudio se derivaron de un cruce de una hembra TX/TX R26rtTA/rtTA 50 con un macho Mus musculus castaneus de acuerdo con las directrices de cuidado animal del Institut Curie (París)51.
1. Diseño de la sonda
- Diseñar una matriz de sondas biotiniladas (oligonucleótidos de ARN de 120 mer) que cubran la región objetivo de interés.
- Mosaice la región de interés con oligonucleótidos superpuestos de modo que, en promedio, cada secuencia dentro del objetivo esté cubierta por dos sondas únicas (cobertura 2x) (Figura 1).
- Excluya secuencias repetitivas de la cobertura de la sonda para evitar el enriquecimiento de interacciones inespecíficas.
NOTA: Para maximizar el enriquecimiento de los fragmentos de ligadura informativos, se definieron regiones que abarcan 300 pb aguas arriba y aguas abajo de cada sitio de restricción DpnII a través del objetivo (ChrX: 102,475,000-105,475,000), y se diseñaron 28,913 sondas biotiniladas de acuerdo con la tecnología de enriquecimiento de objetivos SureSelect DNA a través de la plataforma Sure Design52. De acuerdo con esta estrategia, se permiten hasta un máximo de 40 bases de secuencias repetitivas en cada oligonucleótido para minimizar el enriquecimiento de interacciones inespecíficas. La matriz de sondas fue sintetizada por Agilent. Aquí, DpnII se utiliza como enzima de restricción por dos razones: (1) es un cuatro cortadores utilizado rutinariamente en varios métodos basados en3C 53; y (2) maximiza las posibilidades de capturar polimorfismos informativos de nucleótido único (SNP) en la proximidad de los sitios de corte en comparación con otras enzimas de restricción que se probaron in silico en líneas híbridas F1 utilizadas en este estudio (C57BL / 6J x CASTEi / J).
2. Procedimiento experimental
- Preparación celular
- Sembrar el número apropiado de células en una o varias placas de cultivo celular para lograr un número total de células de ≥ 5 x 107 células el día de la fijación.
NOTA: En este estudio se utilizaron células madre embrionarias de ratón (mESCs). Las mESCs se colocan en placas de cultivo celular gelatinizadas (0,1% de gelatina en 1x PBS - o/n a 37 °C, incubadora de CO2 al 5%) en medio de mESCs que contiene 2i + LIF y suero fetal de ternera probado por lotes (DMEM, 15% FBS, 0,1 mM β-mercaptoetanol, 1.000 U/mL−1 factor inhibidor de la leucemia (LIF), CHIR99021 (3 μM) y PD0325901 (1 μM)). Para este tipo de célula, una placa de 10 cm confluente al 80% contiene aproximadamente 2 x 107 células. - Prepare una placa de cultivo celular adicional para el recuento celular.
NOTA: Se puede utilizar una placa de cultivo celular más pequeña para reducir el uso de medios. En este caso, el número de células a sembrar en la placa más pequeña debe ajustarse en consecuencia (por ejemplo, 3 veces menos celdas en una placa de 10 cm en comparación con una placa de 15 cm).
- Sembrar el número apropiado de células en una o varias placas de cultivo celular para lograr un número total de células de ≥ 5 x 107 células el día de la fijación.
- Fijación de formaldehído
- Calcule el número total de celdas que se entrecruzarán.
- Antes de comenzar la reacción de reticulación, tripsinizar y contar las células de la placa de control preparada específicamente para el recuento de células utilizando un contador de células automatizado de acuerdo con las instrucciones del fabricante.
- Incluir una tinción de viabilidad (por ejemplo, azul de tripano) para determinar el porcentaje de células viables54. A partir de este recuento de células, estime el número total de células en la(s) placa(s) preparada(s) para la reticulación.
- Retirar el medio de cultivo de las placas preparadas para la reticulación y sustituirlo por la cantidad adecuada de solución de fijación (formaldehído al 2% en medio de cultivo celular). Use 10 ml en una placa de 10 cm (por ejemplo, ~ 20 ml para una placa de 15 cm).
NOTA: Agregue un volumen exacto de solución de fijación. Si la fijación de células adherentes no es posible, este paso puede adaptarse a células tripsinizadas y realizarse en 30 ml de solución de fijación en tubos de centrífuga cónica de 50 ml. El formaldehído no debe tener más de 1 año de edad. Se prefiere utilizar viales de un solo uso. La solución de fijación debe llevarse a temperatura ambiente (RT) antes de su uso.
PRECAUCIÓN: El formaldehído es peligroso y debe manipularse de acuerdo con las normas de salud y seguridad apropiadas. - Fijar durante 10 minutos a RT bajo una mezcla suave en una coctelera.
- Apagar la reacción de fijación mediante la adición de 2,5 M de glicina-1x PBS a una concentración final de 0,125 M. Añadir 530 μL de 2,5 M de glicina-1x PBS a 10 ml en una placa de 10 cm (p. ej., 1060 μL a 20 ml en una placa de 15 cm).
NOTA: Si las células se fijaron en solución, apagar la reacción de fijación con 1590 μL de 2,5 M glicina-1x PBS. - Incubar durante 5 minutos en RT, mezclando suavemente en una coctelera.
- Transfiera los platos al hielo e incube durante 15 minutos adicionales en hielo mientras se mezclan suavemente en una coctelera.
NOTA: A partir de ahora, las celdas deben mantenerse en hielo, y los tampones deben enfriarse previamente para evitar una mayor reticulación. Muévase a una cámara frigorífica si es necesario procesar muchas placas. - Retire la solución de fijación de las células virtiéndolo en un vaso de precipitados para garantizar un manejo rápido.
NOTA: Asegúrese de eliminar los residuos líquidos que contienen formaldehído de acuerdo con las normas de salud y seguridad apropiadas. - Enjuague la placa de 10 cm rápidamente dos veces con 5 ml de glicina-1x PBS fría de 0,125 M (8 ml para una placa de 15 cm) para lavar los residuos y las células muertas. Retire el líquido de la placa virtiéndolo en un vaso de precipitados para garantizar un manejo rápido.
- Agregue 5 ml de glicina-1x PBS 0.125 M fría a la placa de 10 cm (10 ml para una placa de 15 cm) y raspe rápidamente las células de la placa con un raspador de células de plástico.
- Transfiera la suspensión celular a un tubo de centrífuga cónica de 50 ml preenfriado utilizando una pipeta serológica.
- Enjuague la placa dos veces con 5 ml de glicina-1x PBS 0.125 M fría y agregue la suspensión celular al tubo de centrífuga cónica.
- Girar a 480 x g durante 10 min a 4 °C.
NOTA: Si las celdas se fijaron en solución, transfiera la celda a un tubo de centrífuga cónica preenfriado y gire hacia abajo a 480 x g durante 10 min a 4 °C. Retire la solución de fijación vertiéndola en un vaso de precipitados y lave tres veces en 10 ml de glicina-1x PBS fría 0.125 M. Asegúrese de volver a suspender las celdas en cada paso de lavado. - Retire el sobrenadante aspirando con un sistema de aspiración de sobremesa. Resuspender las células en 500 μL de 1x PBS por 1 x 107 células pipeteando cuidadosamente hacia arriba y hacia abajo con una pipeta P1000. Para resuspender células en el volumen exacto, consúltese la estimación del número total de células obtenida en 2.2.1.
- Alícuota 500 μL de la suspensión celular en el número calculado de tubos de microcentrífuga de 1,5 ml (1 x 107 células /tubo).
- Girar a 480 x g durante 10 min a 4 °C.
- Retire el sobrenadante con un sistema de aspiración de sobremesa y congele rápidamente los gránulos de células en nitrógeno líquido. Conservar los gránulos de células secas a -80 °C.
NOTA: Las muestras se pueden almacenar durante al menos 1 año.
- Calcule el número total de celdas que se entrecruzarán.
- Lisis celular
- Descongele la(s) bolita(s) congelada(s) en hielo.
- Preparar 1,5 ml de tampón de lisis en H 2 0 por muestra: añadir 10 mM de Tris-HCl, pH 8,0, 10 mM de NaCl y NP40 al0,2%.
- Añadir 600 μL del tampón de lisis fría y resuspender bien sobre hielo.
- Incubar en hielo durante 15 minutos para dejar que las células se hinchen.
- Girar hacia abajo a 2655 x g durante 5 min a 4 °C y retirar el sobrenadante mediante un sistema de aspiración de sobremesa.
- Para eliminar los residuos, vuelva a suspender el pellet en 1 ml del tampón de lisis en frío, gire hacia abajo a 2655 x g durante 5 min a 4 °C y retire el sobrenadante.
- Girar de nuevo brevemente a 2655 x g y 4 °C y retirar la mayor cantidad posible de sobrenadante restante utilizando un sistema de aspiración de sobremesa equipado con una punta P200.
- Resuspender en 100 μL de 0,5% (vol/vol) SDS.
- Incubar en un termomezclador a 62 °C, girando a 1400 rpm durante 10 min.
- Añadir 290 μL deH2O+ 50 μL de TritonX-100 al 10% y mezclar bien, evitando burbujas de aire.
- Incubar en un termomezclador a 37 °C, girando a 1400 rpm durante 15 min.
- Agregue 50 μL de tampón Dpnll 10x e invierta el tubo para mezclar.
- Tome 50 μL de ADN no digerido para el control de calidad en un tubo separado. No olvide tomar la muestra de control no digerida.
- Digestión DpnII
- Añadir 10 μL de Dpnll de alta concentración (500 U en total) y mezclar invirtiendo.
- Incubar las muestras y el control no digerido en un termomezclador a 37 °C, girando a 1400 rpm durante >4 h.
- Añadir 10 μL de Dpnll de alta concentración (500 U en total) al final del día.
- Incubar las muestras y el control no digerido a 37 °C, girando a 1400 rpm durante la noche.
- Añadir 10 μL de Dpnll alta concentración (500 U en total) al comienzo del día siguiente a las muestras.
- Incubar las muestras y el control no digerido en un termomezclador a 37 °C, girando a 1400 rpm durante 4 h.
- Ligadura e inversión de la reticulación
- Incubar los tubos a 65 °C durante 20 min a 1400 rpm.
NOTA: No agregue SDS en este momento. La idea es preservar la integridad nuclear, por lo que la ligadura se lleva a cabo dentro de los núcleos, evitando la necesidad de una dilución extrema. - Enfriar las muestras en hielo durante un máximo de 5-10 min. Para evitar la precipitación SDS, no deje las muestras en hielo más tiempo que esto.
- Tome 50 μL del ADN digerido no ligado para el control de calidad en un tubo separado. Conservar los controles no digeridos y no ligados a -20 °C.
NOTA: No olvide tomar la muestra de control no ligada. - Añadir 800 μL de cóctel de ligadura: 122 μL de tampón ligasa 10x, 8 μL de ligasa T4 (30 U/μL) y 670 μL deH20.
- Incubar a 16 °C, girando a 1000 rpm durante la noche.
- Añadir 7,5 μL de proteinasa K (20 mg/ml) a las muestras y 2 μL a los controles.
- Incubar a 65 °C durante 4 h a 1000 rpm.
- Incubar los tubos a 65 °C durante 20 min a 1400 rpm.
- Purificación del ADN
- Transfiera las muestras en hielo a tubos de centrífuga cónica de 15 ml preenfriados y agregue 2 ml de agua, 10,5 ml de EtOH helado y 583 μL de NaAC de 3 M.
NOTA: El agua adicional tiene como objetivo evitar el arrastre de TDT en el pellet. - Agregue 200 μL de EtOH helado, 10.8 μL de NaAC y 1 μL del coprecipitante a los controles de calidad no digeridos y no ligados.
- Incubar a -80 °C durante al menos 4 h hasta toda la noche.
- Girar los tubos de 15 ml a 2200 x g a 4 °C durante 45 min.
- Girar los tubos de control de 1,5 ml a 20.500 x g a 4 °C durante 30 min.
- Lavar una vez con 3 mL (muestras) y 1 mL (controles) de EtOH al 70% helado.
- Centrifugar a 2200 x g (muestras) o 20.500 x g (controles) a 4 °C durante 10 min.
- Retire cuidadosamente el EtOH y seque al aire en RT durante 10-15 min; No seque demasiado.
- Resuspender las muestras y los controles en 100 μL y 20 μL de H20, respectivamente.
- Añadir 1 μL de RNAseA e incubar a 37 °C, girando a 1400 rpm durante 30 min.
- Transfiera las muestras en hielo a tubos de centrífuga cónica de 15 ml preenfriados y agregue 2 ml de agua, 10,5 ml de EtOH helado y 583 μL de NaAC de 3 M.
- Control de calidad de la preparación de plantillas 3C
- Cuantifique cada muestra y controle utilizando un kit de fluorómetro para mediciones de concentración de ADN de alta sensibilidad.
- Cargar 100-200 ng de cada muestra y cada control en una agarosa al 1% / 1x gel TBE.
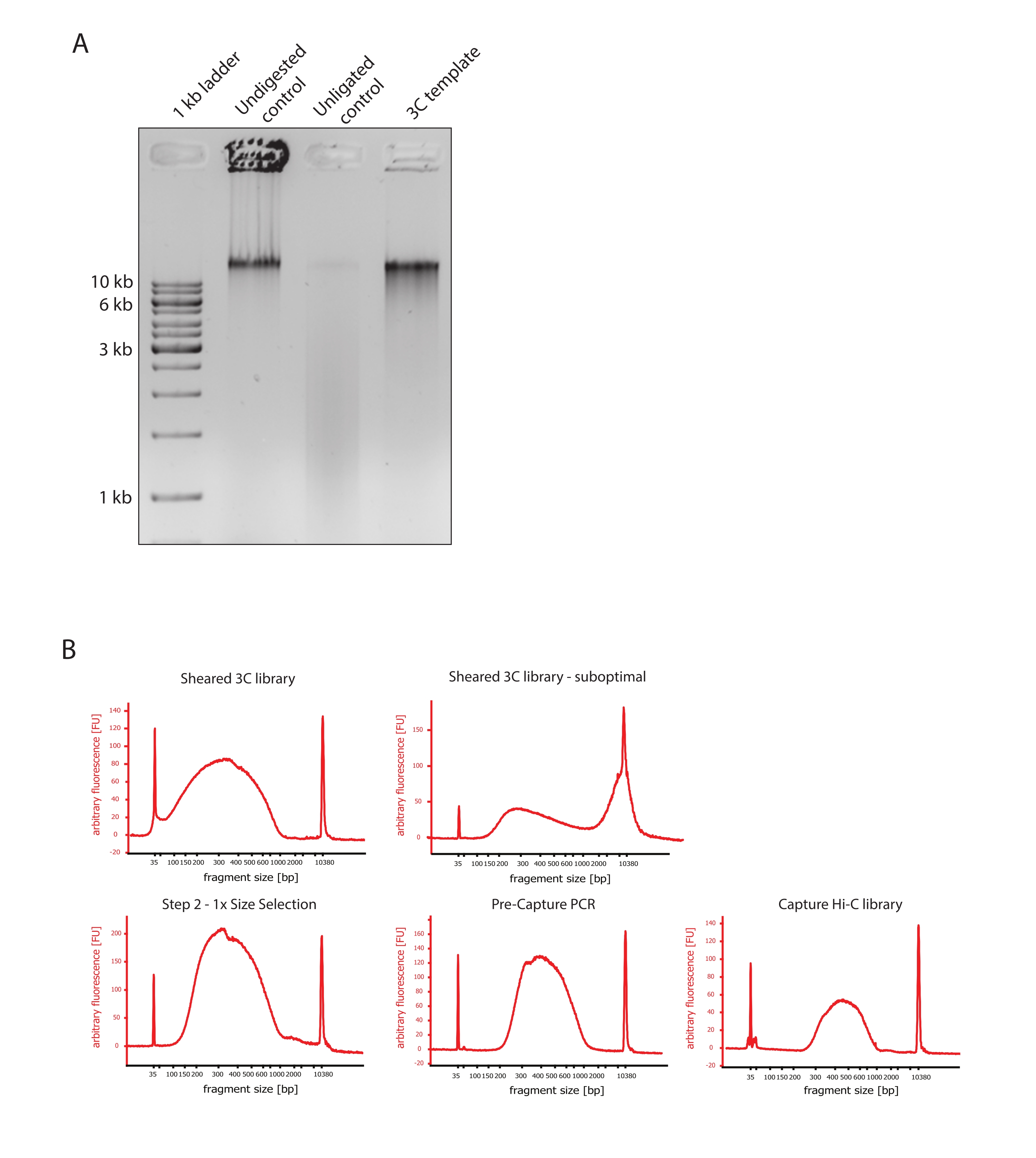
- Verifique que la imagen de gel muestre el resultado esperado comparando las diferencias en el tamaño de los fragmentos de ADN de los controles y la plantilla 3C como se muestra en la Figura 2A.
- Conservar las muestras y los controles a -20 °C.
- Hibridación, captura y procesamiento de muestras para secuenciación multiplexada
- Para hibridar la matriz de sondas de ARN biotiniladas a la plantilla 3C, capture los fragmentos de ligadura dirigidos y prepare las muestras para la secuenciación multiplexada de acuerdo con el sistema de enriquecimiento de objetivos utilizado en este estudio para la secuenciación multiplexada de extremo pareado (ver Tabla de materiales). Siga el protocolo de acuerdo con las instrucciones del fabricante al introducir las siguientes modificaciones menores:
- Sección 2 del protocolo del fabricante: Preparación de la muestra
- Siga las instrucciones para el enriquecimiento del objetivo a partir de 3 μg de entrada de ADNg.
- Corte el ADN en un sonicador utilizando las siguientes especificaciones: ciclo de trabajo del 10%, intensidad 4, 200 cyc/ráfaga y 130 s. Comience con 4 μg de plantilla 3C resuspendida en 130 μL de agua para cada reacción de captura para garantizar suficiente material para continuar la preparación de la muestra con 3 μg del ADN cortado.
- Evaluar la calidad del ADN cortado. Ejecute 1 μL del ADN cortado en un bioanalizador de ADN de acuerdo con el protocolo de alta sensibilidad. Espere una distribución del tamaño del fragmento entre 150-700 pb (Figura 2).
- Purificar la muestra utilizando perlas de inmovilización reversible en fase sólida (SPRI). Agregue 124 μL de perlas SPRI a 124 μL de la muestra de ADN para realizar una selección de tamaño del lado izquierdo 1: 1 de acuerdo con las instrucciones del fabricante y eluya en 25 μL de agua libre de nucleasas. Este paso de purificación eliminará fragmentos más cortos para enriquecer fragmentos de alrededor de 300 pb (Figura 2).
NOTA: La cantidad de muestras y perlas SPRI utilizadas en este paso tiene en cuenta la pérdida de volumen que se produjo al transferir las muestras a nuevos tubos y ejecutar los controles de calidad en el Bioanalyzer. Todos los pasos posteriores de selección de tamaño se realizan de acuerdo con las proporciones recomendadas por el protocolo del fabricante. La elución del ADN de las perlas SPRI se realiza en RT durante todo el protocolo. - Evaluar la calidad del ADN cortado seleccionado por tamaño. Ejecute 1 μL del ADN cortado en el bioanalizador de ADN de acuerdo con el protocolo de alta sensibilidad (HS). Espere una distribución de tamaños de fragmentos con el mayor enriquecimiento a 300 pb (Figura 2). Continúe con la cuantificación del ADN cortado si el cizallamiento fue exitoso.
- Cuantifique el ADN cortado con un kit de fluorómetro para mediciones de concentración de ADN HS.
NOTA: Si el cizallamiento de ADN da como resultado un rendimiento de ADN de <3 μg, realice una segunda ronda de corte de ADN con otros 4 μg de ADN y combine las muestras de ADN cortadas después del primer paso de purificación de perlas SPRI para lograr un total de 3 μg de ADN cortado. - Agregue agua libre de nucleasas a la muestra de ADN limpia seleccionada por el tamaño (3 μg en total) hasta un volumen final de 48 μL y proceda con la reacción de reparación final de acuerdo con el protocolo del fabricante.
- Después de la ligadura de los adaptadores emparejados, amplíe la biblioteca realizando cinco ciclos de PCR previa a la captura de acuerdo con las instrucciones del fabricante (las condiciones para la PCR y los cebadores se proporcionan en el kit).
- Sección 4 del protocolo del fabricante: Hibridación y captura
- Para hibridar las muestras de ADN preparadas con las sondas de ARN específicas del objetivo, diluya 750 ng de muestras de ADN en un volumen final de 3,4 μL, lo que resulta en una concentración inicial de 221 ng / μL. Para muestras de ADN diluidas en volúmenes más grandes, use un concentrador de vacío de velocidad para reducir al volumen final. Una concentración de velocidad-vacío (250 x g; ≤45 °C) durante 15-20 min es normalmente suficiente para las muestras resuspendidas en 10 μL. Asegúrese de tener el mismo volumen de entrada para cada muestra antes de encender el concentrador de velocidad-vacío.
- Incubar la mezcla de hibridación durante 16-18 h a 65 °C con una tapa calentada a 105 °C según las instrucciones del fabricante.
- Sección 5 del protocolo del fabricante: Indexación y procesamiento de muestras para secuenciación multiplexada
- Para amplificar las bibliotecas capturadas con cebadores de indexación, realice 12 ciclos de PCR posterior a la captura de acuerdo con las instrucciones del fabricante (las condiciones para la PCR y los cebadores se proporcionan en el kit).
- Sección 2 del protocolo del fabricante: Preparación de la muestra
- Para hibridar la matriz de sondas de ARN biotiniladas a la plantilla 3C, capture los fragmentos de ligadura dirigidos y prepare las muestras para la secuenciación multiplexada de acuerdo con el sistema de enriquecimiento de objetivos utilizado en este estudio para la secuenciación multiplexada de extremo pareado (ver Tabla de materiales). Siga el protocolo de acuerdo con las instrucciones del fabricante al introducir las siguientes modificaciones menores:
- Secuenciación de próxima generación
- Para ejecutar varias bibliotecas Hi-C de captura en la misma celda de flujo, prepare una mezcla equimolar de las bibliotecas de captura y secuencie lecturas de 100-120 M por biblioteca.
- Si se necesita el análisis alelo específico, secuencie 150 pb pareado para garantizar una cobertura de SNP suficiente.
3. Análisis de datos
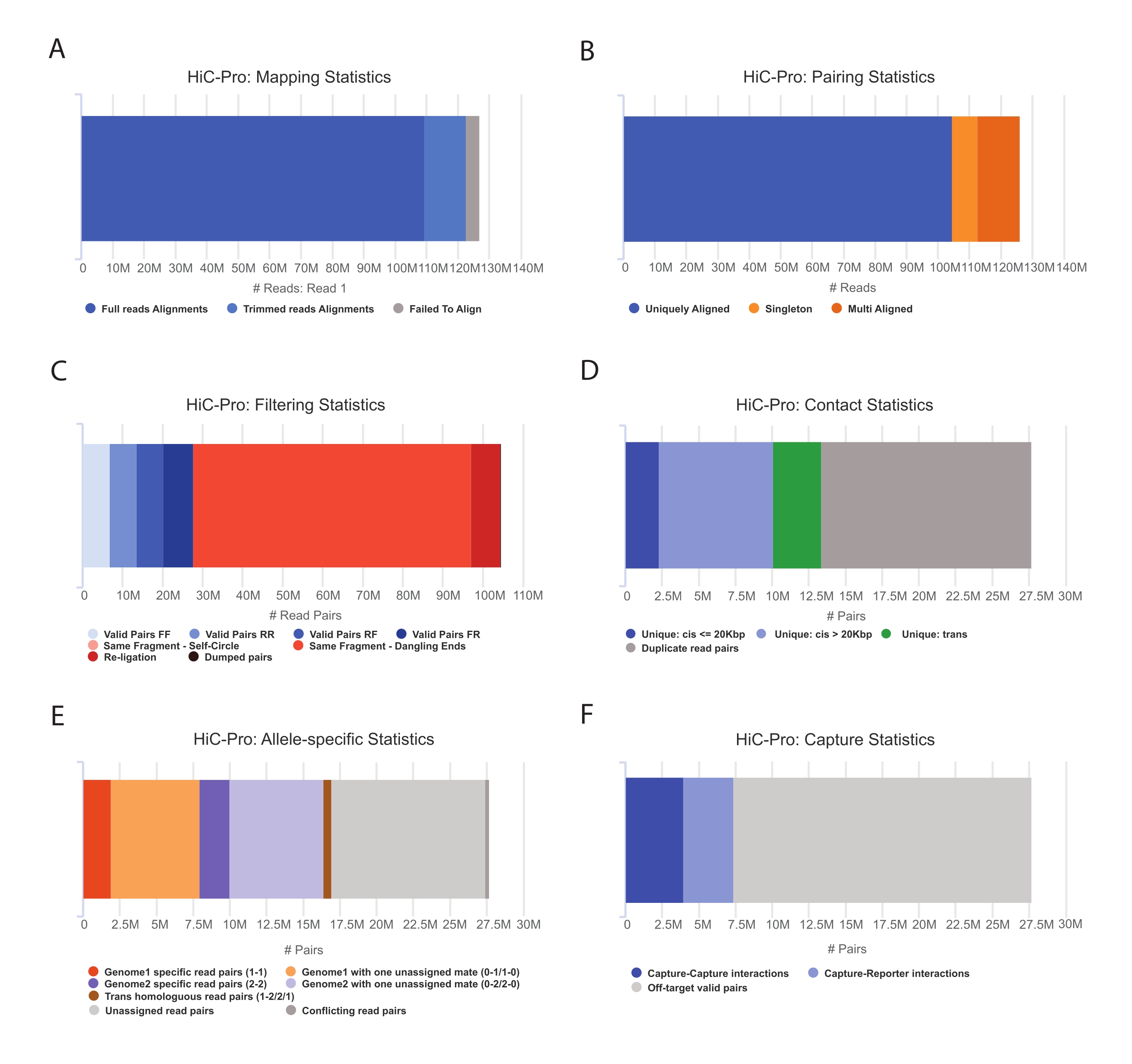
- Aplique la canalización HiC-Pro para realizar el análisis de datos Capture Hi-C55. HiC-Pro proporciona controles de calidad en cada paso del procesamiento, incluyendo (Figura 3):
i) La tasa de alineación en el genoma de referencia que especifica la fracción de lecturas que abarca un sitio de ligadura, así como el número de pares y singletons.
ii) La fracción de productos de ligadura válidos y pares de lectura no informativos (extremo colgante, autoligadura, etc.).
iii) La fracción de contactos corto/largo alcance e intra/intercromosómicos.
iv) La fracción de contactos en el destino para Capture Hi-C.
v) La fracción de alelo específico se lee si se especifica.
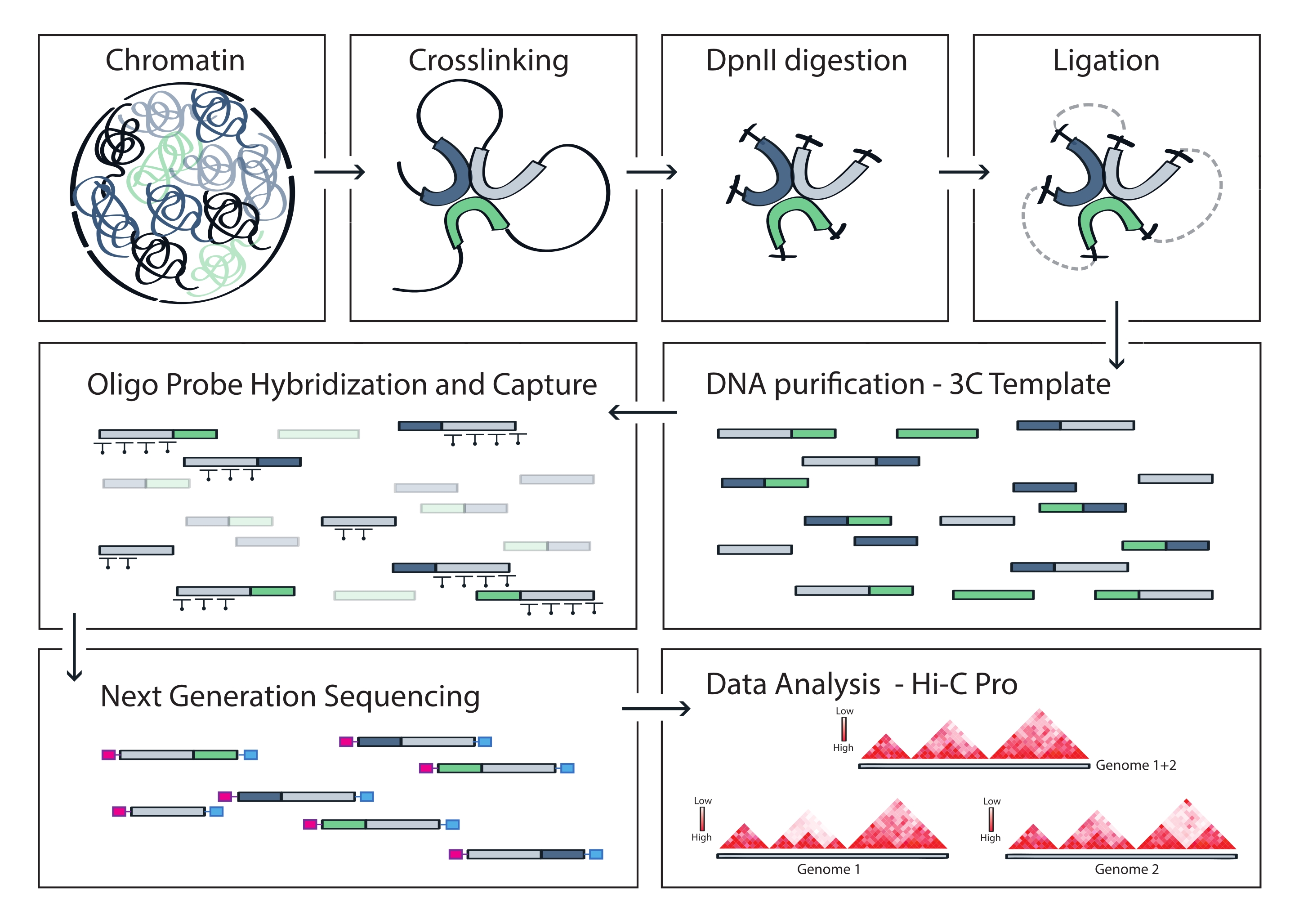
NOTA: HiC-Pro admite una amplia gama de protocolos, incluidos Hi-C in situ y Capture Hi-C. En este último caso, el usuario simplemente necesita especificar la región de destino (formato BED) en el archivo de configuración. Una vez que se procesan los datos, las salidas HiC-Pro se pueden convertir fácilmente en un objeto más frío para el análisis posterior56. En este paso, los mapas de contacto en varias resoluciones se normalizan utilizando el método ICE descrito previamente por Imakaev y colegas57. Luego se pueden ejecutar varios análisis para llamar a compartimentos cromosómicos, TAD o bucles de cromatina (para la revisión58). El flujo de trabajo del protocolo se muestra en la figura 4. Aquí, el conjunto 'cooltools' se aplica para calcular la puntuación de aislamiento y los límites de los TAD, como se ilustra en la Figura 5 y la Figura 659.
Resultados
El protocolo Capture Hi-C descrito se basa en la preparación de la plantilla 3C de todo el genoma utilizando un cortador de cuatro bases (DpnII). El enriquecimiento posterior de fragmentos de ligadura a través de la región genómica de interés se obtiene mediante la hibridación de una matriz de sondas de ARN de mosaico y su captura basada en estreptavidina de acuerdo con el sistema de enriquecimiento objetivo utilizado en este estudio (Figura 1). Se seleccionaron sondas de ARN biotiniladas ya que muestran una afinidad de unión más estrecha a sus objetivos en comparación con las sondas de ADN52,60. Las bibliotecas capturadas se indexan y agrupan para una secuenciación multiplexada de alto rendimiento. Los datos de captura de Hi-C se pueden visualizar como mapas de interacción Hi-C de alta resolución, pero también como mapas de contacto de punto de vista único similares a 4C para visualizar específicamente las interacciones de secuencias más pequeñas, como promotores o potenciadores, dentro de toda la región capturada. El flujo de trabajo del protocolo se muestra en la figura 4. Los controles de calidad previos a la secuenciación se muestran en la Figura 2 e incluyen la evaluación de la digestión y religación adecuadas de la plantilla 3C y su cizallamiento y purificación eficientes en los diferentes pasos del protocolo. Se espera que el ADN de la plantilla 3C cortado funcione entre 150 y 700 pb, y no se debe detectar ningún enriquecimiento de fragmentos >2 kb. Durante los siguientes pasos, se realizan varios pasos de limpieza y selección de tamaño de ADN basados en perlas, primero después del cizallamiento, luego después de las PCR previas y posteriores a la captura. Las bibliotecas limpias muestran un perfil de enriquecimiento de fragmentos distinto como se visualiza en un bioanalizador de ADN de alta sensibilidad (Figura 2). El tamaño medio del fragmento aumenta en el transcurso de la preparación de la biblioteca debido a la ligadura de adaptadores, secuenciación e indexación de cebadores. Los controles de calidad posteriores a la secuenciación se obtienen a través de Hi-C Pro y se muestran en la Figura 3. Se han propuesto muchas aplicaciones de software bioinformático diferentes para el procesamiento y análisis de datos similares a 3C. Entre ellas, el pipeline HiC-Pro es una de las soluciones más populares, permitiendo el procesamiento de datos de secuenciación sin procesar a los mapas de contacto finales en varias resoluciones55. HiC-Pro utiliza una estrategia de mapeo de dos pasos para alinear las lecturas de secuenciación en el genoma de referencia. Los productos 3C se reconstruyen y filtran para eliminar pares de contactos no informativos y generar los mapas de contacto. Además, es capaz de utilizar una lista de polimorfismos conocidos para realizar análisis alelo-específicos y separar los contactos procedentes de los dos alelos parentales en distintos mapas de contacto. Más recientemente, HiC-Pro se ha incluido y extendido al marco nf-core (nf-core-hic), proporcionando una canalización impulsada por la comunidad altamente escalable y reproducible61,62.
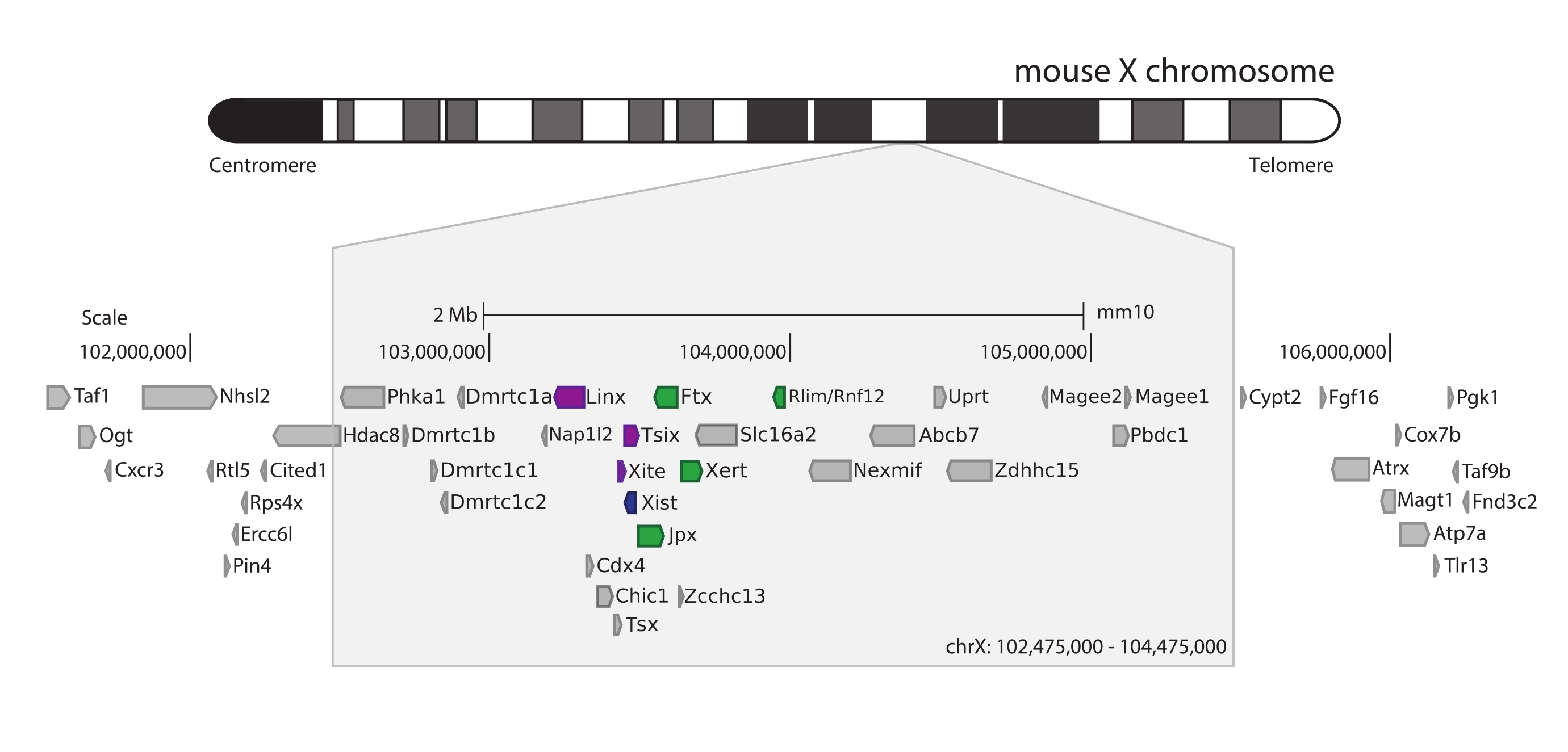
Para capturar el ratón Xic, se diseñó una matriz de 28.913 sondas de ARN en mosaico de 3 Mb del cromosoma X. Esta región incluye el jugador clave en XCI, el gen largo no codificante Xist, y su conocido panorama regulatorio de ~800 kb (Figura 5). Esta región de ~800 kb se divide en dos TAD: uno que incluye el promotor Xist y sus reguladores positivos conocidos (es decir, las transcripciones no codificantes Ftx, Jpx y Xert y el gen codificador de proteínas Rnf12), y el TAD vecino que abarca los reguladores cis negativos de Xist (es decir, su transcripción antisentido Tsix, el elemento potenciador Xite y la transcripción no codificante Linx) (para la revisión44, 45).
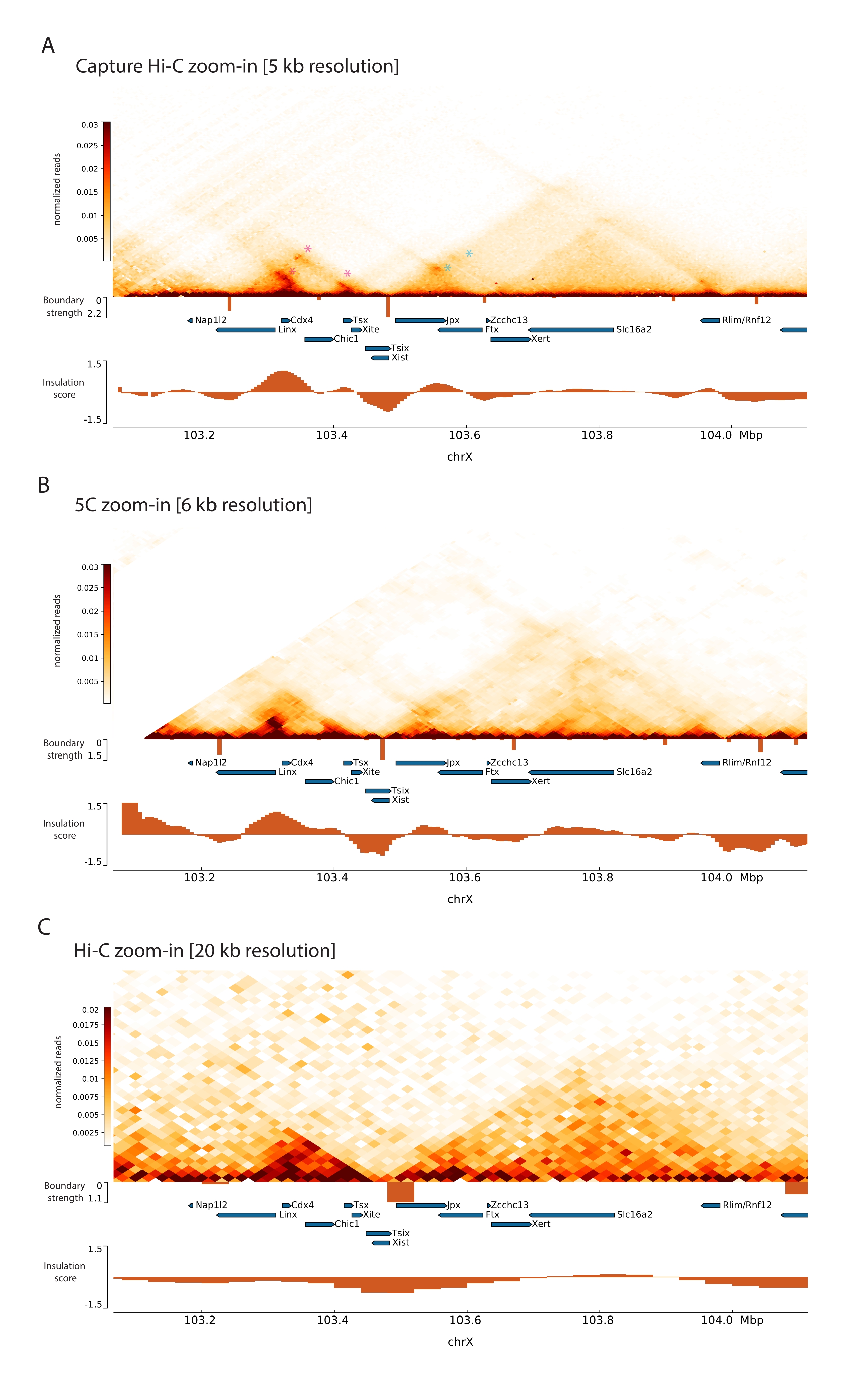
Al aplicar el protocolo Capture Hi-C descrito al Xic, la organización topológica de este locus se obtuvo a una resolución sin precedentes (Figura 6 y Figura 7). Esto es particularmente claro cuando se compara el perfil Capture Hi-C con 5C47 publicado anteriormente (Figura 6 y Figura 7; Cuadro complementario 1) y Hi-C61 (Figura 6 y Figura 7; Cuadro complementario 1) Perfiles. Por ejemplo, las estructuras sub-TAD son más evidentes: el TAD que contiene el promotor Xist ( Xist-TAD ) se subdivide claramente en dos dominios más pequeños (Figura 6A, punta de flecha azul). Anteriormente, esto solo se podía "adivinar" visualmente a partir del perfil 5C (Figura 6B), aunque la detección de un límite en esta región utilizando el algoritmo de puntuación de aislamiento. Asimismo, la resolución del perfil Capture Hi-C permite la identificación de dos dominios más pequeños en el TAD vecino (Figura 6A, B), que contiene el promotor del locus Tsix (Tsix-TAD ); esto no se lograba anteriormente con 5C (Figura 6B). Cabe destacar que los límites topológicos determinados por la puntuación de aislamiento de los datos Capture Hi-C y 5C generalmente se detectan en ubicaciones ligeramente diferentes y con diferentes resistencias relativas.
Además, otras estructuras sub-TAD como los bucles de contacto son claramente visibles a partir de los datos de Capture Hi-C, como el bucle entre Xist y Ftx (Figura 7A), previamente identificado con Capture-C63, y el bucle entre Xist y Xert (Figura 7B), recientemente identificado utilizando un protocolo similar para Capture Hi-C48. Otros contactos también se pueden mapear con mayor precisión debido a la mayor resolución de los perfiles Capture Hi-C, como los que forman los puntos de contacto conocidos dentro del Tsix-TAD entre los loci Linx, Chic1 y Xite (Figura 7A).
En comparación con los datos de Hi-C que se muestran en la Figura 7, Capture Hi-C permitió un aumento de cuatro veces en la resolución, sin embargo, requirió solo una cuarta parte de la profundidad de secuenciación (es decir, 126 M lecturas frente a 571 M) (Tabla complementaria 1). Este aumento en la resolución permite la detección de subTADs e interacciones de bucle que Hi-C no pudo detectar a la profundidad de secuenciación que se muestra en la Figura 6 y la Figura 7. El protocolo descrito para Capture Hi-C permite una caracterización mucho más detallada y de alta resolución de una gran región genómica de interés, en comparación con los enfoques anteriores.

Figura 1: Diseño de la sonda. Representación esquemática de la estrategia utilizada para el diseño de sondas. Se seleccionaron regiones de 300 pb aguas arriba y aguas abajo de cada sitio de restricción de DpnII en la región objetivo de 3 Mb y se embalsaron con sondas de ARN biotiniladas superpuestas. Se muestra una de estas regiones seleccionadas, chrX: 102,474,805-102,475,500. No se permiten más de 40 bases de secuencias repetitivas en cada sonda. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Capture los controles de calidad de presecuenciación Hi-C . (A) Ejemplo representativo de controles de calidad de plantillas 3C. Se cargaron 200 ng de ADN en un gel de agarosa al 1%. Carril 1: escalera de 1 kb. Carril 2: La cromatina no digerida, reticulada e intacta funciona como una banda afilada a >10 kb. Carril 3: La cromatina reticulada digerida por DpnII se ejecuta como un frotis de entre 1 kb y 3 kb de tamaño. Carril 4: Biblioteca o plantilla 3C final; los extremos libres de los fragmentos de ADN reticulados digeridos se vuelven a ligar. El frotis de ADN de menor tamaño molecular es casi indetectable, y el producto de ligadura se detecta como una banda de >10 kb. (B) Ejemplos representativos de perfiles de ADN bioanalizadores de alta sensibilidad. Arriba a la izquierda: biblioteca 3C cortada con éxito que muestra una distribución del tamaño del fragmento entre 150 pb y 700 pb. Arriba a la derecha: biblioteca 3C cortada insatisfactoria. El ADN no cortado se detecta como un amplio enriquecimiento de fragmentos >2 kb. (C) Abajo a la izquierda: muestra de ADN cortada siguiendo una selección de tamaño de lado izquierdo 1:1 utilizando perlas SPRI. Se enriquecen fragmentos de ~300 pb. Parte inferior central: perfil de PCR previo a la captura después de la ligadura de adaptadores de extremo emparejado de acuerdo con el protocolo del fabricante. Abajo a la derecha: biblioteca final de Capture Hi-C que incluye adaptadores, secuenciación y cebadores de indexación para secuenciación multiplexada. Abreviaturas: bp = pares de bases, FU = unidad de fluorescencia arbitraria. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: Captura de controles de calidad posteriores a la secuenciación Hi-C con HiC-Pro . (A) Ejemplo de tasa de mapeo en el genoma de referencia para el primer compañero de los pares de secuenciación. La fracción azul claro representa las lecturas alineadas por HiC-Pro y que abarcan una unión de ligadura. Por lo tanto, esta métrica se puede utilizar para validar el paso de ligadura experimental. (B) Una vez que los compañeros de secuenciación están alineados en el genoma, solo se mantienen los pares de lectura alineados de forma única para su análisis. (C) Los pares no válidos (en rojo) como el extremo colgante, el autocírculo o la religación se descartan del análisis. La fracción de pares válidos es un buen indicador de la eficiencia de ligadura y pull-down. (D) Los pares válidos se pueden dividir en contactos intra/intercromosómicos y de corto/largo alcance. Los pares de lectura duplicados que probablemente representen artefactos de PCR se descartan del análisis. (E) Para el análisis específico de alelos, HiC-Pro informa el número de lecturas alélicas apoyadas por uno o dos compañeros para cada genoma parental (es decir, C57BL / 6J x CASTEi / J). Se espera la misma fracción de lecturas asignadas al alelo materno y paterno. (F) Finalmente, solo se seleccionan pares válidos que se superponen a la región de captura para construir los mapas de contacto. Los pares captura-captura representan contactos dentro de la región objetivo, mientras que los pares captura-reportero implican la interacción entre la región objetivo y una fuera del objetivo. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4: Flujo de trabajo del protocolo Capture Hi-C. Representación esquemática de diferentes pasos del protocolo. Para generar la plantilla 3C de todo el genoma, la cromatina se reticula primero con formaldehído y luego se digiere con la enzima de restricción DpnII. Los extremos libres de ADN se vuelven a ligar, la reticulación se invierte y el ADN se purifica. Para enriquecer los fragmentos que abarcan la región objetivo, una serie de sondas de ARN biotiniladas se hibridan a la plantilla 3C y se capturan mediante una reducción mediada por estreptavidina. Las bibliotecas de captura se procesan para la secuenciación multiplexada, y los fragmentos de ligadura válidos se cuantifican para inferir la frecuencia de los contactos de cromatina a través del objetivo, que se visualizan como mapas de interacción de alta resolución. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5: Descripción general de la región que abarca el Xic en el cromosoma X del ratón. Representación esquemática del cromosoma X del ratón y zoom de la región capturada de 3 Mb (ChrX: 102.475.000-105.475.000). La región objetivo incluye ~800 kb de ADN correspondiente al Xic, el locus regulador maestro de XCI. El Xic incluye los genes largos no codificantes, Xist, un jugador clave de XCI, y su panorama regulatorio. Los reguladores positivos de Xist se muestran en verde y los reguladores negativos en púrpura. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 6: Capture mapas de interacción Hi-C, 5C y Hi-C en la región capturada de 3 Mb. (A) Capturar el mapa de interacción Hi-C del objetivo de 3 Mb que abarca el Xic del ratón con una resolución de 10 kb (este estudio). (B) Mapa de interacción 5C de la misma región objetivo que en A a una resolución de 6 kb (datos reprocesados a partir de47). Las regiones repetitivas no incluidas en los análisis están enmascaradas en blanco. Los datos 5C requieren su propio procesamiento bioinformático (ver47). Después de la limpieza y alineación, los mapas 5C en la resolución de cebador se agrupan utilizando una mediana de ejecución (ventana = 30 kb, paso = 5) para alcanzar una resolución final de 6 kb. (C) Mapa de interacción Hi-C de la misma región genómica que en A y B a una resolución de 40 kb (datos reprocesados a partir de64). Todos los mapas de interacción se generaron a partir de ESC de ratón. La puntuación de aislamiento se calculó utilizando cooltools y se representa como histogramas con mínimos de aislamiento en los límites TAD. Los límites TAD se muestran como líneas verticales debajo del mapa. La altura de cada línea indica la fuerza límite. Los genes se muestran como flechas que apuntan en la dirección de la transcripción. Los límites sub-TAD que se detectan exclusivamente o con mayor precisión en los mapas Capture Hi-C se indican con puntas de flecha magenta y azul para los sub-TAD en los TAD Tsix y Xist, respectivamente. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 7: Capture mapas de interacción Hi-C, 5C y Hi-C en 1 Mb dentro de la región capturada. (A) Capturar el mapa de interacción Hi-C de la región genómica de 1 Mb que abarca el ratón Xic a una resolución de 5 kb (este estudio). (B) Mapa de interacción 5C de la misma región genómica que en A. A una resolución de 6 KB (datos reprocesados a partir de47). Las regiones repetitivas no incluidas en los análisis están enmascaradas en blanco. Cabe destacar que los datos de 5C requieren su propio procesamiento bioinformático (ver47). Después de la limpieza y alineación, los mapas 5C en la resolución de cebador se agrupan utilizando una mediana de ejecución (ventana = 30 kb, paso = 5) para alcanzar una resolución final de 6 kb. (C) Mapa de interacción Hi-C de la misma región genómica que en A y B de Hi-C a una resolución de 20 kb (datos reprocesados a partir de64). Todos los mapas de interacción se generaron a partir de mESCs. La puntuación de aislamiento se calculó utilizando cooltools y se representa como histogramas con mínimos de aislamiento en los límites TAD. Los límites TAD se muestran como líneas verticales debajo del mapa. La altura de cada línea indica la fuerza límite. Los genes se muestran como flechas que apuntan a la dirección de la transcripción. Los bucles de contacto que se detectan exclusivamente o con mayor precisión en Capture Hi-C se indican con asteriscos magenta y azul para bucles en los TAD Tsix y Xist, respectivamente. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Tabla complementaria 1: Estadísticas posteriores a la secuenciación para los conjuntos de datos utilizados en este manuscrito: Capture Hi-C (este estudio), Hi-C64 y 5C47. Haga clic aquí para descargar este archivo.
Discusión
Aquí describimos un protocolo Capture Hi-C relativamente rápido y fácil para caracterizar la organización de orden superior de regiones genómicas de tamaño megabase a una resolución de 5-10 kb. Capture Hi-C pertenece a la familia de tecnologías Capture-C que están diseñadas para enriquecer las interacciones de cromatina dirigidas a partir de plantillas 3C o Hi-C de todo el genoma. Hasta la fecha, la gran mayoría de las aplicaciones de Capture-C se han explotado para mapear contactos de cromatina de elementos reguladores relativamente pequeños dispersos por todo el genoma. En el primer protocolo Capture-C, se utilizaron múltiples sondas biotiniladas de ARN superpuestas para capturar >400 promotores preseleccionados en bibliotecas 3C preparadas a partir de células eritroides31. La misma estrategia se mejoró posteriormente en Next Generation (NG) y Nuclear Titrated (NuTi) Capture-C para lograr perfiles de interacción de alta resolución de >8,000 promotores mediante el uso de cebos de ADN únicos de 120 pb que abarcan sitios de restricción individuales y dos rondas secuenciales de captura para maximizar el enriquecimiento de fragmentos de ligadura informativos32,40. Estas estrategias condujeron a la disección funcional de elementos de acción cis en muchos contextos diferentes, incluido el desarrollo embrionario de ratón, la diferenciación celular, la inactivación del cromosoma X y la mala regulación génica en condiciones patológicas 46,63,65,66,67,68,69,70,71.
En Promoter Capture Hi-C (PCHi-C), >22.000 promotores anotados que contenían fragmentos de restricción fueron extraídos de las bibliotecas Hi-C mediante hibridación de sondas biotiniladas de ARN 120-mer individuales en uno o ambos extremos del fragmento de restricción34,72. Este método permitió la disección del interactoma de miles de promotores en un número cada vez mayor de tipos de células, incluyendo células madre embrionarias de ratón, células hepáticas fetales y adipocitos 34,35,72,73, pero también líneas linfoblastoides humanas, progenitores hematopoyéticos, queratinocitos epidérmicos y células pluripotentes 37,74,75,76,77 .
En comparación con estas tecnologías de enriquecimiento de objetivos, Capture Hi-C se dirige a regiones genómicas contiguas hasta la escala de megabase, abarcando así uno o más TAD y abarcando paisajes reguladores de genes. Toda la región de interés debe estar embaldosada con una serie de sondas biotiniladas que abarquen cada sitio de restricción de DpnII dentro del objetivo. La hibridación de la matriz biotinilada a la plantilla 3C, su posterior captura basada en estreptavidina y el procesamiento para la secuenciación multiplexada se realiza utilizando un sistema de enriquecimiento de objetivos para la secuenciación multiplexada Illumina Paired-End. Todo el protocolo es rápido, ya que se puede realizar en 1 semana desde la preparación de la biblioteca 3C hasta la secuenciación NGS, y solo requiere adaptaciones menores y / o solución de problemas específicos personalizados.
El protocolo también proporciona ventajas en comparación con otros métodos basados en 3C. Para obtener mapas de interacción a una resolución de 5-10 kb, secuenciamos lecturas de extremo pareado de 100-120 M. Como comparación, utilizamos aquí un conjunto de datos Hi-C de 571 M lecturas para alcanzar una resolución de 20 kb64 (GSM2053973), y se necesitarían al menos 1.000 millones de lecturas para alcanzar una resolución de 5 kb con Hi-C22 en todo el cromosoma.
La captura Hi-C utilizada en el presente estudio alcanza una resolución mucho mayor que la 5C publicada anteriormente basada en una enzima de restricción cortadora de 6 pb47 (Tabla suplementaria 1). Es importante destacar que la estrategia diseñada para enriquecer y amplificar las interacciones dirigidas en 5C no permite el análisis alelo específico de las interacciones de la cromatina. Por el contrario, los datos de Capture Hi-C pueden ser mapeados alelo específicamente, permitiendo la disección de los paisajes estructurales 3D de pares de cromosomas homólogos, por ejemplo en células humanas o en líneas celulares híbridas F1 derivadas mediante el cruce de cepas de ratón genéticamente diferentes78. Para generar mapas de interacción Capture Hi-C específicos de alelos a una resolución de 5 kb, secuenciamos lecturas de extremo pareado de 150 pb para aumentar la cobertura de SNP. Se pueden aplicar enfoques alelo-específicos similares a líneas celulares humanas, para las cuales la anotación de SNPs está disponible22.
Es importante destacar que, aunque Capture Hi-C generalmente garantiza una alta resolución al tiempo que mejora la asequibilidad de los costos de secuenciación, la producción de oligonucleótidos biotinilados personalizados tiene un impacto en el costo total de este método. Por lo tanto, la elección del método 3C más adecuado diferirá para diferentes aplicaciones, y dependerá de la cuestión biológica que se esté abordando y la resolución requerida, así como del tamaño de la región de interés. Otros protocolos Capture Hi-C desarrollados comparten características clave con el protocolo descrito aquí. Por ejemplo, se aplicó una estrategia Capture Hi-C para caracterizar regiones genómicas de ~50 kb a 1 Mb que abarcan variantes no codificantes asociadas con el riesgo de cáncer de mama y colorrectal; en este protocolo, las regiones objetivo se retiraron de las bibliotecas Hi-C hibridando cebos de ARN de 120 mer en mosaico de las regiones objetivo a una cobertura 33,38,79. Del mismo modo, HYbrid Capture Hi-C (Hi-C 2) se utilizó para orientar las interacciones dentro de las regiones de interés de hasta2 Mb80. En ambos protocolos, el uso de una plantilla Hi-C enriquecida para fragmentos de ligadura extraídos de biotina aumentó el porcentaje de lecturas informativas totales en comparación con nuestro protocolo. Por ejemplo, en el conjunto de datos Hi-C que utilizamos aquí para la comparación64 (GSM2053973), el porcentaje de pares válidos después de la eliminación de duplicados es 4,8 veces mayor que los pares válidos obtenidos en Capture Hi-C como se describe en la Figura 3 y la Tabla suplementaria 1. Sin embargo, la extracción consecutiva de fragmentos ligados biotinilados y sondas hibridadas hace que el protocolo sea significativamente más complejo y lento, al tiempo que posiblemente disminuya la complejidad de la región capturada.
Otro método disponible para enriquecer las plantillas 3C con sondas de mosaico es Tiled-C, que se aplicó para estudiar la arquitectura de la cromatina a alta resolución espacial y temporal durante la diferenciación eritroide del ratón43. En Tiled-C, se utiliza un panel de sondas biotiniladas de 70 pb para enriquecer los contactos dentro de regiones a gran escala en dos rondas consecutivas de captura para generar mapas de muy alta resolución de interacciones específicas43,81. El doble enriquecimiento de captura también hace que el protocolo sea más largo y complejo en comparación con Capture Hi-C. Sin embargo, a diferencia de las estrategias Capture-C dirigidas a sitios de restricción individuales, en Tiled-C la segunda ronda de captura no parece aumentar significativamente la eficiencia de captura y, por lo tanto, probablemente se pueda omitir43. Finalmente, se aplicó un enfoque de mosaico similar basado en la misma estrategia de enriquecimiento objetivo utilizada en este estudio a la disección de paisajes regulatorios que abarcan variantes estructurales descritas en pacientes con malformaciones congénitas y rediseñadas en ratones transgénicos41,42. En este caso, la matriz de mosaico de sondas se diseñó en todo el objetivo en lugar de en la proximidad de los sitios de restricción DpnII41. Sin embargo, este trabajo fue fundamental para resaltar la sensibilidad y el poder de esta estrategia para lograr la caracterización de alta resolución de grandes regiones genómicas en diferentes contextos41,42,48.
En conclusión, el protocolo descrito aquí representa una estrategia fácil, robusta y poderosa para la caracterización 3D de alta resolución de cualquier región genómica de interés. La aplicación de este enfoque a diferentes sistemas modelo, tipos de células, paisajes de cromatina regulados por el desarrollo y regulación génica en condiciones saludables y patológicas es probable que facilite nuestra comprensión de la interacción y la causalidad entre la topología del genoma y la regulación génica, una de las preguntas abiertas fundamentales en el campo de la epigenética. Además, la aplicación de Capture Hi-C para mapear interacciones de largo alcance y plegamiento de cromatina de orden superior de variantes de riesgo identificadas por estudios GWAS tiene el potencial de revelar la relevancia funcional de los loci genómicos no codificantes asociados con enfermedades humanas en diferentes contextos, proporcionando así nuevos conocimientos sobre los procesos potencialmente subyacentes a la patogénesis.
Divulgaciones
Kai Hauschulz es Field Application Scientist en Agilent Technologies - Diagnostic and Genomics Group. Todos los demás autores declaran no tener intereses en conflicto.
Agradecimientos
El trabajo en el laboratorio Heard fue apoyado por un premio Advanced Investigator del Consejo Europeo de Investigación (XPRESS - AdG671027). A.L. cuenta con el apoyo de una beca individual Marie Skłodowska-Curie Actions de la Unión Europea (IF-838408). A.H. cuenta con el apoyo de ITN Innovative and Interdisciplinary Network ChromDesign, en virtud del acuerdo de subvención Marie Skłodowska-Curie 813327. Los autores agradecen a Daniel Ibrahim (MPI for Molecular Genetics, Berlín) por su útil asesoramiento técnico, a la plataforma NGS en el Institut Curie (París), y a Vladimir Benes y al Genomics Core Facility en EMBL (Heidelberg), por su apoyo y asistencia.
Materiales
| Name | Company | Catalog Number | Comments |
| 10x PBS pH 7.4 | Gibco | 10010-023 | |
| 37% (vol/vol) paraformaldehyde solution | Electron Microscopy Sciences | 15686 | single use glass-vials; do not reuse |
| 50 mL PP conical tube | Falcon | 352070 | |
| Agarose | Sigma | A9539-500g | |
| Bioanalyzer | Agilent | G2939BA | |
| Cell Scrapers - 25 cm Handle and 3.0 cm Blade | Falcon | 353089 | |
| CHIR99021 | Axon Medchem BV | Axon 1386 | |
| cOmplete Mini, Protease inhibitor cocktail (EDTA-free) | Merck | 11836170001 | |
| Countess Cell Counting Chamber Slides | Invitrogen | C10228 | |
| Countess II FL | Invitrogen | ZGEXSCCOUNTESS2FL | Automated cell counter |
| Covaris S2 | Covaris | 500217 | Sonicator |
| DNA LoBind tube, 1.5 mL | Eppendorf | 30108051 | |
| DpnII (50000 units/mL) | New England Biolabs | R0543M | |
| Dulbecco's Modified Eagle Medium (DMEM) | Merck | D6429 | |
| Ethanol (100%) | Merck | 1.00983.2500 | |
| Fetal Bovine Serum (FBS) | Thermo Scientific | 10270106 | |
| gelatine from porcine skin | Sigma | G1890 | |
| GeneRuler 1 kb Plus DNA Ladder | Thermo Scientific | SM0313 | |
| GlycoBlue | Thermo Scientific | AM9516 | Coprecipitant |
| High-Sensitivity Bioanlayzer chips | Agilent | 5067-4626 | |
| Large Cooling Centrifuge 5920 R | Eppendorf | 5948000018 | |
| leukaemia inhibitory factor (LIF) | Merck | ESG1107 | |
| Liquiport | KNF | NF300 | Benchtop aspiration system |
| Low-binding filter tips | Biozym | VT0260U, VT0240, VT0220, VT0200U | |
| Molecular biology grade water | Merck | W3500-6x500ML | |
| Next Seq 500 | Illumina | SY-415-1001 | |
| Next Seq 500 High Output v2 Kit (300 cycles) | Illumina | FC-404-2004 | |
| Nonidet P40 Substitute (NP40) | Merck | 11332473001 | |
| PD0325901 | Axon Medchem BV | Axon 1408 | |
| Protease inhibitor cocktail (EDTA-free) | Merck | 11873580001 | |
| Proteinase K - recombinant, PCR-grade (20 mg/mL) | Thermo Scientific | EO0491 | |
| Qubit 2.0 | Thermo Scientific | Q32871 | |
| Qubit assay tubes | Thermo Scientific | Q32856 | |
| Qubit dsDNA High Sensitivity kit | Thermo Scientific | Q32851 | |
| RNase A (10 mg/mL) | Thermo Scientific | EN0531 | |
| Sodium acetate pH 5.2 (3M) | Merck | S7899 | |
| speed vacuum concentrator | Eppendorf | EP5305000100-1EA | |
| Agencourt AMPureXP | Beckman Coulter | A63881 | SPRI beads |
| SureSelect Target Enrichment Box 1 | Agilent | 5190-8645 | |
| SureSelect Target Enrichment Kit ILM Indexing Hyb Module Box 2 | Agilent | 5190-4455 | |
| SureSelect XT Library Prep Kit ILM | Agilent | 5500-0132 | |
| T4 ligase (30 units/µL) | Thermo Scientific | EL0013 | |
| table-top Centrifuge 5427 R | Eppendorf | 5409000012 | |
| Triton-X-100 (500 mL) | Merck | X100-500ML | |
| Trypan Blue | Invitrogen | T10282 | |
| Trypsine | Thermo Scientific | 25300054 | |
| UltraPure Glycine | Thermo Scientific | 15527013 | |
| β-mercaptoethanol | Thermo Scientific | 31350010 |
Referencias
- Ibrahim, D. M., Mundlos, S. The role of 3D chromatin domains in gene regulation: a multi-facetted view on genome organization. Current Opinion in Genetics & Development. 61, 1-8 (2020).
- Bolt, C. C., Duboule, D. The regulatory landscapes of developmental genes. Development. 147 (3), (2020).
- Glaser, J., Mundlos, S. 3D or not 3D: Shaping the genome during development. Cold Spring Harbor Perspectives in Biology. 14 (5), 040188(2021).
- Denker, A., De Laat, W. The second decade of 3C technologies: detailed insights into nuclear organization. Genes & Development. 30 (12), 1357-1382 (2016).
- Kempfer, R., Pombo, A. Methods for mapping 3D chromosome architecture. Nature Reviews Genetics. 21 (4), 207-226 (2020).
- McCord, R. P., Kaplan, N., Giorgetti, L. Chromosome conformation capture and beyond: Toward an integrative view of chromosome structure and function. Molecular Cell. 77 (4), 688-708 (2020).
- Jerkovic, I., Cavalli, G. Understanding 3D genome organization by multidisciplinary methods. Nature ReviewsMolecular Cell Biology. 22 (8), 511-528 (2021).
- Hsieh, T. -H. S., et al. Mapping nucleosome resolution chromosome folding in yeast by Micro-C. Cell. 162 (1), 108-119 (2015).
- Krietenstein, N., et al. Ultrastructural details of mammalian chromosome architecture. Molecular Cell. 78 (3), 554-565 (2020).
- Dekker, J., Rippe, K., Dekker, M., Kleckner, N. Capturing chromosome conformation. Science. 295 (5558), 1306-1311 (2002).
- Naumova, N., Smith, E. M., Zhan, Y., Dekker, J. Analysis of long-range chromatin interactions using Chromosome Conformation Capture. Methods. 58 (3), 192-203 (2012).
- Simonis, M., et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nature Genetics. 38 (11), 1348-1354 (2006).
- Zhao, Z., et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra-and interchromosomal interactions. Nature Genetics. 38 (11), 1341-1347 (2006).
- Würtele, H., Chartrand, P. Genome-wide scanning of HoxB1-associated loci in mouse ES cells using an open-ended Chromosome Conformation Capture methodology. Chromosome Research. 14 (5), 477-495 (2006).
- De Wit, E., De Laat, W. A decade of 3C technologies: insights into nuclear organization. Genes & Development. 26 (1), 11-24 (2012).
- Dostie, J., et al. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Research. 16 (10), 1299-1309 (2006).
- Splinter, E., et al. The inactive X chromosome adopts a unique three-dimensional conformation that is dependent on Xist RNA. Genes & Development. 25 (13), 1371-1383 (2011).
- Ferraiuolo, M. A., Sanyal, A., Naumova, N., Dekker, J., Dostie, J. From cells to chromatin: capturing snapshots of genome organization with 5C technology. Methods. 58 (3), 255-267 (2012).
- Kim, J. H., et al. 5C-ID: Increased resolution Chromosome-Conformation-Capture-Carbon-Copy with in situ 3C and double alternating primer design. Methods. 142, 39-46 (2018).
- Lieberman-Aiden, E., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326 (5950), 289-293 (2009).
- Zhang, Y., et al. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell. 148 (5), 908-921 (2012).
- Rao, S. S. P., et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 159 (7), 1665-1680 (2014).
- Dixon, J. R., et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 485 (7398), 376-380 (2012).
- Nora, E. P., et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 485 (7398), 381-385 (2012).
- Krefting, J., Andrade-Navarro, M. A., Ibn-Salem, J. Evolutionary stability of topologically associating domains is associated with conserved gene regulation. BMC Biology. 16 (1), 87(2018).
- Galupa, R., Heard, E. Topologically associating domains in chromosome architecture and gene regulatory landscapes during development, disease, and evolution. Cold Spring Harbor Symposia on Quantitative Biology. 82, 267-278 (2017).
- Tena, J. J., Santos-Pereira, J. M. Topologically associating domains and regulatory landscapes in development, evolution and disease. Frontiers in Cell and Developmental Biology. 9, 702787(2021).
- Lupiáñez, D. G., Spielmann, M., Mundlos, S. Breaking TADs: How alterations of chromatin domains result in disease. Trends in Genetics. 32 (4), 225-237 (2016).
- Davidson, I. F., Peters, J. -M. Genome folding through loop extrusion by SMC complexes. Nature Reviews Molecular Cell Biology. 22 (7), 445-464 (2021).
- Schmitt, A. D., Hu, M., Ren, B. Genome-wide mapping and analysis of chromosome architecture. Nature Reviews Molecular Cell Biology. 17 (12), 743-755 (2016).
- Hughes, J. R., et al. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nature Genetics. 46 (2), 205-212 (2014).
- Davies, J. O. J., et al. Multiplexed analysis of chromosome conformation at vastly improved sensitivity. Nature Methods. 13 (1), 74-80 (2016).
- Jäger, R., et al. Capture Hi-C identifies the chromatin interactome of colorectal cancer risk loci. Nature Communications. 6, 6178(2015).
- Schoenfelder, S., et al. The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Research. 25 (4), 582-597 (2015).
- Sahlén, P., et al. Genome-wide mapping of promoter-anchored interactions with close to single-enhancer resolution. Genome Biology. 16, 156(2015).
- Joshi, O., et al. Dynamic reorganization of extremely long-range promoter-promoter interactions between two states of pluripotency. Cell Stem Cell. 17 (6), 748-757 (2015).
- Mifsud, B., et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nature Genetics. 47 (6), 598-606 (2015).
- Dryden, N. H., et al. Unbiased analysis of potential targets of breast cancer susceptibility loci by Capture Hi-C. Genome Research. 24 (11), 1854-1868 (2014).
- Oudelaar, A. M., Davies, J. O. J., Downes, D. J., Higgs, D. R., Hughes, J. R. Robust detection of chromosomal interactions from small numbers of cells using low-input Capture-C. Nucleic Acids Research. 45 (22), 184(2017).
- Oudelaar, A. M., et al. Single-allele chromatin interactions identify regulatory hubs in dynamic compartmentalized domains. Nature Genetics. 50 (12), 1744-1751 (2018).
- Franke, M., et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature. 538 (7624), 265-269 (2016).
- Despang, A., et al. Functional dissection of the Sox9-Kcnj2 locus identifies nonessential and instructive roles of TAD architecture. Nature Genetics. 51 (8), 1263-1271 (2019).
- Oudelaar, A. M., et al. Dynamics of the 4D genome during in vivo lineage specification and differentiation. Nature Communications. 11 (1), 1-12 (2020).
- Galupa, R., Heard, E. X-chromosome inactivation: A crossroads between chromosome architecture and gene regulation. Annual Review of Genetics. 52, 535-566 (2018).
- Loda, A., Collombet, S., Heard, E. Gene regulation in time and space during X-chromosome inactivation. Nature Reviews. Molecular Cell Biology. 23 (4), 231-249 (2022).
- van Bemmel, J. G., et al. The bipartite TAD organization of the X-inactivation center ensures opposing developmental regulation of Tsix and Xist. Nature Genetics. 51 (6), 1024-1034 (2019).
- Galupa, R., et al. A conserved noncoding locus regulates random monoallelic Xist expression across a topological boundary. Molecular Cell. 77 (2), 352-367 (2020).
- Gjaltema, R. A. F., et al. Distal and proximal cis-regulatory elements sense X chromosome dosage and developmental state at the Xist locus. Molecular Cell. 82 (1), 190-208 (2022).
- Galupa, R., et al. Inversion of a topological domain leads to restricted changes in its gene expression and affects inter-domain communication. Development. 149 (9), (2022).
- Savarese, F., Flahndorfer, K., Jaenisch, R., Busslinger, M., Wutz, A. Hematopoietic precursor cells transiently reestablish permissiveness for X inactivation. Molecular and Cellular Biology. 26 (19), 7167-7177 (2006).
- Schulz, E. G., et al. The two active X chromosomes in female ESCs block exit from the pluripotent state by modulating the ESC signaling network. Cell Stem Cell. 14 (2), 203-216 (2014).
- Gnirke, A., et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nature Biotechnology. 27 (2), 182-189 (2009).
- Akgol Oksuz, B., et al. Systematic evaluation of chromosome conformation capture assays. Nature Methods. 18 (9), 1046-1055 (2021).
- Piccinini, F., Tesei, A., Arienti, C., Bevilacqua, A. Cell counting and viability assessment of 2D and 3D Cell cultures: Expected reliability of the trypan blue assay. Biological Procedures Online. 19 (1), 8(2017).
- Servant, N., et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biology. 16, 259(2015).
- Abdennur, N., Mirny, L. A. Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics. 36 (1), 311-316 (2020).
- Imakaev, M., et al. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nature Methods. 9 (10), 999-1003 (2012).
- Forcato, M., et al. Comparison of computational methods for Hi-C data analysis. Nature Methods. 14 (7), 679-685 (2017).
- Venev, S., et al. open2c/cooltools: v0.4.1. , (2021).
- Wages, J. M. NUCLEIC ACIDS | Immunoassays. Encyclopedia of Analytical Science. , Elsevier. 408-417 (2005).
- Ewels, P. A., et al. The nf-core framework for community-curated bioinformatics pipelines. Nature Biotechnology. 38 (3), 276-278 (2020).
- Servant, N., Peltzer, A. nf-core/hic: Initial release of nf-core/hic. Zenodo. , (2019).
- Furlan, G., et al. The Ftx noncoding locus controls X chromosome inactivation independently of its RNA products. Molecular Cell. 70 (3), 462-472 (2018).
- Giorgetti, L., et al. Structural organization of the inactive X chromosome in the mouse. Nature. 535 (7613), 575-579 (2016).
- Simon, C. S., et al. Functional characterisation of cis-regulatory elements governing dynamic Eomes expression in the early mouse embryo. Development. 144 (7), 1249-1260 (2017).
- Williams, R. M., et al. Reconstruction of the global neural crest gene regulatory network in vivo. Developmental Cell. 51 (2), 255-276 (2019).
- Godfrey, L., et al. DOT1L inhibition reveals a distinct subset of enhancers dependent on H3K79 methylation. Nature Communications. 10 (1), 2803(2019).
- Hanssen, L. L. P., et al. Tissue-specific CTCF-cohesin-mediated chromatin architecture delimits enhancer interactions and function in vivo. Nature Cell Biology. 19 (8), 952-961 (2017).
- Larke, M. S. C., et al. Enhancers predominantly regulate gene expression during differentiation via transcription initiation. Molecular Cell. 81 (5), 983-997 (2021).
- Oudelaar, A. M., et al. A revised model for promoter competition based on multi-way chromatin interactions at the α-globin locus. Nature Communications. 10 (1), 5412(2019).
- Long, H. K., et al. Loss of extreme long-range enhancers in human neural crest drives a craniofacial disorder. Cell Stem Cell. 27 (5), 765-783 (2020).
- Schoenfelder, S., Javierre, B. -M., Furlan-Magaril, M., Wingett, S. W., Fraser, P. Promoter Capture Hi-C: High-resolution, genome-wide profiling of promoter interactions. Journal of Visualized Experiments. (136), e57320(2018).
- Siersbæk, R., et al. Dynamic rewiring of promoter-anchored chromatin loops during adipocyte differentiation. Molecular Cell. 66 (3), 420-435 (2017).
- Rubin, A. J., et al. Lineage-specific dynamic and pre-established enhancer-promoter contacts cooperate in terminal differentiation. Nature Genetics. 49 (10), 1522-1528 (2017).
- Freire-Pritchett, P., et al. Global reorganisation of cis-regulatory units upon lineage commitment of human embryonic stem cells. eLife. 6, 21926(2017).
- Javierre, B. M., et al. Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell. 167 (5), 1369-1384 (2016).
- Miguel-Escalada, I., et al. Human pancreatic islet three-dimensional chromatin architecture provides insights into the genetics of type 2 diabetes. Nature Genetics. 51 (7), 1137-1148 (2019).
- Keane, T. M., et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 477 (7364), 289-294 (2011).
- Baxter, J. S., et al. Capture Hi-C identifies putative target genes at 33 breast cancer risk loci. Nature Communications. 9 (1), 1028(2018).
- Sanborn, A. L., et al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proceedings of the National Academy of Sciences. 112 (47), 6456-6465 (2015).
- Owens, D. D. G., et al. Dynamic Runx1 chromatin boundaries affect gene expression in hematopoietic development. Nature Communications. 13 (1), 773(2022).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados