
Method Article
Entschlüsselung der hochauflösenden 3D-Chromatinorganisation über Capture Hi-C
In diesem Artikel
Zusammenfassung
Dieses Protokoll beschreibt die Capture Hi-C-Methode, die verwendet wird, um die 3D-Organisation von megabasierten Zielgenomregionen mit hoher Auflösung zu charakterisieren, einschließlich der Grenzen topologisch assoziierender Domänen (TADs) und langreichweitiger Chromatininteraktionen zwischen regulatorischen und anderen DNA-Sequenzelementen.
Zusammenfassung
Die räumliche Organisation des Genoms trägt zu seiner Funktion und Regulation in vielen Kontexten bei, einschließlich Transkription, Replikation, Rekombination und Reparatur. Das Verständnis der genauen Kausalität zwischen Genomtopologie und -funktion ist daher von entscheidender Bedeutung und zunehmend Gegenstand intensiver Forschung. Technologien zur Erfassung von Chromosomenkonformationen (3C) ermöglichen es, auf die 3D-Struktur des Chromatins zu schließen, indem die Häufigkeit der Interaktionen zwischen einer beliebigen Region des Genoms gemessen wird. Hier beschreiben wir ein schnelles und einfaches Protokoll zur Durchführung von Capture Hi-C, einer 3C-basierten Target-Anreicherungsmethode, die die allelspezifische 3D-Organisation von megabasierten genomischen Targets mit hoher Auflösung charakterisiert. Bei Capture Hi-C werden die Zielregionen mit einer Reihe von biotinylierten Sonden erfasst, bevor eine nachgeschaltete Hochdurchsatzsequenzierung erfolgt. Dadurch wird eine höhere Auflösung und Allelspezifität erreicht und gleichzeitig die Zeiteffektivität und Erschwinglichkeit der Technologie verbessert. Um seine Stärken zu demonstrieren, wurde das Capture Hi-C-Protokoll auf das X-Inaktivierungszentrum (Xic) der Maus angewendet, den wichtigsten regulatorischen Ort der X-Chromosomen-Inaktivierung (XCI).
Einleitung
Das lineare Genom enthält alle Informationen, die ein Organismus benötigt, um die Embryonalentwicklung zu durchlaufen und das gesamte Erwachsenenalter zu überleben. Die Anweisung genetisch identischer Zellen, unterschiedliche Funktionen auszuführen, ist jedoch von grundlegender Bedeutung, um genau zu kontrollieren, welche Informationen in bestimmten Kontexten verwendet werden, einschließlich verschiedener Gewebe und/oder Entwicklungsstadien. Es wird angenommen, dass die dreidimensionale Organisation des Genoms an dieser genauen räumlich-zeitlichen Regulation der Genaktivität beteiligt ist, indem sie die physikalische Interaktion zwischen regulatorischen Elementen, die im linearen Genom durch mehrere hundert Kilobasen voneinander getrennt sein können, erleichtert oder verhindert (für Übersichtsarbeiten 1,2,3). In den letzten 20 Jahren hat sich unser Verständnis des Zusammenspiels zwischen Genomfaltung und -aktivität rapide verbessert, was vor allem auf die Entwicklung von Technologien zur Erfassung von Chromosomenkonformationen (3C) zurückzuführen ist (für Review 4,5,6,7). Diese Methoden messen die Häufigkeit von Interaktionen zwischen beliebigen Regionen des Genoms und beruhen auf der Ligation von DNA-Sequenzen, die sich in unmittelbarer 3D-Nähe innerhalb des Zellkerns befinden. Die gebräuchlichsten 3C-Protokolle beginnen mit der Fixierung von Zellpopulationen mit einem Vernetzungsmittel wie Formaldehyd. Das vernetzte Chromatin wird dann mit einem Restriktionsenzym verdaut, obwohl auch der MNase-Verdau verwendet wurde 8,9. Nach dem Verdauen werden freie DNA-Enden in unmittelbarer räumlicher Nähe religiert und die Vernetzung rückgängig gemacht. Dieser Schritt führt zur 3C-"Bibliothek" oder "Vorlage", einem gemischten Pool von Hybridfragmenten, in dem Sequenzen, die sich in 3D-Nähe zum Zellkern befanden, eine höhere Wahrscheinlichkeit haben, im selben DNA-Fragment ligiert zu werden. Die nachgelagerte Quantifizierung dieser Hybridfragmente ermöglicht es, auf die 3D-Konformation von Genomregionen zu schließen, die im linearen Genom Tausende von Basenpaaren voneinander entfernt liegen, aber im 3D-Raum interagieren könnten.
Es wurden viele verschiedene Ansätze entwickelt, um die 3C-Bibliothek zu charakterisieren, die sich sowohl darin unterscheiden, welche Untergruppen von Ligationsfragmenten analysiert werden, als auch in der Technologie, die für ihre nachgelagerte Quantifizierung verwendet wird. Das ursprüngliche 3C-Protokoll stützte sich auf die Auswahl von zwei Regions of Interest und die Quantifizierung ihrer "Eins-gegen-Eins"-Interaktionshäufigkeit mittels PCR10,11. Der 4C-Ansatz (Circular Chromosome Conformation Capture) misst die Wechselwirkungen zwischen einem einzelnen Locus von Interesse (d. h. dem "View-Point") und dem Rest des Genoms ("one versus all")12,13,14. In 4C durchläuft die 3C-Bibliothek eine zweite Runde des Aufschlusses und der Religation, um kleine zirkuläre DNA-Moleküle zu erzeugen, die durch View-Point-spezifische Primer mittels PCR amplifiziert werden15. 5C (Chromosome Conformation Capture Carbon Copy) ermöglicht die Charakterisierung von 3D-Wechselwirkungen über größere Regionen von Interesse hinweg und liefert Einblicke in die Chromatinfaltung höherer Ordnung innerhalb dieser Region ("viele gegen viele")16. In 5C wird die 3C-Bibliothek zu einem Pool von Oligonukleotiden hybridisiert, die sich mit Restriktionsstellen überlappen, die anschließend durch Multiplex-PCR mit Universalprimern15 amplifiziert werden können. Sowohl in 4C als auch in 5C wurden die informativen DNA-Fragmente zunächst durch Microarrays und später durch Next-Generation-Sequencing (NGS) quantifiziert17,18,19. Diese Strategien charakterisieren bestimmte Regionen von Interesse, können aber nicht auf die Kartierung genomweiter Interaktionen angewendet werden. Letzteres Ziel wird mit Hi-C erreicht, einer 3C-basierten Hochdurchsatzstrategie, bei der eine massiv parallele Sequenzierung des 3C-Templates die unverzerrte Charakterisierung der Chromatinfaltung auf genomweiter Ebene ("all versus all") ermöglicht20. Das Hi-C-Protokoll umfasst den Einbau eines biotinylierten Rests an den Enden der verdauten Fragmente, gefolgt von einem Herunterziehen von Ligationsfragmenten mit Streptavidin-Kügelchen, um die Rückgewinnung ligierter Fragmentezu erhöhen 20.
Hi-C zeigte, dass die Genome von Säugetieren im 3D-Zellkern auf mehreren Ebenen strukturell organisiert sind. Auf der Megabasen-Skala wird das Genom in Regionen mit aktivem und inaktivem Chromatin, die A- bzw. B-Kompartimente, unterteilt20,21. Die Existenz weiterer Subkompartimente, die durch unterschiedliche Chromatin- und Aktivitätszustände repräsentiert werden, wurde ebenfalls nachträglich gezeigt22. Bei höherer Auflösung wird das Genom weiter in selbstinteragierende Sub-Megabasen-Domänen unterteilt, die als topologisch assoziierende Domänen (TADs) bezeichnet werden, die erstmals durch Hi-C- und 5C-Analysen der Genome von Mensch und Maus aufgedeckt wurden23,24. Im Gegensatz zu Kompartimenten, die gewebespezifisch variieren, sind TADs tendenziell konstant (obwohl es viele Ausnahmen gibt). Wichtig ist, dass die TAD-Grenzen über Spezies25 hinweg erhalten bleiben. In Säugetierzellen umfassen TADs häufig Gene, die die gleiche regulatorische Landschaft teilen, und es hat sich gezeigt, dass sie einen strukturellen Rahmen darstellen, der die Koregulation von Genen erleichtert und gleichzeitig die Interaktionen mit benachbarten regulatorischen Domänen begrenzt (für Review 3,26,27,28). Darüber hinaus können innerhalb von TADs Wechselwirkungen aufgrund von CTCF-Stellen an der Basis von Cohesin-extrudierten Schleifen die Wahrscheinlichkeit von Promotor-Enhancer- oder Enhancer-Enhancer-Wechselwirkungen erhöhen (für Review29).
In Hi-C können Kompartimente und TADs mit einer Auflösung von 1 Mb bis 40 kb detektiert werden, aber eine höhere Auflösung kann erreicht werden, um Kontakte auf kleinerer Skala zu charakterisieren, wie z. B. Schleifenwechselwirkungen zwischen distalen Elementen auf der Skala von 5-10 kb. Die Erhöhung der Auflösung, um solche Schleifen effizient durch HiC detektieren zu können, erfordert jedoch eine deutliche Erhöhung der Sequenziertiefe und damit der Sequenzierungskosten. Dies wird noch verschärft, wenn die Analyse allelspezifisch sein muss. In der Tat erfordert eine X-fache Erhöhung der Auflösung eine X2-fache Erhöhung der Sequenzierungstiefe, was bedeutet, dass hochauflösende und allelspezifische genomweite Ansätze unerschwinglich teuer sein können30.
Um die Kosteneffizienz und Erschwinglichkeit zu verbessern und gleichzeitig eine hohe Auflösung beizubehalten, können die Zielregionen von Interesse nach ihrer Hybridisierung mit komplementären Biotin-markierten Oligonukleotidsonden vor der nachgelagerten Sequenzierung physisch aus genomweiten 3C- oder Hi-C-Bibliotheken gezogen werden. Diese Strategien zur Target-Anreicherung werden als Capture-C-Methoden bezeichnet und ermöglichen die Untersuchung von Interaktionen von Hunderten von Zielorten, die über das Genom verstreut sind (z. B. Promoter Capture (PC) Hi-C; Capture-C der nächsten Generation (NG); Low Input (LI) Capture-C; Kerntitrierte (NuTi) Capture-C; Tri-C)31,32,33,34,35,36,37,38,39,40 oder über Regionen hinweg, die sich über mehrere Megabasen erstrecken (z. B. Capture HiC; HYbrid Capture Hi-C (Hi-C2); Kachel-C)41,42,43. Zwei Aspekte können bei Capture-basierten Methoden variieren: (1) die Art und das Design von biotinylierten Oligonukleotiden (d. h. RNA oder DNA, einzelne Oligos, die disperse genomische Ziele erfassen, oder mehrere Oligos, die eine Region von Interesse bedecken); und (2) die Vorlage, die zum Herunterziehen von Zielen verwendet wird, bei denen es sich um die 3C- oder Hi-C-Bibliothek handeln kann, wobei letztere aus biotinylierten Restriktionsfragmenten besteht, die aus der 3C-Bibliothek heruntergezogen wurden.
Hier wird ein Capture Hi-C-Protokoll beschrieben, das auf der Anreicherung von Zielkontakten aus der 3C-Bibliothek basiert. Das Protokoll beruht auf dem Design eines maßgeschneiderten Kachelarrays von biotinylierten RNA-Sonden und kann in 1 Woche von der Präparation der 3C-Bibliothek bis zur NGS-Sequenzierung durchgeführt werden. Das Protokoll ist schnell, einfach und ermöglicht die Charakterisierung der 3D-Organisation höherer Ordnung von Regions of Interest in Megabase-Größe bei einer Auflösung von 5 kb bei gleichzeitiger Verbesserung der Zeiteffektivität und Erschwinglichkeit im Vergleich zu anderen 3C-Methoden. Das Capture Hi-C-Protokoll wurde auf den regulatorischen Master-Locus der X-Chromosomen-Inaktivierung (XCI), das X-Inaktivierungszentrum (Xic), angewendet, das die nicht-kodierende Xist-RNA beherbergt. Der Xic war bereits Gegenstand umfangreicher Struktur- und Funktionsanalysen (für Review44,45). Bei Säugetieren kompensiert XCI die Dosis von X-chromosomalen Genen zwischen Weibchen (XX) und Männchen (XY) und beinhaltet das transkriptionelle Silencing fast der gesamten beiden X-Chromosomen in weiblichen Zellen. Das Xic stellt einen leistungsstarken Goldstandard-Locus für Studien zur 3D-Genomtopologie und dem Zusammenspiel mit der Genregulationdar 44. Die 5C-Analyse des Xic in embryonalen Stammzellen (mESCs) der Maus führte zur Entdeckung und Benennung von TADs und lieferte damit erste Einblicke in die funktionelle Relevanz der topologischen Partitionierung und der Koregulation von Genen24. In der Folge konnte gezeigt werden, dass die topologische Organisation des Xic entscheidend am geeigneten Entwicklungszeitpunkt der Xist-Hochregulation und XCI 46 beteiligt ist, und auch im Xic47,48,49 wurden kürzlich unerwartete cis-regulatorische Elemente entdeckt, die die Genaktivität innerhalb und zwischen TADs beeinflussen können. Die Anwendung von Capture Hi-C auf 3 MB des X-Chromosoms der Maus, das den Xic überspannt, demonstriert die Leistungsfähigkeit dieses Ansatzes bei der Analyse großflächiger Chromatinfaltung mit hoher Auflösung. Es wird ein detailliertes und leicht verständliches Protokoll bereitgestellt, beginnend mit dem Design des Arrays biotinylierter Sonden an jeder DpnII-Restriktionsstelle innerhalb der interessierenden Region über die Generierung der genomweiten 3C-Bibliothek, die Hybridisierung und Erfassung von Zielkontakten bis hin zur nachgelagerten Datenanalyse. Ein Überblick über die geeigneten Qualitätskontrollen und die erwarteten Ergebnisse wird ebenfalls gegeben, und sowohl die Stärken als auch die Grenzen des Ansatzes werden im Lichte ähnlicher bestehender Methoden diskutiert.
Protokoll
Die in dieser Studie verwendeten embryonalen Stammzellen (mES-Zellen) der Maus wurden aus einer Kreuzung eines TX/TX R26 rtTA/ rtTA-Weibchens50 mit einem Mus musculus castaneus-Männchen gemäß den Tierpflegerichtlinien des Institut Curie (Paris)51 gewonnen.
1. Sonden-Design
- Entwerfen Sie eine Reihe von biotinylierten Sonden (120-mer-RNA-Oligonukleotide), die die Zielregion von Interesse abdecken.
- Kacheln Sie den interessierenden Bereich mit überlappenden Oligonukleotiden, so dass im Durchschnitt jede Sequenz innerhalb des Ziels von zwei eindeutigen Sonden abgedeckt wird (2-fache Abdeckung) (Abbildung 1).
- Schließen Sie sich wiederholende Sequenzen aus der Sondenabdeckung aus, um eine Anreicherung unspezifischer Interaktionen zu vermeiden.
HINWEIS: Um die Anreicherung der informativen Ligationsfragmente zu maximieren, wurden Regionen definiert, die sich über 300 bp stromaufwärts und stromabwärts jeder DpnII-Restriktionsstelle über das Ziel erstrecken (ChrX: 102.475.000-105.475.000), und 28.913 biotinylierte Sonden wurden gemäß der SureSelect DNA-Target-Anreicherungstechnologie über die Sure Design-Plattformentworfen 52. Nach dieser Strategie sind in jedem Oligonukleotid bis zu maximal 40 Basen repetitiver Sequenzen erlaubt, um die Anreicherung unspezifischer Wechselwirkungen zu minimieren. Das Sonden-Array wurde von Agilent synthetisiert. Hier wird DpnII aus zwei Gründen als Restriktionsenzym verwendet: (1) Es ist ein Vierschneider, der routinemäßig in mehreren 3C-basierten Methoden verwendet wird53; und (2) es maximiert die Chancen, informative Einzelnukleotid-Polymorphismen (SNPs) in der Nähe der Schnittstellen im Vergleich zu anderen Restriktionsenzymen zu erfassen, die in silico in F1-Hybridlinien getestet wurden, die in dieser Studie verwendet wurden (C57BL/6J x CASTEi/J).
2. Versuchsablauf
- Zellvorbereitung
- Säen Sie die entsprechende Anzahl von Zellen auf eine oder mehrere Zellkulturplatten, um am Tag der Fixierung eine Gesamtzellzahl von ≥ 5 x 107 Zellen zu erreichen.
HINWEIS: In dieser Studie wurden embryonale Stammzellen (mES-Zellen) der Maus verwendet. mES-Zellen werden auf gelatinierten (0,1 % Gelatine in 1x PBS - o/n bei 37 °C, 5 % CO2-Inkubator) Zellkulturplatten in mES-Medium, das 2i + LIF und chargengeprüftes fetales Kälberserum (DMEM, 15 % FBS, 0,1 mM β-Mercaptoethanol, 1.000 U/ml−1 leukämiehemmender Faktor (LIF), CHIR99021 (3 μM) und PD0325901 (1 μM)) enthält, plattiert. Für diesen Zelltyp enthält eine 10 cm große Platte mit 80 % Konfluenz ca. 2 x 107 Zellen. - Bereiten Sie eine zusätzliche Zellkulturplatte für die Zellzählung vor.
HINWEIS: Eine kleinere Zellkulturplatte kann verwendet werden, um den Medienverbrauch zu reduzieren. In diesem Fall muss die Anzahl der Zellen, die auf der kleineren Platte ausgesät werden sollen, entsprechend angepasst werden (z. B. 3x weniger Zellen auf einer 10-cm-Platte im Vergleich zu einer 15-cm-Platte).
- Säen Sie die entsprechende Anzahl von Zellen auf eine oder mehrere Zellkulturplatten, um am Tag der Fixierung eine Gesamtzellzahl von ≥ 5 x 107 Zellen zu erreichen.
- Formaldehyd-Fixierung
- Schätzen Sie die Gesamtzahl der Zellen, die vernetzt werden sollen.
- Bevor Sie mit der Vernetzungsreaktion beginnen, trypsinisieren und zählen Sie die Zellen von der speziell für die Zellzählung vorbereiteten Steuerplatte mit einem automatisierten Zellzähler gemäß den Anweisungen des Herstellers.
- Fügen Sie eine Viabilitätsfärbung (z. B. Trypanblau) hinzu, um den Prozentsatz der lebensfähigen Zellenzu bestimmen 54. Schätzen Sie anhand dieser Zellzahl die Gesamtzahl der Zellen in der/den Platte(n), die für die Vernetzung vorbereitet sind.
- Entfernen Sie das Nährmedium von den für die Vernetzung vorbereiteten Platten und ersetzen Sie es durch die entsprechende Menge Fixierlösung (2 % Formaldehyd im Zellkulturmedium). Verwenden Sie 10 ml auf einer 10-cm-Platte (z. B. ~20 ml für eine 15-cm-Platte).
Anmerkungen: Fügen Sie ein genaues Volumen der Fixierlösung hinzu. Wenn die Fixierung adhärenter Zellen nicht möglich ist, kann dieser Schritt an trypsinisierte Zellen angepasst und in 30 ml Fixierlösung in konischen 50-ml-Zentrifugenröhrchen durchgeführt werden. Formaldehyd darf nicht älter als 1 Jahr sein. Es wird bevorzugt, Einwegfläschchen zu verwenden. Die Fixierlösung muss vor der Anwendung auf Raumtemperatur (RT) gebracht werden.
VORSICHT: Formaldehyd ist gefährlich und muss gemäß den entsprechenden Gesundheits- und Sicherheitsvorschriften gehandhabt werden. - 10 min bei RT unter leichtem Mischen auf einem Shaker fixieren.
- Die Fixierungsreaktion wird durch Zugabe von 2,5 M Glycin-1x PBS bis zu einer Endkonzentration von 0,125 M abgeschreckt. 530 μl 2,5 M Glycin-1x PBS auf 10 mL auf einer 10 cm Platte (z. B. 1060 μL bis 20 mL auf einer 15 cm Platte) zugeben.
Anmerkungen: Wenn die Zellen in Lösung fixiert wurden, löschen Sie die Fixierungsreaktion mit 1590 μl 2,5 M Glycin-1x PBS. - 5 Minuten bei RT inkubieren, dabei vorsichtig auf einem Shaker mischen.
- Legen Sie die Platten auf Eis und inkubieren Sie sie weitere 15 Minuten auf Eis, während Sie sie vorsichtig auf einem Shaker mischen.
HINWEIS: Von nun an müssen die Zellen auf Eis gelagert und die Puffer vorgekühlt werden, um eine weitere Vernetzung zu vermeiden. Gehen Sie in einen Kühlraum, wenn viele Platten verarbeitet werden müssen. - Entfernen Sie die Fixierlösung aus den Zellen, indem Sie sie in ein Becherglas gießen, um eine schnelle Handhabung zu gewährleisten.
Anmerkungen: Stellen Sie sicher, dass Sie die formaldehydhaltigen flüssigen Abfälle gemäß den entsprechenden Gesundheits- und Sicherheitsvorschriften entsorgen. - Spülen Sie die 10-cm-Platte zweimal schnell mit 5 ml kaltem 0,125 M Glycin-1x PBS (8 ml für eine 15-cm-Platte) aus, um die Ablagerungen und abgestorbenen Zellen abzuwaschen. Entfernen Sie die Flüssigkeit aus dem Teller, indem Sie sie in ein Becherglas gießen, um eine schnelle Handhabung zu gewährleisten.
- Geben Sie 5 ml kaltes 0,125 M Glycin-1x PBS auf die 10-cm-Platte (10 ml für eine 15-cm-Platte) und kratzen Sie die Zellen mit einem Kunststoff-Zellschaber schnell von der Platte ab.
- Die Zellsuspension wird mit einer serologischen Pipette in ein vorgekühltes konisches 50-ml-Zentrifugenröhrchen überführt.
- Spülen Sie die Platte zweimal mit 5 mL kaltem 0,125 M Glycin-1x PBS und geben Sie die Zellsuspension in das konische Zentrifugenröhrchen.
- Bei 480 x g für 10 min bei 4 °C herunterschleudern.
Anmerkungen: Wenn die Zellen in Lösung fixiert wurden, überführen Sie die Zelle in ein vorgekühltes konisches Zentrifugenröhrchen und schleudern Sie sie 10 Minuten lang bei 4 °C bei 480 x g herunter. Entfernen Sie die Fixierlösung, indem Sie sie in ein Becherglas gießen und dreimal in 10 ml kaltem 0,125 M Glycin-1x PBS waschen. Achten Sie darauf, die Zellen bei jedem Waschschritt neu zu suspendieren. - Entfernen Sie den Überstand, indem Sie mit einem Tischabsaugsystem absaugen. Resuspendieren Sie die Zellen in 500 μL 1x PBS pro 1 x 107 Zellen, indem Sie vorsichtig mit einer P1000-Pipette auf und ab pipettieren. Um Zellen im genauen Volumen zu resuspendieren, beziehen Sie sich auf die Schätzung der Gesamtzahl der Zellen in 2.2.1.
- Aliquotieren Sie 500 μl der Zellsuspension in die berechnete Anzahl von 1,5 ml Mikrozentrifugenröhrchen (1 x 10,7 Zellen/Röhrchen).
- Bei 480 x g für 10 min bei 4 °C herunterschleudern.
- Entfernen Sie den Überstand mit einem Tischabsaugsystem und frieren Sie die Zellpellets in flüssigem Stickstoff ein. Lagern Sie die Trockenzellpellets bei -80 °C.
HINWEIS: Proben können mindestens 1 Jahr gelagert werden.
- Schätzen Sie die Gesamtzahl der Zellen, die vernetzt werden sollen.
- Zelllyse
- Tauen Sie die gefrorenen Pellets auf Eis auf.
- Bereiten Sie 1,5 ml Lysepuffer in H 2 0 pro Probe vor: Fügen Sie 10 mM Tris-HCl, pH 8,0, 10 mM NaCl und0,2% NP40 hinzu.
- 600 μl des Kaltlysepuffers zugeben und auf Eis gut resuspendieren.
- 15 Minuten auf Eis inkubieren, damit die Zellen aufquellen können.
- Bei 2655 x g für 5 min bei 4 °C herunterdrehen und den Überstand mit einem Tischabsaugsystem entfernen.
- Um Schmutz zu entfernen, resuspendieren Sie das Pellet in 1 ml des Kaltlysepuffers, schleudern Sie es 5 Minuten lang bei 4 °C bei 2655 x g herunter und entfernen Sie den Überstand.
- Schleudern Sie erneut kurz bei 2655 x g und 4 °C und entfernen Sie so viel Restüberstand wie möglich mit einem Tischabsaugsystem, das mit einer P200-Spitze ausgestattet ist.
- Resuspendierung in 100 μl 0,5 % (vol/vol) SDS.
- In einem Thermomischer bei 62 °C unter 1400 U/min 10 min inkubieren.
- Fügen Sie 290 μlH2O+ 50 μl 10 % TritonX-100 hinzu und mischen Sie gut, wobei Luftblasen vermieden werden.
- Inkubation in einem Thermomischer bei 37 °C unter Schwenken bei 1400 U/min für 15 min.
- Fügen Sie 50 μL 10x Dpnll-Puffer hinzu und drehen Sie das Röhrchen um, um es zu mischen.
- Nehmen Sie 50 μl unverdaute DNA zur Qualitätskontrolle in ein separates Röhrchen. Vergessen Sie nicht, die unverdaute Kontrollprobe zu entnehmen.
- DpnII-Verdauung
- Fügen Sie 10 μl Dpnll hohe Konzentration (insgesamt 500 U) hinzu und mischen Sie durch Invertieren.
- Inkubieren Sie die Proben und die unverdaute Kontrolle in einem Thermomischer bei 37 °C und schwenken Sie bei 1400 U/min für >4 h.
- Fügen Sie am Ende des Tages 10 μl Dpnll hohe Konzentration (insgesamt 500 U) hinzu.
- Inkubieren Sie die Proben und die unverdaute Kontrolle bei 37 °C und schwenken Sie sie über Nacht bei 1400 U/min.
- Zu Beginn des nächsten Tages werden 10 μl Dpnll mit hoher Konzentration (insgesamt 500 U) zu den Proben gegeben.
- Inkubieren Sie die Proben und die unverdaute Kontrolle in einem Thermomischer bei 37 °C und schwenken Sie 4 Stunden lang bei 1400 U/min.
- Ligation und Umkehrung der Vernetzung
- Inkubieren Sie die Röhrchen bei 65 °C für 20 min bei 1400 U/min.
HINWEIS: Fügen Sie an dieser Stelle keine Sicherheitsdatenblätter hinzu. Die Idee ist, die Kernintegrität zu erhalten, so dass die Ligatur innerhalb der Kerne durchgeführt wird, wodurch die Notwendigkeit einer extremen Verdünnung umgangen wird. - Kühlen Sie die Proben maximal 5-10 Minuten auf Eis ab. Um SDS-Ausfällungen zu vermeiden, lassen Sie die Proben nicht länger auf Eis.
- Nehmen Sie 50 μl der unligierten verdauten DNA zur Qualitätskontrolle in einem separaten Röhrchen. Lagern Sie die unverdauten und unverdauten Kontrollen bei -20 °C.
HINWEIS: Vergessen Sie nicht, die unligierte Kontrollprobe zu entnehmen. - Fügen Sie 800 μl Ligationscocktail hinzu: 122 μl 10x Ligasepuffer, 8 μl T4-Ligase (30 U/μl) und 670 μl H20.
- Inkubieren bei 16 °C, Schwenken bei 1000 U/min über Nacht.
- Fügen Sie 7,5 μl Proteinase K (20 mg/ml) zu den Proben und 2 μl zu den Kontrollen hinzu.
- Inkubieren bei 65 °C für 4 h bei 1000 U/min.
- Inkubieren Sie die Röhrchen bei 65 °C für 20 min bei 1400 U/min.
- DNA-Aufreinigung
- Übertragen Sie die Proben auf Eis in vorgekühlte konische 15-ml-Zentrifugenröhrchen und fügen Sie 2 ml Wasser, 10,5 ml eiskaltes EtOH und 583 μl 3 M NaAC hinzu.
Anmerkungen: Zusätzliches Wasser soll verhindern, dass DVB-T in das Pellet übergeht. - Fügen Sie 200 μl eiskaltes EtOH, 10,8 μl NaAC und 1 μl des Kopräzipitationsmittels zu den unverdauten und ungebundenen Qualitätskontrollen hinzu.
- Bei -80 °C mindestens 4 h bis über Nacht inkubieren.
- Schleudern Sie die 15-ml-Röhrchen 45 Minuten lang bei 2200 x g bei 4 °C.
- Schleudern Sie die 1,5-ml-Kontrollröhrchen 30 Minuten lang bei 20.500 x g bei 4 °C.
- Einmal mit 3 ml (Proben) und 1 ml (Kontrollen) eiskaltem 70 % EtOH waschen.
- Schleudern Sie 10 Minuten lang bei 2200 x g (Proben) oder 20.500 x g (Kontrollen) bei 4 °C.
- Entfernen Sie EtOH vorsichtig und lassen Sie es 10-15 Minuten lang bei RT an der Luft trocknen. Nicht übertrocknen.
- Resuspendieren Sie die Proben und Kontrollen in 100 μl bzw. 20 μl H20.
- Fügen Sie 1 μl RNAseA hinzu und inkubieren Sie bei 37 °C, wobei Sie 30 Minuten lang bei 1400 U/min schwenken.
- Übertragen Sie die Proben auf Eis in vorgekühlte konische 15-ml-Zentrifugenröhrchen und fügen Sie 2 ml Wasser, 10,5 ml eiskaltes EtOH und 583 μl 3 M NaAC hinzu.
- Qualitätskontrolle der 3C-Schablonenvorbereitung
- Quantifizieren Sie jede Probe und kontrollieren Sie sie mit einem Fluorometer-Kit für hochempfindliche DNA-Konzentrationsmessungen.
- Laden Sie 100-200 ng jeder Probe und jeder Kontrolle auf ein 1%iges Agarose/1x FSME-Gel.
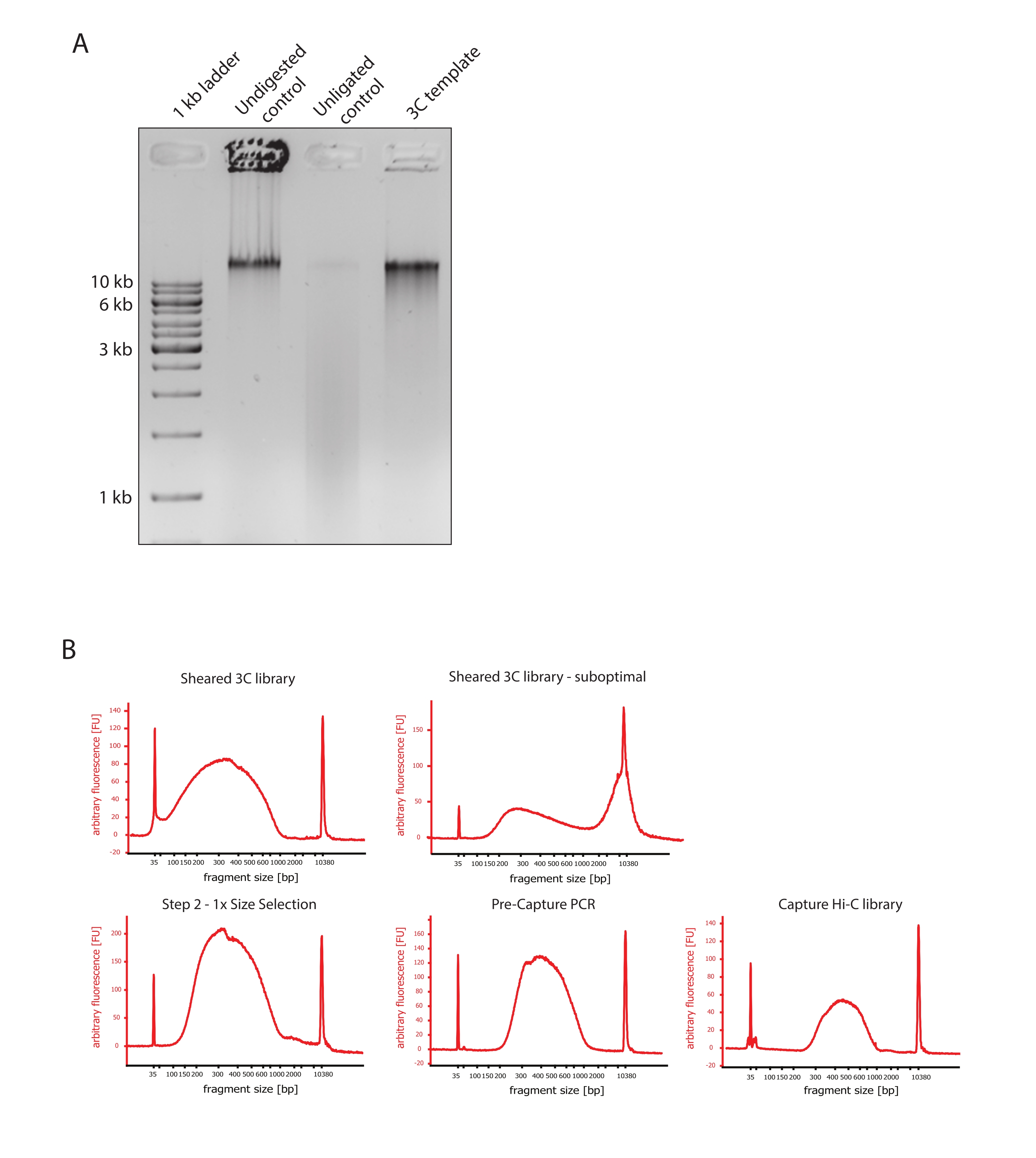
- Vergewissern Sie sich, dass das Gelbild das erwartete Ergebnis zeigt, indem Sie die Unterschiede in der Größe der DNA-Fragmente der Kontrollen und der 3C-Vorlage vergleichen, wie in Abbildung 2A dargestellt.
- Lagern Sie die Proben und Kontrollen bei -20 °C.
- Hybridisierung, Erfassung und Probenverarbeitung für Multiplex-Sequenzierung
- Hybridisierung des Arrays biotinylierter RNA-Sonden mit dem 3C-Template, Erfassung der Zielligationsfragmente und Vorbereitung der Proben für die Multiplex-Sequenzierung gemäß dem in dieser Studie verwendeten Zielanreicherungssystem für die Paired-End-Multiplex-Sequenzierung (siehe Tabelle der Materialien). Befolgen Sie das Protokoll gemäß den Anweisungen des Herstellers und führen Sie die folgenden geringfügigen Änderungen ein:
- Abschnitt 2 des Protokolls des Herstellers: Probenvorbereitung
- Befolgen Sie die Anweisungen zur Target-Anreicherung ab 3 μg gDNA-Input.
- Scheren Sie die DNA in einem Ultraschallgerät mit den folgenden Spezifikationen: 10 % Einschaltdauer, 4 Intensitäten, 200 cyc/Burst und 130 s. Beginnen Sie mit 4 μg 3C-Template, das in 130 μl Wasser für jede Einfangreaktion resuspendiert wird, um sicherzustellen, dass genügend Material vorhanden ist, um die Probenvorbereitung mit 3 μg der gescherten DNA fortzusetzen.
- Beurteilen Sie die Qualität der gescherten DNA. Führen Sie 1 μl der gescherten DNA auf einem DNA-Bioanalysator gemäß dem Hochempfindlichkeitsprotokoll durch. Erwarten Sie eine Verteilung der Fragmentgröße zwischen 150 und 700 bp (Abbildung 2).
- Reinigen Sie die Probe mit SPRI-Beads (Reversible Immobilisierung) für die Festphase. Fügen Sie 124 μl SPRI-Beads zu 124 μl der DNA-Probe hinzu, um eine 1:1-Größenauswahl auf der linken Seite gemäß den Anweisungen des Herstellers durchzuführen, und eluieren Sie in 25 μl nukleasefreiem Wasser. In diesem Reinigungsschritt werden kürzere Fragmente entfernt, um Fragmente von etwa 300 bp anzureichern (Abbildung 2).
HINWEIS: Die Menge der Proben und SPRI-Kügelchen, die in diesem Schritt verwendet werden, berücksichtigt den Volumenverlust, der beim Transfer der Proben in neue Röhrchen und beim Ausführen der Qualitätskontrollen am Bioanalysator aufgetreten ist. Alle nachfolgenden Schritte zur Größenauswahl werden gemäß den vom Protokoll des Herstellers empfohlenen Verhältnissen durchgeführt. Die DNA-Elution aus SPRI-Beads wird während des gesamten Protokolls bei RT durchgeführt. - Beurteilen Sie die Qualität der größenselektierten gescherten DNA. Lassen Sie 1 μl der gescherten DNA gemäß dem Hochempfindlichkeitsprotokoll (HS) auf dem DNA-Bioanalysator laufen. Erwarten Sie eine Verteilung der Fragmentgrößen mit der höchsten Anreicherung bei 300 bp (Abbildung 2). Fahren Sie mit der Quantifizierung der gescherten DNA fort, wenn die Scherung erfolgreich war.
- Quantifizieren Sie die gescherte DNA mit einem Fluorometer-Kit für HS-DNA-Konzentrationsmessungen.
HINWEIS: Wenn die DNA-Scherung zu einer DNA-Ausbeute von <3 μg führt, führen Sie eine zweite Runde der DNA-Scherung mit weiteren 4 μg DNA durch und kombinieren Sie die gescherten DNA-Proben nach dem ersten SPRI-Bead-Reinigungsschritt, um insgesamt 3 μg gescherte DNA zu erhalten. - Geben Sie nukleasefreies Wasser in die gereinigte DNA-Probe (insgesamt 3 μg) bis zu einem Endvolumen von 48 μl und fahren Sie mit der Endreparaturreaktion gemäß dem Protokoll des Herstellers fort.
- Nach der Ligation der Paired-End-Adapter wird die Bibliothek amplifiziert, indem Sie fünf Zyklen der Pre-Capture-PCR gemäß den Anweisungen des Herstellers durchführen (die Bedingungen für die PCR und die Primer sind im Kit enthalten).
- Abschnitt 4 des Protokolls des Herstellers: Hybridisierung und Abscheidung
- Um die präparierten DNA-Proben mit den zielspezifischen RNA-Sonden zu hybridisieren, werden 750 ng DNA-Proben in einem Endvolumen von 3,4 μl verdünnt, was zu einer Anfangskonzentration von 221 ng/μl führt. Verwenden Sie für DNA-Proben, die in größeren Volumina verdünnt wurden, einen Schnellvakuumkonzentrator, um sie auf das endgültige Volumen zu reduzieren. Eine Schnellvakuumkonzentration (250 x g; ≤45 °C) für 15-20 min ist in der Regel ausreichend für die in 10 μL resuspendierten Proben. Stellen Sie sicher, dass Sie für jede Probe das gleiche Eingangsvolumen haben, bevor Sie den Speed-Vakuum-Konzentrator starten.
- Inkubieren Sie die Hybridisierungsmischung 16-18 h bei 65 °C mit einem erhitzten Deckel bei 105 °C gemäß den Anweisungen des Herstellers.
- Abschnitt 5 des Protokolls des Herstellers: Indizierung und Probenverarbeitung für die Multiplex-Sequenzierung
- Um die erfassten Bibliotheken mit Indizierungsprimern zu amplifizieren, führen Sie 12 Zyklen der Post-Capture-PCR gemäß den Anweisungen des Herstellers durch (die Bedingungen für die PCR und die Primer sind im Kit enthalten).
- Abschnitt 2 des Protokolls des Herstellers: Probenvorbereitung
- Hybridisierung des Arrays biotinylierter RNA-Sonden mit dem 3C-Template, Erfassung der Zielligationsfragmente und Vorbereitung der Proben für die Multiplex-Sequenzierung gemäß dem in dieser Studie verwendeten Zielanreicherungssystem für die Paired-End-Multiplex-Sequenzierung (siehe Tabelle der Materialien). Befolgen Sie das Protokoll gemäß den Anweisungen des Herstellers und führen Sie die folgenden geringfügigen Änderungen ein:
- Next-Generation-Sequenzierung
- Um mehrere Hi-C-Erfassungsbibliotheken auf derselben Durchflusszelle auszuführen, bereiten Sie eine äquimolare Mischung der Erfassungsbibliotheken vor und sequenzieren Sie 100-120 Mio. Lesevorgänge pro Bibliothek.
- Wenn die allelspezifische Analyse erforderlich ist, sequenzieren Sie 150 bp paired-end, um eine ausreichende SNP-Abdeckung zu gewährleisten.
3. Datenanalyse
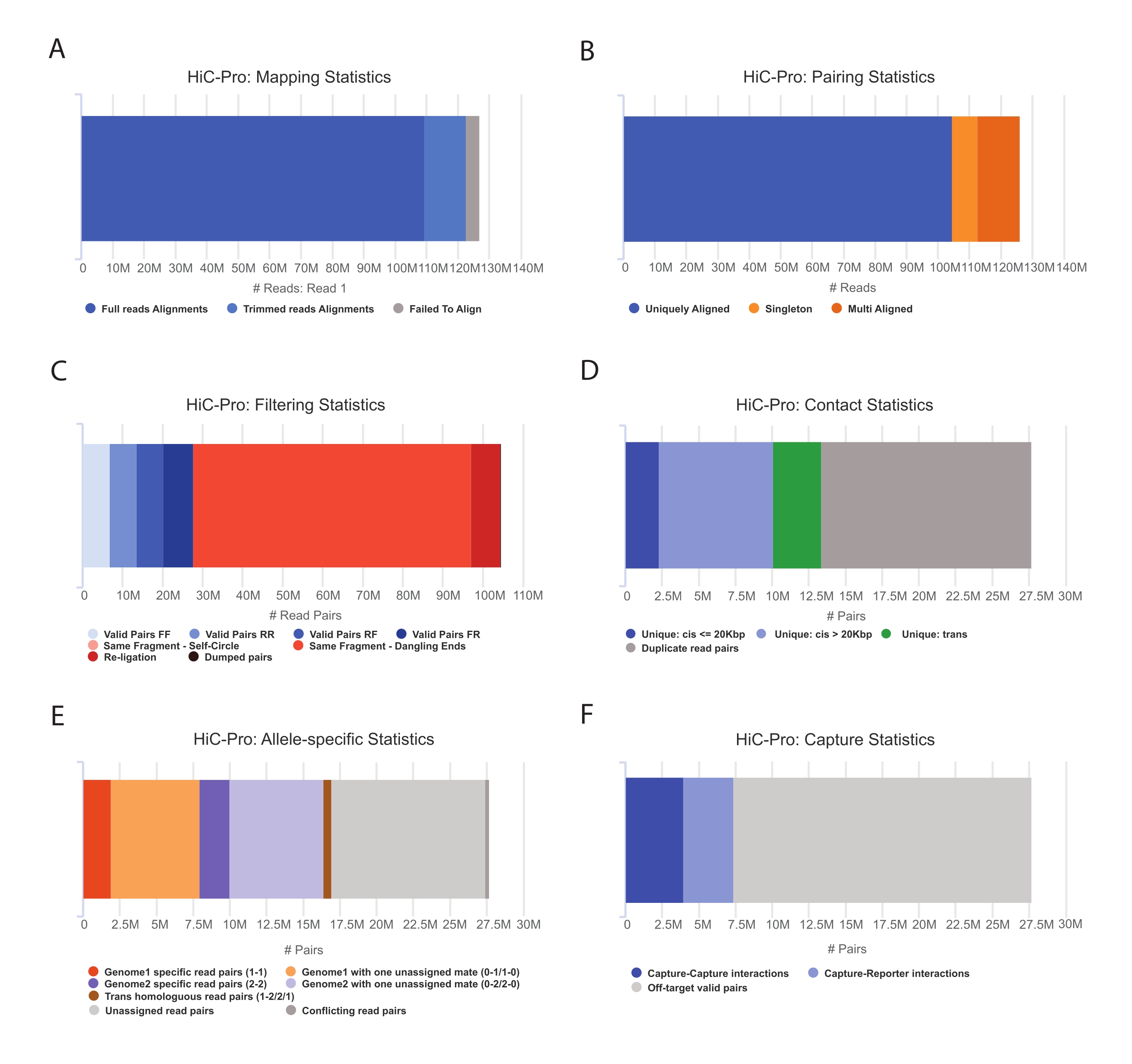
- Wenden Sie die HiC-Pro-Pipeline an, um eine Capture Hi-C-Datenanalysedurchzuführen 55. HiC-Pro bietet Qualitätskontrollen bei jedem Schritt der Verarbeitung, einschließlich (Abbildung 3):
(i) Die Alignment-Rate auf dem Referenzgenom, die den Anteil der Reads angibt, die sich über eine Ligationsstelle erstrecken, sowie die Anzahl der Paare und Singletons.
ii) Der Anteil gültiger Ligationsprodukte und nicht-informativer Lesepaare (dangling-end, self-ligation, etc.).
iii) Der Anteil der kurz-/langreichweitigen und intra-/interchromosomalen Kontakte.
iv) Der Anteil der On-Target-Kontakte für Capture Hi-C.
v) Der Anteil der allelspezifischen Lesevorgänge, falls angegeben.
HINWEIS: HiC-Pro unterstützt eine Vielzahl von Protokollen, einschließlich In-situ-Hi-C und Capture Hi-C. Im letzteren Fall muss der Benutzer lediglich die Zielregion (BED-Format) in der Konfigurationsdatei angeben. Sobald die Daten verarbeitet sind, können die HiC-Pro-Ausgänge leicht in ein kühleres Objekt für die nachgelagerte Analyse56 umgewandelt werden. In diesem Schritt werden die Kontaktkarten mit verschiedenen Auflösungen unter Verwendung der ICE-Methode normalisiert, die zuvor von Imakaev und Kollegen beschrieben wurde57. Anschließend können mehrere Analysen durchgeführt werden, um Chromosomenkompartimente, TADs oder Chromatinschleifen zu nennen (für Review58). Der Workflow des Protokolls ist in Abbildung 4 dargestellt. Hier wird die "cooltools"-Suite angewendet, um den Isolationswert und die TADs-Grenzen zu berechnen, wie in Abbildung 5 und Abbildung 659 dargestellt.
Ergebnisse
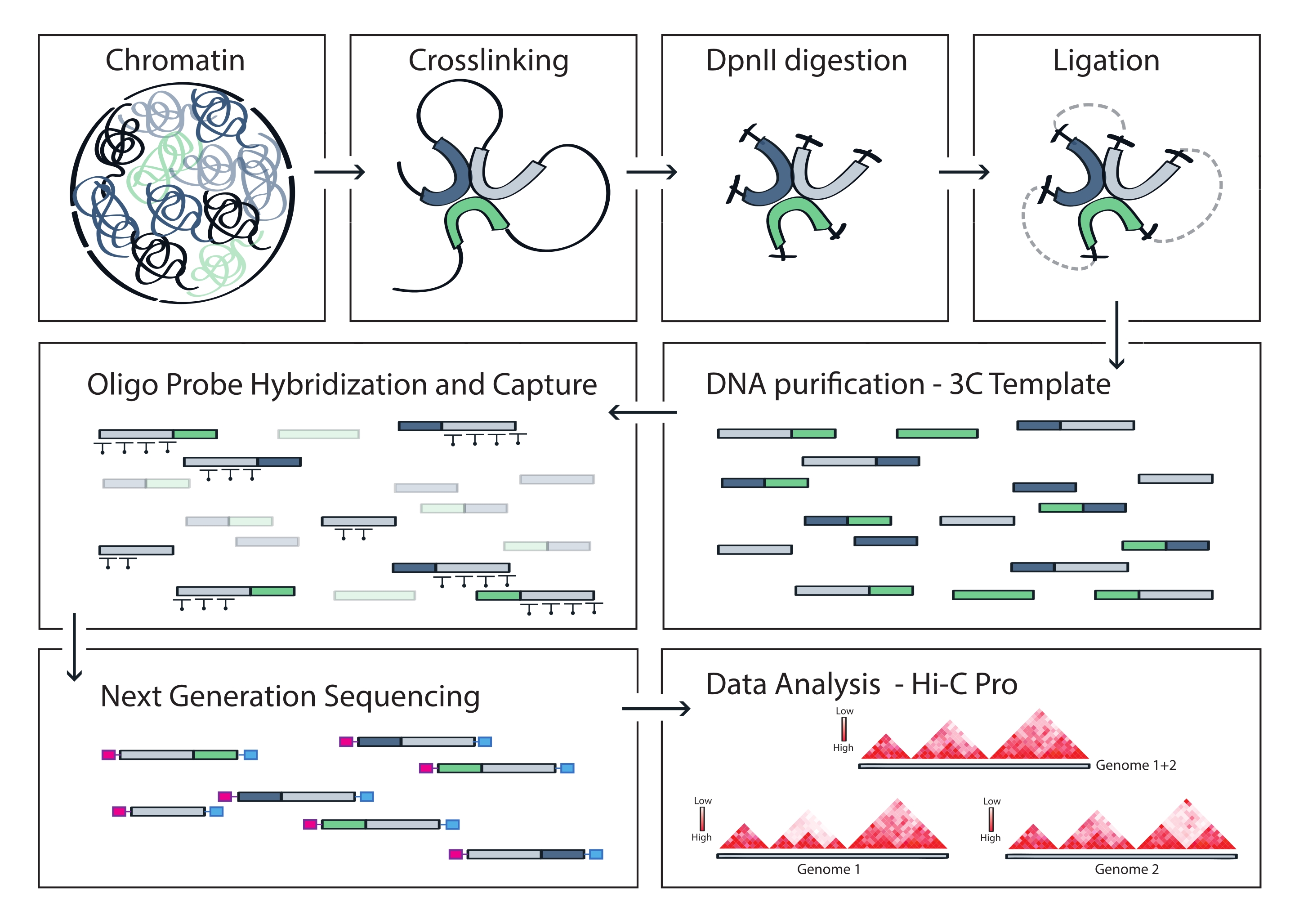
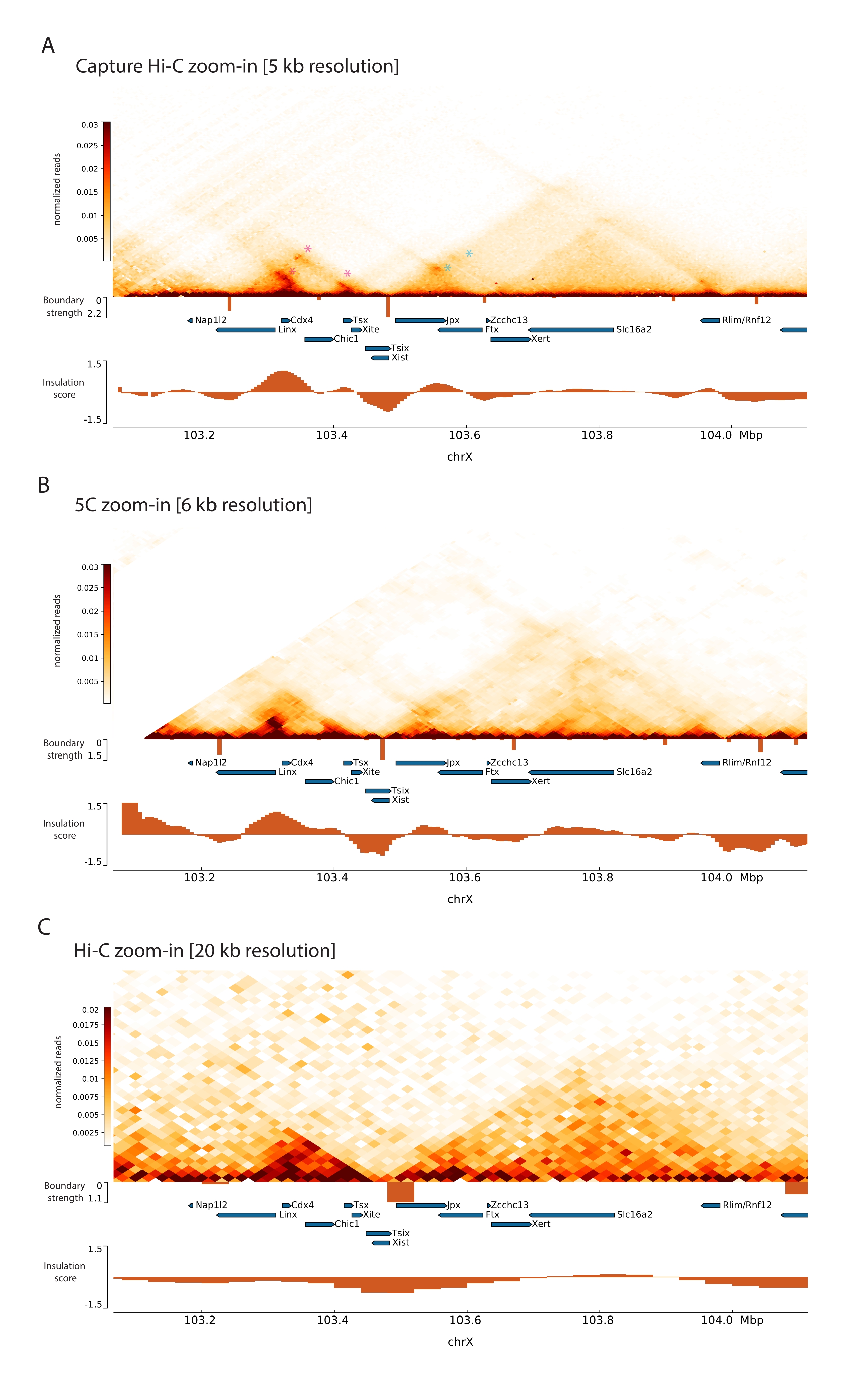
Das beschriebene Capture Hi-C Protokoll basiert auf der Präparation des genomweiten 3C-Templates mit Hilfe eines Four-Basen-Cutters (DpnII). Die anschließende Anreicherung von Ligationsfragmenten über die interessierende genomische Region wird durch Hybridisierung eines Arrays von Kachel-RNA-Sonden und deren Streptavidin-basierter Erfassung gemäß dem in dieser Studie verwendeten Zielanreicherungssystem erreicht (Abbildung 1). Biotinylierte RNA-Sonden wurden ausgewählt, da sie im Vergleich zu DNA-Sonden eine engere Bindungsaffinität zu ihren Zielen aufweisen52,60. Die erfassten Bibliotheken werden dann indiziert und für die gemultiplexte Hochdurchsatzsequenzierung zusammengefasst. Capture-Hi-C-Daten können als hochauflösende Hi-C-Interaktionskarten, aber auch als 4C-ähnliche Single-View-Point-Kontaktkarten visualisiert werden, um die Wechselwirkungen kleinerer Sequenzen wie Promotoren oder Enhancer innerhalb der gesamten erfassten Region gezielt zu visualisieren. Der Workflow des Protokolls ist in Abbildung 4 dargestellt. Die Qualitätskontrollen vor der Sequenzierung sind in Abbildung 2 dargestellt und umfassen die Bewertung des ordnungsgemäßen Aufschlusses und der Religation des 3C-Templates sowie seiner effizienten Abscherung und Reinigung über die verschiedenen Schritte des Protokolls hinweg. Es wird erwartet, dass die gescherte 3C-Templat-DNA zwischen 150 und 700 bp verläuft, und es sollte keine Anreicherung von Fragmenten >2 kb nachgewiesen werden. In den folgenden Schritten werden mehrere Bead-basierte DNA-Aufreinigungs- und Größenauswahlschritte durchgeführt, zuerst nach dem Scheren, dann nach den Pre-Capture- und Post-Capture-PCRs. Die gereinigten Bibliotheken zeigen ein deutliches Fragmentanreicherungsprofil, wie es auf einem hochempfindlichen DNA-Bioanalysator visualisiert wurde (Abbildung 2). Die mittlere Fragmentgröße nimmt im Laufe der Bibliothekspräparation aufgrund der Ligation von Adaptoren, Sequenzierungs- und Indexierungsprimern zu. Die Qualitätskontrollen nach der Sequenzierung werden über Hi-C Pro durchgeführt und sind in Abbildung 3 dargestellt. Viele verschiedene Bioinformatik-Softwareanwendungen wurden für die 3C-ähnliche Datenverarbeitung und -analyse vorgeschlagen. Unter ihnen ist die HiC-Pro-Pipeline eine der beliebtesten Lösungen, die die Verarbeitung von Rohsequenzierungsdaten zu den endgültigen Kontaktkarten mit verschiedenen Auflösungen ermöglicht55. HiC-Pro verwendet eine zweistufige Kartierungsstrategie, um die Sequenzierungs-Reads auf das Referenzgenom abzustimmen. Die 3C-Produkte werden dann rekonstruiert und herausgefiltert, um nicht-informative Kontaktpaare zu entfernen und die Kontaktkarten zu erstellen. Darüber hinaus ist es in der Lage, eine Liste bekannter Polymorphismen zu verwenden, um eine allelspezifische Analyse durchzuführen und die Kontakte, die von den beiden elterlichen Allelen stammen, in unterschiedlichen Kontaktkarten zu trennen. In jüngerer Zeit wurde HiC-Pro in das nf-core-Framework (nf-core-hic) aufgenommen und erweitert, wodurch eine hochgradig skalierbare und reproduzierbare Community-gesteuerte Pipeline bereitgestelltwird 61,62.
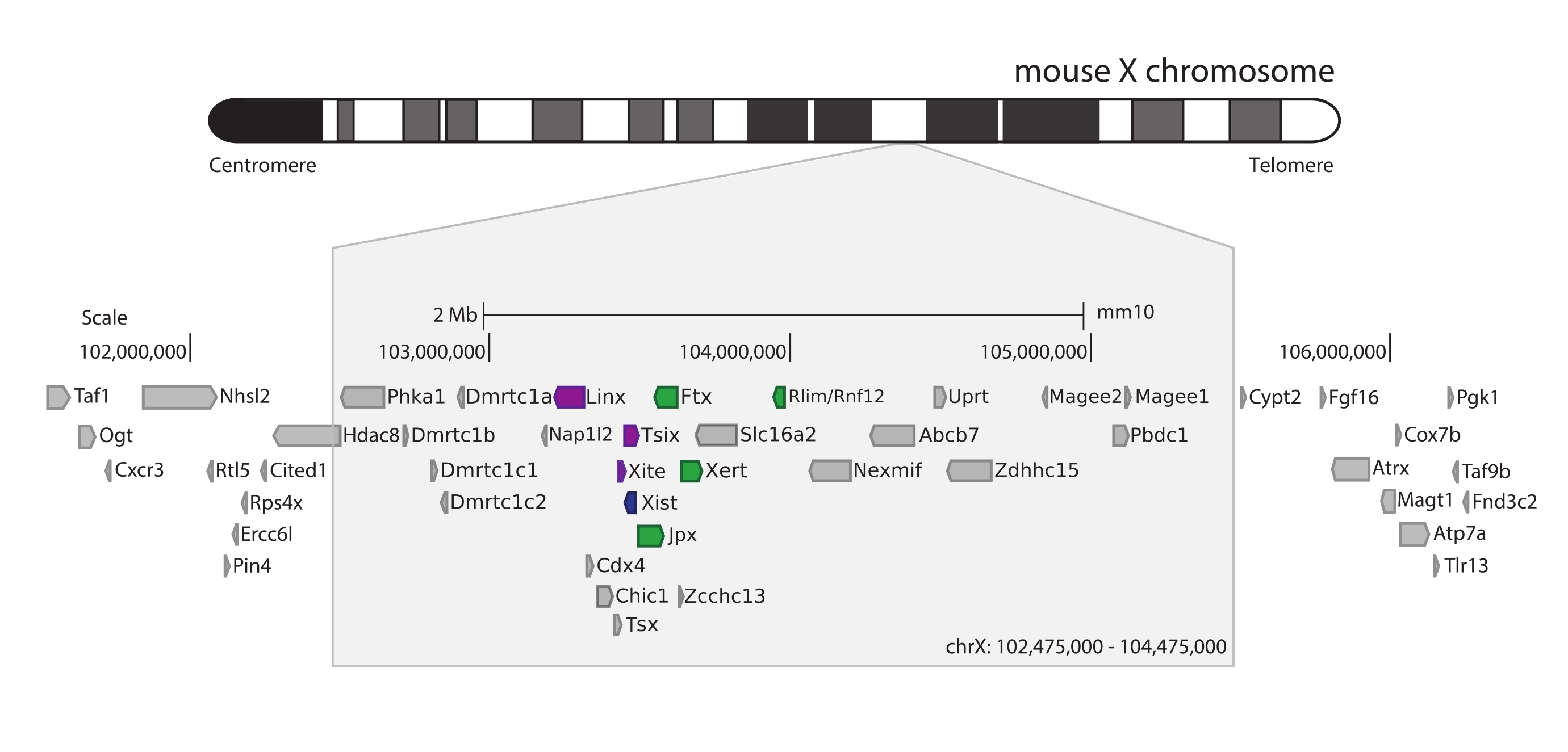
Um den Maus-Xic zu erfassen, wurde ein Array von 28.913 RNA-Sonden entworfen, die 3 Mb des X-Chromosoms kacheln. Diese Region umfasst den Hauptakteur in XCI, das lange nicht-kodierende Gen Xist, und seine bekannte regulatorische Landschaft von ~800 kb (Abbildung 5). Diese ~800 kb große Region ist in zwei TADs unterteilt: eine, die den Xist-Promotor und seine bekannten positiven Regulatoren (d.h. die nicht-kodierenden Transkripte Ftx, Jpx und Xert und das proteinkodierende Gen Rnf12) enthält, und die benachbarte TAD, die die negativen cis-Regulatoren von Xist umfasst (d.h. das Antisense-Transkript Tsix, das Enhancer-Element Xite und das nicht-kodierende Transkript Linx) (für Review44, 45).
Durch die Anwendung des beschriebenen Capture Hi-C-Protokolls auf den Xic konnte die topologische Organisation dieses Locus mit bisher unerreichter Auflösung ermittelt werden (Abbildung 6 und Abbildung 7). Dies wird besonders deutlich, wenn man das Capture Hi-C-Profil mit dem zuvor veröffentlichten 5C47 vergleicht (Abbildung 6 und Abbildung 7; Ergänzende Tabelle 1) und Hi-C61 (Abbildung 6 und Abbildung 7; Ergänzende Tabelle 1) Profile. Zum Beispiel sind Sub-TAD-Strukturen deutlicher – die TAD, die den Xist-Promotor ( Xist-TAD ) enthält, ist deutlich in zwei kleinere Domänen unterteilt (Abbildung 6A, blaue Pfeilspitze). Bisher konnte dies nur visuell aus dem 5C-Profil "erahnt" werden (Abbildung 6B), allerdings durch die Erkennung einer Grenze in diesem Bereich mit dem Isolations-Score-Algorithmus. Ebenso ermöglicht die Auflösung des Capture Hi-C-Profils die Identifizierung zweier kleinerer Domänen in der benachbarten TAD (Abbildung 6A, B), die den Promotor des Tsix-Locus ( Tsix-TAD ) enthält; Dies wurde bisher mit 5C nicht erreicht (Abbildung 6B). Bemerkenswert ist, dass topologische Grenzen, die durch den Isolationswert aus den Capture Hi-C- und 5C-Daten bestimmt werden, im Allgemeinen an leicht unterschiedlichen Positionen und mit unterschiedlichen relativen Stärken erkannt werden.
Darüber hinaus sind andere Sub-TAD-Strukturen, wie z. B. Kontaktschleifen, aus den Capture Hi-C-Daten deutlich sichtbar, wie z. B. die Schleife zwischen Xist und Ftx (Abbildung 7A), die zuvor mit Capture-C63 identifiziert wurde, und die Schleife zwischen Xist und Xert (Abbildung 7B), die kürzlich mit einem ähnlichen Protokoll für Capture Hi-C48 identifiziert wurde. Aufgrund der höheren Auflösung der Capture Hi-C-Profile können auch andere Kontakte genauer abgebildet werden, wie z. B. diejenigen, die die bekannten Kontakt-Hotspots innerhalb des Tsix-TAD zwischen den Linx-, Chic1- und Xit-Loci bilden (Abbildung 7A).
Im Vergleich zu den in Abbildung 7 gezeigten Hi-C-Daten ermöglichte Capture Hi-C eine vierfache Erhöhung der Auflösung, benötigte jedoch nur ein Viertel der Sequenziertiefe (d. h. 126 Mio. Lesevorgänge gegenüber 571 M) (ergänzende Tabelle 1). Diese Erhöhung der Auflösung ermöglicht die Detektion von SubTADs und Schleifenwechselwirkungen, die von Hi-C bei der in Abbildung 6 und Abbildung 7 gezeigten Sequenziertiefe nicht detektiert werden konnten. Das beschriebene Protokoll für Capture Hi-C ermöglicht somit eine wesentlich detailliertere, hochauflösende Charakterisierung einer großen genomischen Region von Interesse im Vergleich zu bisherigen Ansätzen.

Abbildung 1: Sondendesign. Schematische Darstellung der Strategie, die für das Sondendesign verwendet wird. Regionen von 300 bp stromaufwärts und stromabwärts jeder DpnII-Restriktionsstelle in der 3-Mb-Zielregion wurden ausgewählt und mit überlappenden biotinylierten RNA-Sonden gekachelt. Eine dieser ausgewählten Regionen wird angezeigt, chrX: 102.474.805-102.475.500. In jeder Sonde sind nicht mehr als 40 Basen mit sich wiederholenden Sequenzen zulässig. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 2: Erfassung von Hi-C-Qualitätskontrollen vor der Sequenzierung . (A) Repräsentatives Beispiel für Qualitätskontrollen von 3C-Vorlagen. 200 ng DNA wurden auf ein 1%iges Agarosegel geladen. Bahn 1: 1 kb Leiter. Bahn 2: Unverdautes, vernetztes und intaktes Chromatin läuft als scharfe Bande bei >10 kb. Spur 3: DpnII-verdautes vernetztes Chromatin läuft als Abstrich zwischen 1 kb und 3 kb groß. Bahn 4: Finale 3C-Bibliothek oder -Vorlage; Freie Enden von verdauten vernetzten DNA-Fragmenten werden religiert. Der DNA-Abstrich mit geringerer Molekulargröße ist fast nicht nachweisbar, und das Ligationsprodukt wird als Bande von >10 kb nachgewiesen. (B) Repräsentative Beispiele für hochempfindliche DNA-Profile von Bioanalysatoren. Oben links: Erfolgreich gescherte 3C-Bibliothek mit einer Verteilung der Fragmentgröße zwischen 150 bp und 700 bp. Oben rechts: unbefriedigende gescherte 3C-Bibliothek. Ungescherte DNA wird als breite Anreicherung von Fragmenten >2 kb nachgewiesen. (C) Unten links: gescherte DNA-Probe nach einer 1:1-Auswahl der linken Seite unter Verwendung von SPRI-Kügelchen. Fragmente von ~300 bp werden angereichert. Unten Mitte: Pre-Capture-PCR-Profil nach Ligation von Paired-End-Adaptern gemäß dem Protokoll des Herstellers. Unten rechts: endgültige Capture Hi-C-Bibliothek mit Adaptern, Sequenzierung und Indizierungsprimern für die Multiplex-Sequenzierung. Abkürzungen: bp = Basenpaare, FU = beliebige Fluoreszenzeinheit. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 3: Erfassung von Hi-C-Qualitätskontrollen nach der Sequenzierung mit HiC-Pro . (A) Beispiel für die Kartierungsrate des ersten Partners der Sequenzierungspaare auf dem Referenzgenom. Der hellblaue Anteil stellt die von HiC-Pro ausgerichteten Reads dar, die sich über eine Ligationsverbindung erstrecken. Diese Metrik kann somit verwendet werden, um den experimentellen Ligationsschritt zu validieren. (B) Sobald die Sequenzierungspartner auf dem Genom ausgerichtet sind, werden nur noch eindeutig ausgerichtete Lesepaare für die Analyse aufbewahrt. (C) Nicht gültige Paare (in rot), wie z. B. baumelndes Ende, Selbstkreis oder Religation, werden aus der Analyse verworfen. Der Anteil der gültigen Paare ist ein guter Indikator für die Ligatur und die Pulldown-Effizienz. (D) Die gültigen Paare können weiter unterteilt werden in intra-/interchromosomale und kurz-/langreichweitige Kontakte. Duplizierte Lesepaare, bei denen es sich wahrscheinlich um PCR-Artefakte handelt, werden aus der Analyse ausgeschlossen. (E) Für die allelspezifische Analyse gibt HiC-Pro die Anzahl der allelischen Reads an, die entweder von einem oder zwei Partnern für jedes elterliche Genom unterstützt werden (d. h. C57BL/6J x CASTEi/J). Es wird der gleiche Anteil an Reads erwartet, die dem mütterlichen und väterlichen Allel zugeordnet sind. (F) Schließlich werden nur gültige Paare ausgewählt, die die Erfassungsregion überlappen, um die Kontaktkarten zu erstellen. Capture-Capture-Paare stellen Kontakte innerhalb der Zielregion dar, während Capture-Reporter-Paare eine Interaktion zwischen der Zielregion und einer Off-Target-Region beinhalten. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 4: Workflow des Capture Hi-C-Protokolls. Schematische Darstellung verschiedener Protokollschritte. Um das genomweite 3C-Template zu erzeugen, wird Chromatin zunächst mit Formaldehyd vernetzt und dann mit dem DpnII-Restriktionsenzym verdaut. Freie DNA-Enden werden dann religiert, die Quervernetzung rückgängig gemacht und die DNA gereinigt. Zur Anreicherung von Fragmenten, die die Zielregion umfassen, wird eine Reihe von biotinylierten RNA-Sonden an die 3C-Matrize hybridisiert und durch Streptavidin-vermittelten Pulldown eingefangen. Capture-Bibliotheken werden für die Multiplex-Sequenzierung verarbeitet und gültige Ligationsfragmente werden quantifiziert, um auf die Häufigkeit von Chromatinkontakten im gesamten Target zu schließen, die als hochauflösende Interaktionskarten visualisiert werden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 5: Übersicht über die Region, die den Xic auf dem X-Chromosom der Maus umfasst. Schematische Darstellung des X-Chromosoms der Maus und Vergrößerung der 3 Mb großen erfassten Region (ChrX: 102.475.000-105.475.000). Die Zielregion umfasst ~800 kb DNA, die dem Xic, dem regulatorischen Hauptlocus von XCI, entspricht. Das Xic umfasst die langen nicht-kodierenden Gene, Xist, einen wichtigen Akteur von XCI, und seine regulatorische Landschaft. Positive Regulatoren von Xist sind grün und negative Regulatoren violett dargestellt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 6: Erfassen von Hi-C-, 5C- und Hi-C-Interaktionskarten in der erfassten 3-MB-Region. (A) Erfassung der Hi-C-Interaktionskarte des 3-MB-Ziels, das den Maus-Xic mit einer Auflösung von 10 kb umfasst (diese Studie). (B) 5C-Interaktionskarte der gleichen Zielregion wie in A mit einer Auflösung von 6 kb (Daten von47 neu verarbeitet). Sich wiederholende Bereiche, die nicht in die Analysen einbezogen werden, werden weiß maskiert. Die 5C-Daten erfordern eine eigene bioinformatische Verarbeitung (siehe47). Nach der Bereinigung und Ausrichtung werden die 5C-Maps mit der Primerauflösung unter Verwendung eines laufenden Medians (Fenster = 30 kb, Schritt = 5) klassifiziert, um eine endgültige Auflösung von 6 kb zu erreichen. (C) Hi-C-Interaktionskarte der gleichen genomischen Region wie in A und B mit einer Auflösung von 40 kb (Daten von64 neu verarbeitet). Alle Interaktionskarten wurden aus Maus-ESCs generiert. Der Isolationswert wurde mit Cooltools berechnet und wird als Histogramme mit Isolationsminimas an TAD-Grenzen dargestellt. TAD-Grenzen werden als vertikale Linien unterhalb der Karte angezeigt. Die Höhe jeder Linie gibt die Begrenzungsstärke an. Gene werden als Pfeile dargestellt, die in Transkriptionsrichtung zeigen. Sub-TAD-Grenzen, die ausschließlich oder genauer in Capture Hi-C-Maps erkannt werden, werden durch magentafarbene bzw. blaue Pfeilspitzen für Sub-TADs in den Tsix- bzw. Xist-TADs angezeigt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 7: Erfassung von Hi-C-, 5C- und Hi-C-Interaktionskarten über 1 MB innerhalb der erfassten Region. (A) Erfassung der Hi-C-Interaktionskarte der 1-MB-Genomregion, die den Maus-Xic mit einer Auflösung von 5 kb umfasst (diese Studie). (B) 5C-Interaktionskarte der gleichen Genomregion wie in A. mit einer Auflösung von 6 kb (Daten von47 neu verarbeitet). Sich wiederholende Bereiche, die nicht in die Analysen einbezogen werden, werden weiß maskiert. Bemerkenswert ist, dass die 5C-Daten eine eigene bioinformatische Verarbeitung erfordern (siehe47). Nach der Bereinigung und Ausrichtung werden die 5C-Maps mit der Primerauflösung unter Verwendung eines laufenden Medians (Fenster = 30 kb, Schritt = 5) klassifiziert, um eine endgültige Auflösung von 6 kb zu erreichen. (C) Hi-C-Interaktionskarte der gleichen Genomregion wie in A und B von Hi-C mit einer Auflösung von 20 kb (Daten von64 weiterverarbeitet). Alle Interaktionskarten wurden aus mESCs generiert. Der Isolationswert wurde mit Cooltools berechnet und wird als Histogramme mit Isolationsminimas an TAD-Grenzen dargestellt. TAD-Grenzen werden als vertikale Linien unterhalb der Karte angezeigt. Die Höhe jeder Linie gibt die Begrenzungsstärke an. Gene werden als Pfeile dargestellt, die in die Richtung der Transkription zeigen. Kontaktschleifen, die ausschließlich oder genauer in Capture Hi-C detektiert werden, werden durch magentafarbene bzw. blaue Sternchen für Schleifen in den Tsix- bzw. Xist-TADs gekennzeichnet. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
Ergänzende Tabelle 1: Post-Sequenzierungsstatistiken für die in diesem Manuskript verwendeten Datensätze: Capture Hi-C (diese Studie), Hi-C64 und 5C47. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Diskussion
Hier beschreiben wir ein relativ schnelles und einfaches Capture Hi-C Protokoll zur Charakterisierung der übergeordneten Organisation von Megabasen-Genomregionen mit einer Auflösung von 5-10 kb. Capture Hi-C gehört zur Familie der Capture-C-Technologien, die entwickelt wurden, um gezielte Chromatin-Interaktionen aus genomweiten 3C- oder Hi-C-Templates anzureichern. Bisher wurde die große Mehrheit der Capture-C-Anwendungen genutzt, um Chromatinkontakte relativ kleiner regulatorischer Elemente zu kartieren, die über das gesamte Genom verstreut sind. Im ersten Capture-C-Protokoll wurden mehrere überlappende RNA-biotinylierte Sonden verwendet, um >400 vorselektierte Promotoren in 3C-Bibliotheken einzufangen, die aus erythroiden Zellen hergestellt wurden31. Die gleiche Strategie wurde später in Next Generation (NG) und Nuclear Titrated (NuTi) Capture-C verbessert, um hochauflösende Interaktionsprofile von >8.000 Promotoren zu erreichen, indem einzelne 120-bp-DNA-Köder verwendet wurden, die einzelne Restriktionsstellen und zwei aufeinanderfolgende Fangrunden umfassten, um die Anreicherung informativer Ligationsfragmente zu maximieren32,40. Diese Strategien führten zur funktionellen Zerlegung von cis-wirkenden Elementen in vielen verschiedenen Kontexten, einschließlich der embryonalen Entwicklung der Maus, der Zelldifferenzierung, der Inaktivierung des X-Chromosoms und der Fehlregulation von Genen unter pathologischen Bedingungen 46,63,65,66,67,68,69,70,71.
Bei Promoter Capture Hi-C (PCHi-C) wurden >22.000 annotierte Promotoren, die Restriktionsfragmente enthielten, durch Hybridisierung einzelner RNA 120-mer biotinylierter Sonden an einem oder beiden Enden des Restriktionsfragments aus Hi-C-Bibliotheken gezogen34,72. Diese Methode ermöglichte die Dissektion des Interaktoms von Tausenden von Promotoren in einer schnell wachsenden Anzahl von Zelltypen, darunter embryonale Stammzellen der Maus, fetale Leberzellen und Adipozyten 34,35,72,73, aber auch menschliche lymphoblastoide Linien, hämatopoetische Vorläuferzellen, epidermale Keratinozyten und pluripotente Zellen 37,74,75,76,77.
Im Vergleich zu diesen Technologien zur Target-Anreicherung zielt Capture Hi-C auf zusammenhängende genomische Regionen bis auf die Megabasen-Skala ab, wobei es sich über einen oder mehrere TADs erstreckt und regulatorische Landschaften von Genen umfasst. Die gesamte interessierende Region muss mit einer Reihe von biotinylierten Sonden gekachelt werden, die jede DpnII-Restriktionsstelle innerhalb des Ziels umfassen. Die Hybridisierung des biotinylierten Arrays mit dem 3C-Template, seine anschließende Streptavidin-basierte Erfassung und Verarbeitung für die Multiplex-Sequenzierung erfolgt unter Verwendung eines Target-Anreicherungssystems für die Illumina Paired-End Multiplex-Sequenzierung. Das gesamte Protokoll ist schnell, da es von der 3C-Bibliotheksvorbereitung bis zur NGS-Sequenzierung in 1 Woche durchgeführt werden kann und nur geringfügige Anpassungen und/oder kundenspezifische Fehlerbehebungen erfordert.
Das Protokoll bietet auch Vorteile im Vergleich zu anderen 3C-basierten Methoden. Um Interaktionskarten mit einer Auflösung von 5-10 kb zu erhalten, sequenzierten wir 100-120 M Paired-End-Reads. Zum Vergleich haben wir hier einen Hi-C-Datensatz von 571 Mio. Lesevorgängen verwendet, um eine Auflösung von 20 kb64 (GSM2053973) zu erreichen, und mindestens 1 Milliarde Lesevorgänge wären erforderlich, um eine Auflösung von 5 kb mit chromosomenweitem Hi-C22 zu erreichen.
Capture Hi-C, wie es in der vorliegenden Studie verwendet wird, erreicht eine viel höhere Auflösung als das zuvor veröffentlichte 5C, das auf einem 6-bp-Cutter-Restriktionsenzym47 basiert (ergänzende Tabelle 1). Wichtig ist, dass die Strategie, die darauf abzielt, gezielte Interaktionen in 5C anzureichern und zu verstärken, keine allelspezifische Analyse von Chromatininteraktionen zulässt. Im Gegenteil, Capture Hi-C-Daten können allelspezifisch kartiert werden, was die Zerlegung der 3D-Strukturlandschaften von Paaren homologer Chromosomen ermöglicht, beispielsweise in menschlichen Zellen oder in F1-Hybridzelllinien, die durch Kreuzung genetisch unterschiedlicher Mausstämme entstandensind 78. Um allelspezifische Capture Hi-C-Interaktionskarten mit einer Auflösung von 5 kb zu erstellen, sequenzierten wir 150 bp Paired-End-Reads, um die SNP-Abdeckung zu erhöhen. Ähnliche Allel-spezifische Ansätze können auf humane Zelllinien angewendet werden, für die die Annotation von SNPs verfügbar ist22.
Wichtig ist, dass, obwohl Capture Hi-C im Allgemeinen eine hohe Auflösung gewährleistet und gleichzeitig die Erschwinglichkeit der Sequenzierungskosten verbessert, die Herstellung maßgeschneiderter biotinylierter Oligonukleotide einen Einfluss auf die Gesamtkosten dieser Methode hat. Daher ist die Wahl der am besten geeigneten 3C-Methode für verschiedene Anwendungen unterschiedlich und hängt von der biologischen Fragestellung und der erforderlichen Auflösung sowie von der Größe der interessierenden Region ab. Andere entwickelte Capture Hi-C-Protokolle haben wichtige Funktionen mit dem hier beschriebenen Protokoll gemeinsam. Zum Beispiel wurde eine Capture Hi-C-Strategie angewendet, um ~50 kb bis 1 Mb Genomregionen zu charakterisieren, die nicht-kodierende Varianten umfassen, die mit dem Brust- und Darmkrebsrisiko assoziiert sind. In diesem Protokoll wurden Zielregionen aus Hi-C-Bibliotheken herausgezogen, indem 120-mer-RNA-Köder hybridisiert wurden, wobei die Zielregionen mit einer 3-fachen Abdeckungvon 33,38,79 gekachelt wurden. In ähnlicher Weise wurde HYbrid Capture Hi-C (Hi-C 2) verwendet, um Interaktionen innerhalb von Regions of Interest bis zu2 Mb80 anzuvisieren. In beiden Protokollen erhöhte die Verwendung eines Hi-C-Templates, das für Biotin-Pulled-Down-Ligationsfragmente angereichert wurde, den Prozentsatz der gesamten informativen Reads im Vergleich zu unserem Protokoll. In dem Hi-C-Datensatz, den wir hier für den Vergleich64 verwendet haben (GSM2053973), ist beispielsweise der Prozentsatz der gültigen Paare nach dem Entfernen von Duplikaten 4,8-mal höher als die in Capture Hi-C erhaltenen gültigen Paare, wie in Abbildung 3 und ergänzender Tabelle 1 beschrieben. Das aufeinanderfolgende Herunterziehen von biotinylierten ligierten Fragmenten und hybridisierten Sonden macht das Protokoll jedoch deutlich komplexer und zeitaufwändiger, während die Komplexität der erfassten Region möglicherweise verringert wird.
Eine weitere verfügbare Methode zur Anreicherung von 3C-Templates mit Kachelsonden ist Tiled-C, die zur Untersuchung der Chromatinarchitektur mit hoher räumlicher und zeitlicher Auflösung während der erythroiden Differenzierung der Maus eingesetzt wurde43. In Tiled-C wird ein Panel von 70 bp biotinylierten Sonden verwendet, um Kontakte in großräumigen Regionen in zwei aufeinanderfolgenden Erfassungsrunden anzureichern, um sehr hochauflösende Karten gezielter Interaktionen zu erstellen43,81. Die doppelte Capture-Anreicherung macht das Protokoll im Vergleich zu Capture Hi-C auch länger und komplexer. Im Gegensatz zu den Capture-C-Strategien, die auf einzelne Restriktionsstellen abzielen, scheint die zweite Erfassungsrunde in Tiled-C die Erfassungseffizienz jedoch nicht signifikant zu erhöhen und kann daher wahrscheinlich weggelassen werden43. Schließlich wurde ein ähnlicher Kachelansatz, der auf der gleichen Zielanreicherungsstrategie basiert, die in dieser Studie verwendet wurde, auf die Analyse von regulatorischen Landschaften angewendet, die strukturelle Varianten umfassen, die bei Patienten mit angeborenen Fehlbildungen beschrieben und in transgenen Mäusen neu entwickelt wurden41,42. In diesem Fall wurde die Kachelanordnung von Sonden über das gesamte Ziel und nicht in der Nähe von DpnII-Restriktionsstellen41 entworfen. Nichtsdestotrotz war diese Arbeit wegweisend, um die Sensitivität und Leistungsfähigkeit dieser Strategie zur hochauflösenden Charakterisierung großer genomischer Regionen in verschiedenen Kontexten hervorzuheben41,42,48.
Zusammenfassend lässt sich sagen, dass das hier beschriebene Protokoll eine einfache, robuste und leistungsfähige Strategie für die hochauflösende 3D-Charakterisierung beliebiger genomischer Regionen von Interesse darstellt. Die Anwendung dieses Ansatzes auf verschiedene Modellsysteme, Zelltypen, entwicklungsregulierte Chromatinlandschaften und Genregulation unter gesunden und pathologischen Bedingungen dürfte unser Verständnis des Zusammenspiels und der Kausalität zwischen Genomtopologie und Genregulation erleichtern, eine der grundlegenden offenen Fragen im Bereich der Epigenetik. Darüber hinaus hat die Anwendung von Capture Hi-C zur Kartierung von langreichweitigen Interaktionen und Chromatinfaltung höherer Ordnung, die in GWAS-Studien identifiziert wurden, das Potenzial, die funktionelle Relevanz von nicht-kodierenden genomischen Loci aufzudecken, die mit menschlichen Krankheiten in verschiedenen Kontexten assoziiert sind, und damit neue Einblicke in die Prozesse zu gewinnen, die möglicherweise der Pathogenese zugrunde liegen.
Offenlegungen
Kai Hauschulz ist Field Application Scientist bei Agilent Technologies - Diagnostic and Genomics Group. Alle anderen Autoren erklären keine Interessenkonflikte.
Danksagungen
Die Arbeit im Heard-Labor wurde durch einen Advanced Investigator Award des Europäischen Forschungsrats (XPRESS - AdG671027) unterstützt. A.L. wird durch ein Marie-Skłodowska-Curie-Stipendium der Europäischen Union (IF-838408) unterstützt. A.H. wird vom ITN Innovative and Interdisciplinary Network ChromDesign im Rahmen der Marie-Skłodowska-Curie-Grant-Vereinbarung 813327 unterstützt. Die Autoren danken Daniel Ibrahim (MPI für molekulare Genetik, Berlin) für hilfreiche technische Ratschläge, der NGS-Plattform am Institut Curie (Paris) und Vladimir Benes und der Genomics Core Facility am EMBL (Heidelberg) für die Unterstützung und Hilfe.
Materialien
| Name | Company | Catalog Number | Comments |
| 10x PBS pH 7.4 | Gibco | 10010-023 | |
| 37% (vol/vol) paraformaldehyde solution | Electron Microscopy Sciences | 15686 | single use glass-vials; do not reuse |
| 50 mL PP conical tube | Falcon | 352070 | |
| Agarose | Sigma | A9539-500g | |
| Bioanalyzer | Agilent | G2939BA | |
| Cell Scrapers - 25 cm Handle and 3.0 cm Blade | Falcon | 353089 | |
| CHIR99021 | Axon Medchem BV | Axon 1386 | |
| cOmplete Mini, Protease inhibitor cocktail (EDTA-free) | Merck | 11836170001 | |
| Countess Cell Counting Chamber Slides | Invitrogen | C10228 | |
| Countess II FL | Invitrogen | ZGEXSCCOUNTESS2FL | Automated cell counter |
| Covaris S2 | Covaris | 500217 | Sonicator |
| DNA LoBind tube, 1.5 mL | Eppendorf | 30108051 | |
| DpnII (50000 units/mL) | New England Biolabs | R0543M | |
| Dulbecco's Modified Eagle Medium (DMEM) | Merck | D6429 | |
| Ethanol (100%) | Merck | 1.00983.2500 | |
| Fetal Bovine Serum (FBS) | Thermo Scientific | 10270106 | |
| gelatine from porcine skin | Sigma | G1890 | |
| GeneRuler 1 kb Plus DNA Ladder | Thermo Scientific | SM0313 | |
| GlycoBlue | Thermo Scientific | AM9516 | Coprecipitant |
| High-Sensitivity Bioanlayzer chips | Agilent | 5067-4626 | |
| Large Cooling Centrifuge 5920 R | Eppendorf | 5948000018 | |
| leukaemia inhibitory factor (LIF) | Merck | ESG1107 | |
| Liquiport | KNF | NF300 | Benchtop aspiration system |
| Low-binding filter tips | Biozym | VT0260U, VT0240, VT0220, VT0200U | |
| Molecular biology grade water | Merck | W3500-6x500ML | |
| Next Seq 500 | Illumina | SY-415-1001 | |
| Next Seq 500 High Output v2 Kit (300 cycles) | Illumina | FC-404-2004 | |
| Nonidet P40 Substitute (NP40) | Merck | 11332473001 | |
| PD0325901 | Axon Medchem BV | Axon 1408 | |
| Protease inhibitor cocktail (EDTA-free) | Merck | 11873580001 | |
| Proteinase K - recombinant, PCR-grade (20 mg/mL) | Thermo Scientific | EO0491 | |
| Qubit 2.0 | Thermo Scientific | Q32871 | |
| Qubit assay tubes | Thermo Scientific | Q32856 | |
| Qubit dsDNA High Sensitivity kit | Thermo Scientific | Q32851 | |
| RNase A (10 mg/mL) | Thermo Scientific | EN0531 | |
| Sodium acetate pH 5.2 (3M) | Merck | S7899 | |
| speed vacuum concentrator | Eppendorf | EP5305000100-1EA | |
| Agencourt AMPureXP | Beckman Coulter | A63881 | SPRI beads |
| SureSelect Target Enrichment Box 1 | Agilent | 5190-8645 | |
| SureSelect Target Enrichment Kit ILM Indexing Hyb Module Box 2 | Agilent | 5190-4455 | |
| SureSelect XT Library Prep Kit ILM | Agilent | 5500-0132 | |
| T4 ligase (30 units/µL) | Thermo Scientific | EL0013 | |
| table-top Centrifuge 5427 R | Eppendorf | 5409000012 | |
| Triton-X-100 (500 mL) | Merck | X100-500ML | |
| Trypan Blue | Invitrogen | T10282 | |
| Trypsine | Thermo Scientific | 25300054 | |
| UltraPure Glycine | Thermo Scientific | 15527013 | |
| β-mercaptoethanol | Thermo Scientific | 31350010 |
Referenzen
- Ibrahim, D. M., Mundlos, S. The role of 3D chromatin domains in gene regulation: a multi-facetted view on genome organization. Current Opinion in Genetics & Development. 61, 1-8 (2020).
- Bolt, C. C., Duboule, D. The regulatory landscapes of developmental genes. Development. 147 (3), (2020).
- Glaser, J., Mundlos, S. 3D or not 3D: Shaping the genome during development. Cold Spring Harbor Perspectives in Biology. 14 (5), 040188(2021).
- Denker, A., De Laat, W. The second decade of 3C technologies: detailed insights into nuclear organization. Genes & Development. 30 (12), 1357-1382 (2016).
- Kempfer, R., Pombo, A. Methods for mapping 3D chromosome architecture. Nature Reviews Genetics. 21 (4), 207-226 (2020).
- McCord, R. P., Kaplan, N., Giorgetti, L. Chromosome conformation capture and beyond: Toward an integrative view of chromosome structure and function. Molecular Cell. 77 (4), 688-708 (2020).
- Jerkovic, I., Cavalli, G. Understanding 3D genome organization by multidisciplinary methods. Nature ReviewsMolecular Cell Biology. 22 (8), 511-528 (2021).
- Hsieh, T. -H. S., et al. Mapping nucleosome resolution chromosome folding in yeast by Micro-C. Cell. 162 (1), 108-119 (2015).
- Krietenstein, N., et al. Ultrastructural details of mammalian chromosome architecture. Molecular Cell. 78 (3), 554-565 (2020).
- Dekker, J., Rippe, K., Dekker, M., Kleckner, N. Capturing chromosome conformation. Science. 295 (5558), 1306-1311 (2002).
- Naumova, N., Smith, E. M., Zhan, Y., Dekker, J. Analysis of long-range chromatin interactions using Chromosome Conformation Capture. Methods. 58 (3), 192-203 (2012).
- Simonis, M., et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nature Genetics. 38 (11), 1348-1354 (2006).
- Zhao, Z., et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra-and interchromosomal interactions. Nature Genetics. 38 (11), 1341-1347 (2006).
- Würtele, H., Chartrand, P. Genome-wide scanning of HoxB1-associated loci in mouse ES cells using an open-ended Chromosome Conformation Capture methodology. Chromosome Research. 14 (5), 477-495 (2006).
- De Wit, E., De Laat, W. A decade of 3C technologies: insights into nuclear organization. Genes & Development. 26 (1), 11-24 (2012).
- Dostie, J., et al. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Research. 16 (10), 1299-1309 (2006).
- Splinter, E., et al. The inactive X chromosome adopts a unique three-dimensional conformation that is dependent on Xist RNA. Genes & Development. 25 (13), 1371-1383 (2011).
- Ferraiuolo, M. A., Sanyal, A., Naumova, N., Dekker, J., Dostie, J. From cells to chromatin: capturing snapshots of genome organization with 5C technology. Methods. 58 (3), 255-267 (2012).
- Kim, J. H., et al. 5C-ID: Increased resolution Chromosome-Conformation-Capture-Carbon-Copy with in situ 3C and double alternating primer design. Methods. 142, 39-46 (2018).
- Lieberman-Aiden, E., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326 (5950), 289-293 (2009).
- Zhang, Y., et al. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell. 148 (5), 908-921 (2012).
- Rao, S. S. P., et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 159 (7), 1665-1680 (2014).
- Dixon, J. R., et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 485 (7398), 376-380 (2012).
- Nora, E. P., et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 485 (7398), 381-385 (2012).
- Krefting, J., Andrade-Navarro, M. A., Ibn-Salem, J. Evolutionary stability of topologically associating domains is associated with conserved gene regulation. BMC Biology. 16 (1), 87(2018).
- Galupa, R., Heard, E. Topologically associating domains in chromosome architecture and gene regulatory landscapes during development, disease, and evolution. Cold Spring Harbor Symposia on Quantitative Biology. 82, 267-278 (2017).
- Tena, J. J., Santos-Pereira, J. M. Topologically associating domains and regulatory landscapes in development, evolution and disease. Frontiers in Cell and Developmental Biology. 9, 702787(2021).
- Lupiáñez, D. G., Spielmann, M., Mundlos, S. Breaking TADs: How alterations of chromatin domains result in disease. Trends in Genetics. 32 (4), 225-237 (2016).
- Davidson, I. F., Peters, J. -M. Genome folding through loop extrusion by SMC complexes. Nature Reviews Molecular Cell Biology. 22 (7), 445-464 (2021).
- Schmitt, A. D., Hu, M., Ren, B. Genome-wide mapping and analysis of chromosome architecture. Nature Reviews Molecular Cell Biology. 17 (12), 743-755 (2016).
- Hughes, J. R., et al. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nature Genetics. 46 (2), 205-212 (2014).
- Davies, J. O. J., et al. Multiplexed analysis of chromosome conformation at vastly improved sensitivity. Nature Methods. 13 (1), 74-80 (2016).
- Jäger, R., et al. Capture Hi-C identifies the chromatin interactome of colorectal cancer risk loci. Nature Communications. 6, 6178(2015).
- Schoenfelder, S., et al. The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Research. 25 (4), 582-597 (2015).
- Sahlén, P., et al. Genome-wide mapping of promoter-anchored interactions with close to single-enhancer resolution. Genome Biology. 16, 156(2015).
- Joshi, O., et al. Dynamic reorganization of extremely long-range promoter-promoter interactions between two states of pluripotency. Cell Stem Cell. 17 (6), 748-757 (2015).
- Mifsud, B., et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nature Genetics. 47 (6), 598-606 (2015).
- Dryden, N. H., et al. Unbiased analysis of potential targets of breast cancer susceptibility loci by Capture Hi-C. Genome Research. 24 (11), 1854-1868 (2014).
- Oudelaar, A. M., Davies, J. O. J., Downes, D. J., Higgs, D. R., Hughes, J. R. Robust detection of chromosomal interactions from small numbers of cells using low-input Capture-C. Nucleic Acids Research. 45 (22), 184(2017).
- Oudelaar, A. M., et al. Single-allele chromatin interactions identify regulatory hubs in dynamic compartmentalized domains. Nature Genetics. 50 (12), 1744-1751 (2018).
- Franke, M., et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature. 538 (7624), 265-269 (2016).
- Despang, A., et al. Functional dissection of the Sox9-Kcnj2 locus identifies nonessential and instructive roles of TAD architecture. Nature Genetics. 51 (8), 1263-1271 (2019).
- Oudelaar, A. M., et al. Dynamics of the 4D genome during in vivo lineage specification and differentiation. Nature Communications. 11 (1), 1-12 (2020).
- Galupa, R., Heard, E. X-chromosome inactivation: A crossroads between chromosome architecture and gene regulation. Annual Review of Genetics. 52, 535-566 (2018).
- Loda, A., Collombet, S., Heard, E. Gene regulation in time and space during X-chromosome inactivation. Nature Reviews. Molecular Cell Biology. 23 (4), 231-249 (2022).
- van Bemmel, J. G., et al. The bipartite TAD organization of the X-inactivation center ensures opposing developmental regulation of Tsix and Xist. Nature Genetics. 51 (6), 1024-1034 (2019).
- Galupa, R., et al. A conserved noncoding locus regulates random monoallelic Xist expression across a topological boundary. Molecular Cell. 77 (2), 352-367 (2020).
- Gjaltema, R. A. F., et al. Distal and proximal cis-regulatory elements sense X chromosome dosage and developmental state at the Xist locus. Molecular Cell. 82 (1), 190-208 (2022).
- Galupa, R., et al. Inversion of a topological domain leads to restricted changes in its gene expression and affects inter-domain communication. Development. 149 (9), (2022).
- Savarese, F., Flahndorfer, K., Jaenisch, R., Busslinger, M., Wutz, A. Hematopoietic precursor cells transiently reestablish permissiveness for X inactivation. Molecular and Cellular Biology. 26 (19), 7167-7177 (2006).
- Schulz, E. G., et al. The two active X chromosomes in female ESCs block exit from the pluripotent state by modulating the ESC signaling network. Cell Stem Cell. 14 (2), 203-216 (2014).
- Gnirke, A., et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nature Biotechnology. 27 (2), 182-189 (2009).
- Akgol Oksuz, B., et al. Systematic evaluation of chromosome conformation capture assays. Nature Methods. 18 (9), 1046-1055 (2021).
- Piccinini, F., Tesei, A., Arienti, C., Bevilacqua, A. Cell counting and viability assessment of 2D and 3D Cell cultures: Expected reliability of the trypan blue assay. Biological Procedures Online. 19 (1), 8(2017).
- Servant, N., et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biology. 16, 259(2015).
- Abdennur, N., Mirny, L. A. Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics. 36 (1), 311-316 (2020).
- Imakaev, M., et al. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nature Methods. 9 (10), 999-1003 (2012).
- Forcato, M., et al. Comparison of computational methods for Hi-C data analysis. Nature Methods. 14 (7), 679-685 (2017).
- Venev, S., et al. open2c/cooltools: v0.4.1. , (2021).
- Wages, J. M. NUCLEIC ACIDS | Immunoassays. Encyclopedia of Analytical Science. , Elsevier. 408-417 (2005).
- Ewels, P. A., et al. The nf-core framework for community-curated bioinformatics pipelines. Nature Biotechnology. 38 (3), 276-278 (2020).
- Servant, N., Peltzer, A. nf-core/hic: Initial release of nf-core/hic. Zenodo. , (2019).
- Furlan, G., et al. The Ftx noncoding locus controls X chromosome inactivation independently of its RNA products. Molecular Cell. 70 (3), 462-472 (2018).
- Giorgetti, L., et al. Structural organization of the inactive X chromosome in the mouse. Nature. 535 (7613), 575-579 (2016).
- Simon, C. S., et al. Functional characterisation of cis-regulatory elements governing dynamic Eomes expression in the early mouse embryo. Development. 144 (7), 1249-1260 (2017).
- Williams, R. M., et al. Reconstruction of the global neural crest gene regulatory network in vivo. Developmental Cell. 51 (2), 255-276 (2019).
- Godfrey, L., et al. DOT1L inhibition reveals a distinct subset of enhancers dependent on H3K79 methylation. Nature Communications. 10 (1), 2803(2019).
- Hanssen, L. L. P., et al. Tissue-specific CTCF-cohesin-mediated chromatin architecture delimits enhancer interactions and function in vivo. Nature Cell Biology. 19 (8), 952-961 (2017).
- Larke, M. S. C., et al. Enhancers predominantly regulate gene expression during differentiation via transcription initiation. Molecular Cell. 81 (5), 983-997 (2021).
- Oudelaar, A. M., et al. A revised model for promoter competition based on multi-way chromatin interactions at the α-globin locus. Nature Communications. 10 (1), 5412(2019).
- Long, H. K., et al. Loss of extreme long-range enhancers in human neural crest drives a craniofacial disorder. Cell Stem Cell. 27 (5), 765-783 (2020).
- Schoenfelder, S., Javierre, B. -M., Furlan-Magaril, M., Wingett, S. W., Fraser, P. Promoter Capture Hi-C: High-resolution, genome-wide profiling of promoter interactions. Journal of Visualized Experiments. (136), e57320(2018).
- Siersbæk, R., et al. Dynamic rewiring of promoter-anchored chromatin loops during adipocyte differentiation. Molecular Cell. 66 (3), 420-435 (2017).
- Rubin, A. J., et al. Lineage-specific dynamic and pre-established enhancer-promoter contacts cooperate in terminal differentiation. Nature Genetics. 49 (10), 1522-1528 (2017).
- Freire-Pritchett, P., et al. Global reorganisation of cis-regulatory units upon lineage commitment of human embryonic stem cells. eLife. 6, 21926(2017).
- Javierre, B. M., et al. Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell. 167 (5), 1369-1384 (2016).
- Miguel-Escalada, I., et al. Human pancreatic islet three-dimensional chromatin architecture provides insights into the genetics of type 2 diabetes. Nature Genetics. 51 (7), 1137-1148 (2019).
- Keane, T. M., et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 477 (7364), 289-294 (2011).
- Baxter, J. S., et al. Capture Hi-C identifies putative target genes at 33 breast cancer risk loci. Nature Communications. 9 (1), 1028(2018).
- Sanborn, A. L., et al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proceedings of the National Academy of Sciences. 112 (47), 6456-6465 (2015).
- Owens, D. D. G., et al. Dynamic Runx1 chromatin boundaries affect gene expression in hematopoietic development. Nature Communications. 13 (1), 773(2022).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten