
Method Article
キャプチャHi-C による 高分解能3Dクロマチン構成の解読
要約
このプロトコルは、トポロジー的会合ドメイン(TAD)の境界や、調節と他のDNA配列要素との間の長距離クロマチン相互作用など、メガベースサイズの標的ゲノム領域の3D組織を高解像度で特徴付けるために使用されるCapture Hi-C法について説明しています。
要約
ゲノムの空間構成は、転写、複製、組換え、修復など、多くの状況でその機能と調節に貢献しています。したがって、ゲノムトポロジーと機能の間の正確な因果関係を理解することは非常に重要であり、ますます集中的な研究の対象となっています。染色体立体構造捕捉技術(3C)は、ゲノムの任意の領域間の相互作用の頻度を測定することにより、クロマチンの3D構造を推測することを可能にする。ここでは、メガベースサイズのゲノムターゲットの対立遺伝子特異的3D組織を高解像度で特徴付ける3Cベースのターゲット濃縮法であるCapture Hi-Cを実行するための高速で簡単なプロトコルについて説明します。Capture Hi-Cでは、ダウンストリームのハイスループットシーケンシングの前に、ターゲット領域がビオチン化プローブのアレイによってキャプチャされます。したがって、より高い分解能と対立遺伝子特異性が達成され、技術の時間効率と手頃な価格が向上します。その強みを実証するために、Capture Hi-Cプロトコルを、X染色体不活性化(XCI)のマスター調節遺伝子座であるマウスX不活性化センター (Xic)に適用しました。
概要
線形ゲノムは、生物が胚発生を経験し、成人期を通して生き残るために必要なすべての情報を保持しています。しかし、遺伝的に同一の細胞に異なる機能を実行するように指示することは、異なる組織や発生段階を含む特定のコンテキストで使用される情報を正確に制御するための基本です。ゲノムの3次元構成は、線状ゲノム内の数百キロベース離れることができる調節要素間の物理的相互作用を促進または防止することにより、遺伝子活性のこの正確な時空間調節に関与していると考えられています(レビュー1,2,3).過去20年間で、ゲノムフォールディングと活性の相互作用に関する私たちの理解は、主に染色体立体構造捕捉技術(3C)の開発により急速に増加しました(レビュー4,5,6,7)。これらの方法は、ゲノムの任意の領域間の相互作用の頻度を測定し、核内で3D近接しているDNA配列のライゲーションに依存しています。最も一般的な3Cプロトコルは、ホルムアルデヒドなどの架橋剤で細胞集団を固定することから始まります。その後、架橋クロマチンは制限酵素で消化されますが、MNase消化も使用されています8,9。消化後、空間的に近接した遊離DNA末端が再ライゲーションされ、架橋が逆転します。このステップにより、核に3D近接していた配列が同じDNA断片にライゲーションされる可能性が高いハイブリッド断片の混合プールである3C「ライブラリ」または「テンプレート」が生じます。これらのハイブリッドフラグメントの下流定量により、線形ゲノム内で数千塩基対離れて位置しているが、3D空間で相互作用する可能性のあるゲノム領域の3Dコンフォメーションを推測できます。
3Cライブラリの特性評価にはさまざまなアプローチが開発されており、ライゲーションフラグメントのサブセットを分析する方法と、その下流の定量に使用する技術の両方が異なります。元の3Cプロトコルは、2つの関心領域の選択と、PCR10,11によるそれらの「1対1」の相互作用頻度の定量化に依存していました。4Cアプローチ(環状染色体コンフォメーションキャプチャ)は、関心のある単一の遺伝子座(「視点」)とゲノムの残りの部分(「1つ対すべて」)の間の相互作用を測定します12,13,14。4Cでは、3Cライブラリーは2回目の消化および再ライゲーションを経て、視点特異的プライマー15によってPCR増幅される小環状DNA分子を生成する。5C(染色体コンフォメーションキャプチャーカーボンコピー)は、より大きな関心領域にわたる3D相互作用の特性評価を可能にし、その領域内の高次クロマチンフォールディング(「多対多」)に関する洞察を提供します16。5Cにおいて、3Cライブラリーは、制限部位と重複するオリゴヌクレオチドのプールにハイブリダイズされ、その後、ユニバーサルプライマー15を用いたマルチプレックスPCRによって増幅され得る。4Cと5Cの両方で、有益なDNA断片は最初にマイクロアレイによって定量され、後に次世代シーケンシング(NGS)によって定量されました17,18,19。これらの戦略は、標的の関心領域を特徴付けますが、ゲノム全体の相互作用のマッピングには適用できません。この後者の目標は、3Cテンプレートの超並列シーケンシングにより、ゲノムワイドレベル(「すべて対すべて」)でのクロマチンフォールディングの偏りのない特性評価を可能にする3Cベースのハイスループット戦略であるHi-Cによって達成されます20。Hi-Cプロトコルには、消化されたフラグメントの末端にビオチン化残基を組み込み、その後、ライゲーションフラグメントをストレプトアビジンビーズでプルダウンして、ライゲーションフラグメントの回収率を高めることが含まれます20。
Hi-Cは、哺乳類のゲノムが3次元核内で複数のスケールで構造的に組織化されていることを明らかにしました。メガベーススケールでは、ゲノムは活性クロマチンと不活性クロマチンの領域、それぞれAコンパートメントとBコンパートメントに分けられます20,21。異なるクロマチンおよび活性状態によって表されるさらなるサブコンパートメントの存在もまた、続いて示された22。より高い解像度では、ゲノムはさらにトポロジー的会合ドメイン(TAD)と呼ばれるサブメガベースの自己相互作用ドメインに分割され、ヒトおよびマウスゲノムのHi-Cおよび5C分析によって最初に明らかにされる23,24。組織特異的に変化するコンパートメントとは異なり、TADは一定である傾向があります(ただし、多くの例外があります)。重要なことに、TAD境界は種全体で保存されています25。哺乳類細胞では、TADは同じ調節ランドスケープを共有する遺伝子を包含することが多く、隣接する調節ドメインとの相互作用を制限しながら遺伝子共制御を促進する構造フレームワークを表すことが示されています(レビュー3,26,27,28)。さらに、TAD内では、コヒーシン押出ループの基部にあるCTCF部位による相互作用は、プロモーター-エンハンサーまたはエンハンサー-エンハンサー相互作用の可能性を高める可能性がある(レビュー29)。
Hi-Cでは、コンパートメントとTADは1 Mb〜40 kbの解像度で検出できますが、5〜10 kbのスケールで遠位エレメント間のループ相互作用などのより小さなスケールの接触を特徴付けるために、より高い分解能を実現できます。ただし、このようなループをHiCで効率的に検出できるように分解能を上げるには、シーケンシングの深さを大幅に増やす必要があり、したがってシーケンシングコストも増加します。分析が対立遺伝子特異的である必要がある場合、これは悪化します。実際、分解能をX倍に高めるには、シーケンシングの深さをX2 増加させる必要があり、高解像度で対立遺伝子特異的なゲノムワイドアプローチは法外に高価になる可能性があります30。
高分解能を維持しながら費用対効果と手頃な価格を向上させるために、ターゲット領域は、ダウンストリームシーケンシングの前に相補的なビオチン標識オリゴヌクレオチドプローブとのハイブリダイゼーション後に、ゲノムワイドな3CまたはHi-Cライブラリから物理的にプルダウンできます。これらの標的濃縮戦略はCapture-C法と呼ばれ、ゲノム全体に散在する数百の標的遺伝子座の相互作用の調査を可能にします(すなわち、プロモーターキャプチャー(PC)Hi-C;次世代(NG)キャプチャ-C;低入力(LI)キャプチャ-C;核滴定(NuTi)キャプチャ-C;Tri-C)31、32、33、34、35、36、37、38、39、40、または複数のメガベースにまたがる地域間(すなわち、Capture HiC;ハイブリッドキャプチャハイC(ハイC2);タイル-C)41,42,43。キャプチャベースの方法では2つの態様が異なる場合があります:(1)ビオチン化オリゴヌクレオチド(すなわち、RNAまたはDNA、分散したゲノムターゲットを捕捉する単一オリゴ、または関心領域をタイリングする複数のオリゴ)の性質および設計;(2)ターゲットをプルダウンするために使用されるテンプレートは、3CまたはHi-Cライブラリであり得、後者は3Cライブラリからプルダウンされたビオチン化制限断片からなる。
ここでは、3Cライブラリからのターゲットコンタクトのエンリッチメントに基づくキャプチャHi-Cプロトコルについて説明します。このプロトコルは、ビオチン化RNAプローブのカスタムメイドのタイリングアレイの設計に依存しており、3Cライブラリの調製からNGSシーケンシングまで1週間で実行できます。このプロトコルは高速でシンプルで、メガベースサイズの関心領域の高次3D構成を5 kbの解像度で特徴付けることができ、他の3Cメソッドと比較して時間効率と手頃な価格が向上します。Capture Hi-Cプロトコルは、X染色体不活性化(XCI)のマスター調節遺伝子座であるX-不活性化センター(Xic)に適用され、XistノンコーディングRNAをホストします。Xicは、以前は広範な構造および機能分析の対象となっていました(レビュー44,45)。哺乳類では、XCIは雌(XX)と雄(XY)の間のX結合遺伝子の投与量を補い、雌細胞内の2つのX染色体のうちの1つのほぼ全体の転写サイレンシングを含みます。Xicは、3Dゲノムトポロジーと遺伝子制御との相互作用の研究のための強力でゴールドスタンダードの遺伝子座を表しています44。マウス胚性幹細胞(mESC)におけるXicの5C分析は、TADの発見と命名につながり、トポロジカル分割と遺伝子共制御の機能的関連性に関する最初の洞察を提供しました24。その後、Xicのトポロジカル構成は、XistのアップレギュレーションとXCI46の適切な発生タイミングに決定的に関与していることが示され、TAD内およびTAD間の遺伝子活性に影響を与える可能性のある疑われないシス調節要素も最近Xic47,48,49内で発見されました。Capture Hi-CをXicにまたがるマウスX染色体の3 Mbに適用すると、大規模なクロマチンフォールディングを高解像度で解剖するこのアプローチの力が実証されます。対象領域内のすべてのDpnII制限部位にわたるビオチン化プローブのアレイの設計から、ゲノムワイドな3Cライブラリの生成、ターゲットコンタクトのハイブリダイゼーションとキャプチャ、およびダウンストリームデータ解析まで、詳細でわかりやすいプロトコルが提供されます。また、適切な品質管理と期待される結果の概要も含まれており、同様の既存の方法に照らして、アプローチの長所と限界の両方について説明します。
プロトコル
この研究で使用されたマウス胚性幹細胞(mESC)は、キュリー研究所(パリ)51の動物飼育ガイドラインに従って、TX / TX R26rtTA / rtTA雌50とMus musculus castaneus雄の交配から得られました。
1. プローブ設計
- 目的のターゲット領域をカバーするビオチン化プローブ(120-mer RNAオリゴヌクレオチド)のアレイを設計します。
- ターゲット内の各配列が平均して2つの固有のプローブ(2倍のカバレッジ)で覆われるように、重複するオリゴヌクレオチドで関心領域をタイル化します(図1)。
- プローブカバレッジから反復配列を除外して、非特異的相互作用の濃縮を回避します。
注:有益なライゲーションフラグメントの濃縮を最大化するために、ターゲット全体の各DpnII制限部位の上流および下流300 bpにまたがる領域が定義され(ChrX:102,475,000-105,475,000)、Sure Designプラットフォームを介してSureSelect DNAターゲット濃縮技術に従って28,913のビオチン化プローブを設計しました52.この戦略によれば、非特異的相互作用の濃縮を最小限に抑えるために、各オリゴヌクレオチドに最大40塩基の反復配列が許可されます。プローブアレイはアジレント社により合成した。ここで、DpnIIは、2つの理由で制限酵素として使用される:(1)それはいくつかの3Cベースの方法で日常的に使用される4カッターである53;(2)この研究で使用されたF1ハイブリッド株(C57BL / 6J x CASTEi / J)の インシリコ でテストされた他の制限酵素と比較して、切断部位の近くで有益な一塩基多型(SNP)をキャプチャする可能性を最大化します。
2. 実験手順
- 細胞調製
- 1つまたは複数の細胞培養プレートに適切な数の細胞を播種し、固定当日に5 x 107 細胞≥の合計細胞数を達成します。
注:この研究では、マウス胚性幹細胞(mESC)を使用しました。mESCは、2i + LIFおよびバッチ試験されたウシ胎児血清(DMEM、15%FBS、0.1 mM β-メルカプトエタノール、1,000 U/mL-1白血病抑制因子(LIF)、CHIR99021(3 μM)、およびPD0325901(1 μM)を含むmESC培地のゼラチン化(37°C、5%CO2インキュベーターで1x PBS - o/n中の0.1%ゼラチン)細胞培養プレートにプレーティングされます。この細胞型の場合、1つの80%コンフルエントな10 cmプレートには、約2 x 107 細胞が含まれています。 - 細胞カウント用に追加の細胞培養プレートを1枚準備します。
注:より小さな細胞培養プレートを使用して、培地の使用量を減らすことができます。この場合、小さい方のプレートに播種する細胞の数は、それに応じて調整する必要があります(例えば、15cmのプレートと比較して、10cmのプレート上の細胞数が3倍少なくなります)。
- 1つまたは複数の細胞培養プレートに適切な数の細胞を播種し、固定当日に5 x 107 細胞≥の合計細胞数を達成します。
- ホルムアルデヒド固定
- 架橋する細胞の総数を推定します。
- 架橋反応を開始する前に、製造元の指示に従って、自動セルカウンターを使用して、細胞カウント用に特別に調製されたコントロールプレートから細胞をトリプシン処理してカウントします。
- 生存率染色(例えば、トリパンブルー)を含めて、生存細胞54の割合を決定する。この細胞数から、架橋用に調製したプレート内の細胞の総数を推定します。
- 架橋用に調製したプレートから培養液を取り出し、適量の固定液(細胞培養液中の2%ホルムアルデヒド)と交換します。10 cmプレートで10 mLを使用します(例:15 cmプレートの場合は~20 mL)。.
注意: 固定液の正確な量を追加します。接着細胞を固定することが不可能な場合、このステップはトリプシン処理された細胞に適合させ、50mLのコニカル遠沈管中の30mLの固定溶液中で行うことができる。ホルムアルデヒドは1歳以上であってはなりません。使い捨てバイアルを使用することが好ましい。固定液は使用前に室温(RT)にする必要があります。
注意: ホルムアルデヒドは危険であり、適切な健康と安全の規制に従って取り扱う必要があります。 - シェーカーで穏やかに混合しながらRTで10分間固定します。
- 2.5 M グリシン-1x PBSを最終濃度0.125 Mまで添加して固定反応をクエンチし、530 μLの2.5 Mグリシン-1x PBSを10 cmプレートで10 mLに加えます(例:15 cmプレートで1060 μLから20 mL)。
注:細胞を溶液中で固定した場合は、1590 μLの2.5 Mグリシン-1x PBSで固定反応をクエンチします。 - RTで5分間インキュベートし、シェーカーで穏やかに混合します。
- プレートを氷に移し、シェーカーで穏やかに混合しながら、氷上でさらに15分間インキュベートします。
注:今後は、細胞を氷上に保持し、さらなる架橋を避けるためにバッファーを事前に冷却する必要があります。多くのプレートを処理する必要がある場合は、冷蔵室に移動します。 - 固定溶液をビーカーに注いで細胞から取り除き、迅速な取り扱いを確保します。
注意: ホルムアルデヒド含有液体廃棄物は、適切な健康と安全の規制に従って廃棄してください。 - 10 cmプレートを5 mLの冷たい0.125 Mグリシン-1x PBS(15 cmプレートの場合は8 mL)で2回すばやくすすぎ、破片と死んだ細胞を洗い流します。液体をビーカーに注ぎ、プレートから液体を取り除き、迅速な取り扱いを確保します。
- 5 mLの冷たい0.125 Mグリシン-1x PBSを10 cmプレート(15 cmプレートの場合は10 mL)に加え、プラスチックセルスクレーパーを使用してプレートから細胞をすばやくこすり落とします。
- 血清学的ピペットを使用して、細胞懸濁液を予冷された50 mLコニカル遠心チューブに移します。
- プレートを5 mLの冷たい0.125 Mグリシン-1x PBSで2回すすぎ、細胞懸濁液を円錐形遠心チューブに加えます。
- 480 x g で4°Cで10分間スピンダウンします。
注:細胞を溶液に固定した場合は、細胞を事前に冷却した円錐形の遠沈管に移し、480 x g で4°Cで10分間スピンダウンします。 固定液をビーカーに注ぎ、10 mLの冷たい0.125 Mグリシン-1x PBSで3回洗浄します。各洗浄ステップで細胞を再懸濁してください。 - ベンチトップ吸引システムで吸引して上清を除去します。P1000ピペットで慎重に上下にピペッティングすることにより、1 x 107 細胞あたり1x PBSの500 μLで細胞を再懸濁します。正確な体積の細胞を再懸濁するには、2.2.1で得られた総細胞数の推定値を参照してください。
- 500 μLの細胞懸濁液を、計算された数の1.5 mLマイクロ遠心チューブ(1 x 107 セル/チューブ)に分注します。
- 480 x g で4°Cで10分間スピンダウンします。
- ベンチトップ吸引システムで上清を除去し、細胞ペレットを液体窒素で急速凍結します。乾燥菌体ペレットを-80°Cで保存する。
注:サンプルは少なくとも1年間保管できます。
- 架橋する細胞の総数を推定します。
- 細胞溶解
- 凍結したペレットを氷上で解凍します。
- サンプルあたり H 2 0 中の 1.5 mL 溶解バッファーを調製します:10 mM トリス塩酸塩、pH 8.0、10 mM NaCl、および0.2% NP40 を追加します。
- 600 μLの低温溶解バッファーを加え、氷上でよく再懸濁します。
- 氷上で15分間インキュベートして、細胞を膨潤させます。
- 2655 x g で4°Cで5分間スピンダウンし、ベンチトップ吸引システムを使用して上清を除去します。
- 破片を除去するには、ペレットを1 mLのコールドリシスバッファーに再懸濁し、2655 x g で4°Cで5分間スピンダウンし、上清を除去します。
- 2655 x g および4°Cで再びスピンし、P200チップを備えたベンチトップ吸引システムを使用して、残りの上清をできるだけ多く除去します。
- 0.5%(容量/容量)SDSの100 μLに再懸濁します。
- 62°Cのサーモミキサーで、1400rpmで10分間旋回しながらインキュベートします。
- 290 μLのH2O + 50 μLの10%TritonX-100を加え、気泡を避けてよく混合します。
- 37°Cのサーモミキサーで、1400rpmで15分間旋回させてインキュベートします。
- 50 μLの10x Dpnllバッファーを加え、チューブを反転させて混合します。
- 品質管理のために50 μLの未消化DNAを別のチューブに入れます。未消化の対照サンプルを採取することを忘れないでください。
- DpnII消化
- 高濃度のDpnll(合計500 U)を10 μL加え、反転させて混合します。
- サンプルと未消化コントロールを37°Cのサーモミキサーでインキュベートし、1400 rpmで>4時間旋回させます。
- 一日の終わりに10 μLのDpnll高濃度(合計500 U)を追加します。
- サンプルと未消化コントロールを37°Cでインキュベートし、1400rpmで一晩旋回させます。
- 翌日初めに高濃度Dpnll(合計500U)を10μLサンプルに加えます。
- サンプルと未消化コントロールを37°Cのサーモミキサーでインキュベートし、1400 rpmで4時間旋回させます。
- 架橋のライゲーションと反転
- チューブを65°Cで1400rpmで20分間インキュベートします。
注: この時点では SDS を追加しないでください。アイデアは核の完全性を維持することであるため、ライゲーションは核内で行われ、極端な希釈の必要性を回避します。 - サンプルを氷上で最大5〜10分間冷却します。SDSの沈殿を避けるために、サンプルをこれより長く氷上に放置しないでください。
- ライゲーションされていない消化DNA50 μLを品質管理のために別のチューブに入れます。未消化および結紮されていないコントロールを-20°Cで保存します。
注:結紮されていない対照サンプルを採取することを忘れないでください。 - 800 μLのライゲーションカクテルを加えます:122 μLの10xリガーゼバッファー、8 μLのT4リガーゼ(30 U / μL)、および670 μLのH20。
- 16°Cでインキュベートし、1000rpmで一晩旋回させます。
- 7.5 μLのプロテイナーゼK(20 mg/mL)をサンプルに、2 μLをコントロールに加えます。
- 65°Cで1000rpmで4時間インキュベートします。
- チューブを65°Cで1400rpmで20分間インキュベートします。
- DNA精製
- 氷上でサンプルを予冷した15 mLコニカル遠心チューブに移し、2 mLの水、10.5 mLの氷冷EtOH、および583 μLの3 M NaACを加えます。
注:追加の水は、ペレットへのDTTのキャリーオーバーを防ぐことを目的としています。 - 200 μLの氷冷EtOH、10.8 μLのNaAC、および1 μLの共集塵剤を未消化および結紮されていない品質管理に加えます。
- -80°Cで少なくとも4時間、一晩インキュベートします。
- 15 mLチューブを2200 x g で4°Cで45分間回転させます。
- 1.5 mL コントロールチューブを 20,500 x g で 4 °C で 30 分間回転させます。
- 3 mL(サンプル)と1 mL(コントロール)の氷冷70%EtOHで1回洗浄します。
- 2200 x g (サンプル) または 20,500 x g (コントロール) で 4 °C で 10 分間スピンします。
- EtOHを慎重に取り外し、RTで10〜15分間風乾します。過度に乾燥させないでください。
- サンプルとコントロールをそれぞれ100 μLと20 μLのH20に再懸濁します。
- 1 μLのRNAseAを添加し、37°Cでインキュベートし、1400 rpmで30分間旋回させます。
- 氷上でサンプルを予冷した15 mLコニカル遠心チューブに移し、2 mLの水、10.5 mLの氷冷EtOH、および583 μLの3 M NaACを加えます。
- 3Cテンプレート調製の品質管理
- 高感度DNA濃度測定用の蛍光光度計キットを使用して、各サンプルとコントロールを定量します。
- 各サンプルと各コントロールの100〜200 ngを1%アガロース/ 1x TBEゲルにロードします。
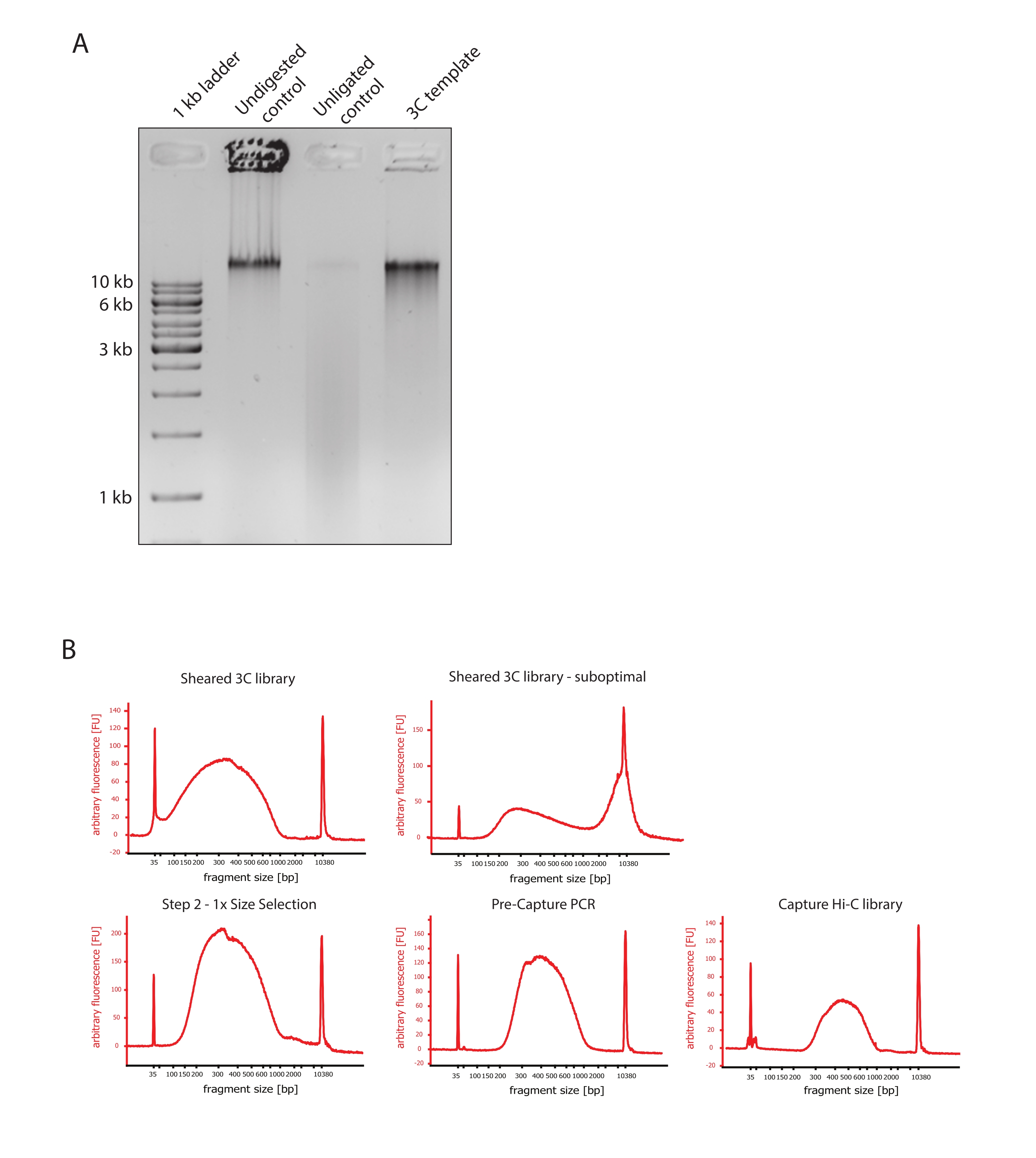
- 図2Aに示すように、コントロールと3CテンプレートのDNAフラグメントサイズの違いを比較することにより、ゲル画像が期待される結果を示していることを確認します。
- サンプルとコントロールは-20°Cで保存してください。
- マルチプレックスシーケンシングのためのハイブリダイゼーション、キャプチャー、サンプル処理
- ビオチン化RNAプローブのアレイを3Cテンプレートにハイブリダイズするには、標的ライゲーションフラグメントを捕捉し、ペアエンドマルチプレックスシーケンシングのためにこの研究で使用されたターゲットエンリッチメントシステムに従ってマルチプレックスシーケンシング用のサンプルを調製します(参照 材料表).製造元の指示に従ってプロトコルに従い、次の小さな変更を導入します。
- メーカーのプロトコルのセクション2:サンプル調製
- 3 μgのgDNAインプットから始まるターゲット濃縮の指示に従ってください。
- 次の仕様を使用して超音波処理器でDNAを剪断します:10%デューティサイクル、4強度、200 cyc /バースト、および130秒。捕捉反応ごとに4 μgの3Cテンプレートを130 μLの水に再懸濁して、3 μgの剪断DNAでサンプル調製を継続するのに十分な材料を確保します。
- せん断されたDNAの品質を評価します。せん断したDNAを1 μL、高感度プロトコルに従ってDNAバイオアナライザーで実行します。フラグメントサイズの分布は150〜700 bpであると予想してください(図2)。
- 固相可逆固定化(SPRI)ビーズを使用してサンプルを精製します。124 μLの DNA サンプルに 124 μL の SPRI ビーズを加え、製造元の指示に従って 1:1 の左側サイズ選択を行い、25 μL のヌクレアーゼフリー水で溶出します。この精製ステップでは、短いフラグメントを除去して、約300 bpのフラグメントを濃縮します(図2)。
注:このステップで使用されるサンプルとSPRIビーズの量は、サンプルを新しいチューブに移し、バイオアナライザーで品質管理を実行している間に発生した体積損失を考慮に入れています。後続のすべてのサイズ選択ステップは、メーカーのプロトコルで推奨されている比率に従って実行されます。SPRIビーズからのDNA溶出は、プロトコル全体を通してRTで行われます。 - サイズ選択せん断DNAの品質を評価します。せん断されたDNAの1 μLを、高感度(HS)プロトコルに従ってDNAバイオアナライザーに流します。300 bpで最も高い濃縮度を持つフラグメントサイズの分布を期待してください(図2)。せん断が成功した場合は、せん断されたDNAの定量に進みます。
- HS DNA濃度測定用の蛍光光度計キットを使用して、せん断されたDNAを定量します。
注:DNAせん断の結果、DNA収量が<3 μgの場合は、さらに4 μgのDNAで2回目のDNAせん断を実行し、最初のSPRIビーズ精製ステップの後にせん断されたDNAサンプルを組み合わせて、合計3 μgのせん断DNAを達成します。 - サイズ選択した洗浄済みDNAサンプル(合計3 μg)にヌクレアーゼフリーの水を加えて最終容量48 μLにし、メーカーのプロトコルに従って最終修復反応を進めます。
- ペアエンドアダプターのライゲーション後、製造元の指示に従って5サイクルのプレキャプチャーPCRを実行してライブラリを増幅します(PCRの条件とプライマーはキットに記載されています)。
- メーカーのプロトコルのセクション4:ハイブリダイゼーションとキャプチャ
- 調製したDNAサンプルをターゲット特異的RNAプローブにハイブリダイズするには、750 ngのDNAサンプルを最終容量3.4 μLで希釈し、初期濃度を221 ng/μLにします。より大きな容量に希釈されたDNAサンプルの場合は、スピード真空濃縮器を使用して最終容量まで減らします。通常、10 μLに再懸濁したサンプルには、15〜20分間の速度真空濃縮(250 x g; ≤45°C)で十分です。 速度真空濃縮器を始動する前に、各サンプルの入力量が同じであることを確認してください。
- ハイブリダイゼーション混合物を、製造元の指示に従って、105°Cで加熱した蓋をして、65°Cで16〜18時間インキュベートします。
- メーカーのプロトコルのセクション5:マルチプレックスシーケンシングのためのインデックス作成とサンプル処理
- キャプチャしたライブラリをインデックスプライマーで増幅するには、製造元の指示に従って12サイクルのポストキャプチャーPCRを実行します(PCRの条件とプライマーはキットに記載されています)。
- メーカーのプロトコルのセクション2:サンプル調製
- ビオチン化RNAプローブのアレイを3Cテンプレートにハイブリダイズするには、標的ライゲーションフラグメントを捕捉し、ペアエンドマルチプレックスシーケンシングのためにこの研究で使用されたターゲットエンリッチメントシステムに従ってマルチプレックスシーケンシング用のサンプルを調製します(参照 材料表).製造元の指示に従ってプロトコルに従い、次の小さな変更を導入します。
- 次世代シーケンシング
- 同じフローセル上で複数のキャプチャHi-Cライブラリを実行するには、キャプチャライブラリの等モル混合物を調製し、ライブラリごとに100〜120 Mの読み取りをシーケンスします。
- 対立遺伝子特異的分析が必要な場合は、十分なSNPカバレッジを確保するために、150 bpペアエンドを配列します。
3.データ分析
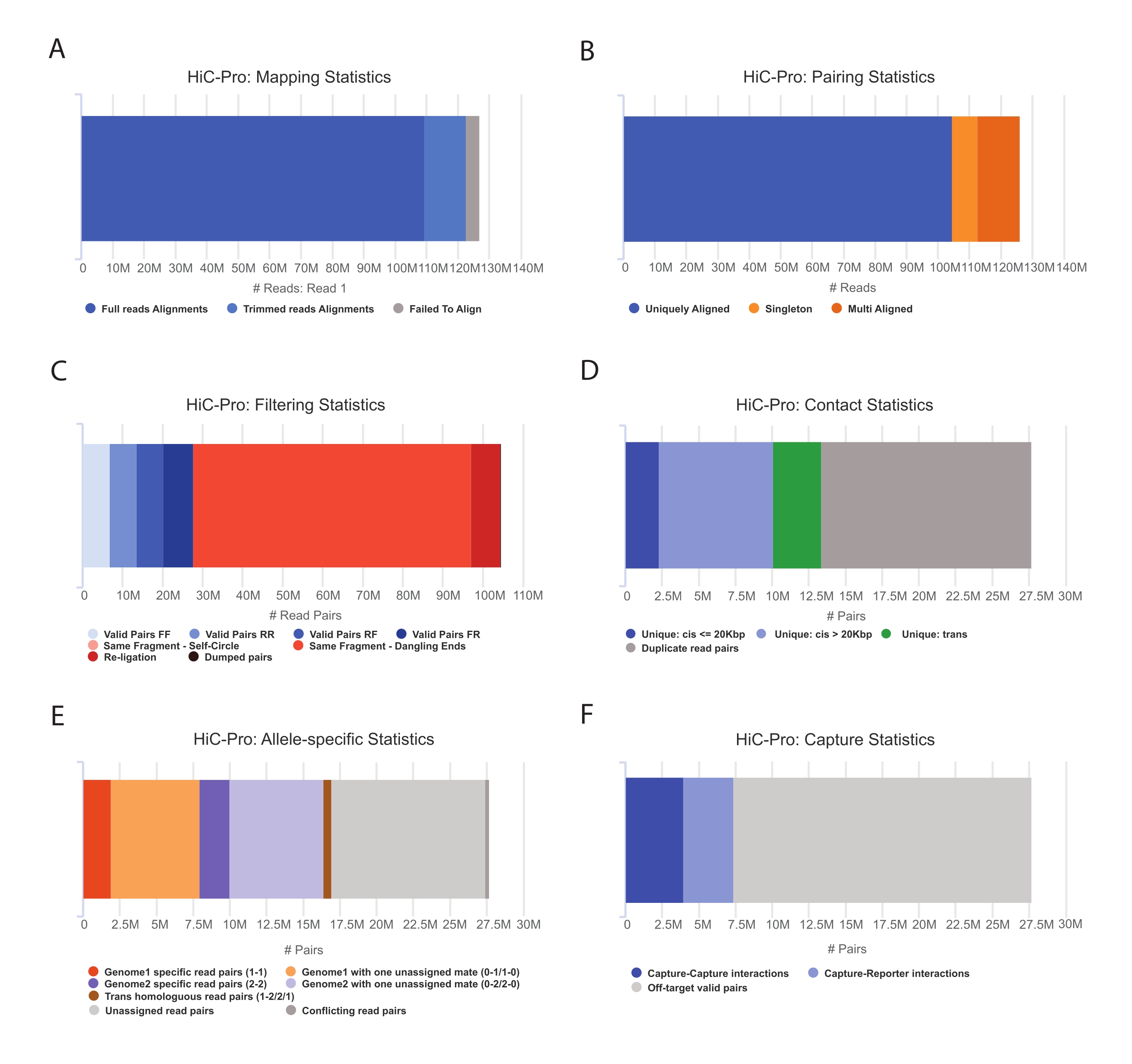
- HiC-Proパイプラインを適用して、キャプチャHi-Cデータ分析55を実行します。HiC-Proは、処理の各ステップで次のような品質管理を提供します(図3)。
(i)ライゲーション部位にまたがるリードの割合、およびペアとシングルトンの数を指定する参照ゲノム上のアラインメント率。
(2)有効なライゲーション製品と非有益なリードペア(ダングリングエンド、セルフライゲーションなど)の割合。
(ウ)短距離/長距離および染色体内/染色体間接触の割合。
(4)キャプチャHi-Cのオンターゲットコンタクトの割合。
(5)対立遺伝子特異的リードの割合(指定されている場合)。
注意: HiC-Proは、in situHi-CやキャプチャHi-Cを含む幅広いプロトコルをサポートしています。後者の場合、ユーザーは構成ファイルでターゲット領域 (BED 形式) を指定するだけで済みます。データが処理されると、HiC−Pro出力は、ダウンストリーム分析のためにより低温の物体に容易に変換することができる56。このステップでは、さまざまな解像度のコンタクトマップが、Imakaevらによって以前に記述されたICE法を使用して正規化されます57。その後、染色体コンパートメント、TAD、またはクロマチンループを呼び出すためにいくつかの分析を実行できます(レビュー58の場合)。プロトコルのワークフローを図 4 に示します。ここでは、「cooltools」スイートを適用して、図5および図6に示すように、絶縁スコアとTADの境界を計算します59。
結果
記載されたCapture Hi−Cプロトコルは、4塩基カッター(DpnII)を用いたゲノムワイド3Cテンプレートの調製に基づいている。その後の目的のゲノム領域にわたるライゲーションフラグメントの濃縮は、この研究で使用された標的濃縮システムに従って、タイリングRNAプローブのアレイとそれらのストレプトアビジンベースのキャプチャーのハイブリダイゼーションによって得られます(図1)。ビオチン化RNAプローブは、DNAプローブと比較して標的に対してより強い結合親和性を示すため、選択されました52,60。キャプチャされたライブラリは、多重化された高スループットシーケンシングのためにインデックス化およびプールされます。キャプチャーHi-Cデータは、高解像度のHi-C相互作用マップとして視覚化できるだけでなく、4Cのような単一視点接触マップとして視覚化して、キャプチャ領域全体におけるプロモーターやエンハンサーなどの小さな配列の相互作用を具体的に視覚化することもできます。プロトコルのワークフローを図 4 に示します。プレシーケンシングの品質管理を図2に示し、3Cテンプレートの適切な消化と再ライゲーションの評価、およびプロトコルのさまざまなステップにわたる効率的なせん断と精製が含まれています。せん断された3CテンプレートDNAは150〜700 bpで実行されると予想され、>2 kbのフラグメントの濃縮は検出されません。以下のステップでは、ビーズベースのDNAクリーンアップおよびサイズ選択ステップが、最初にせん断後、次にプリキャプチャーおよびポストキャプチャーPCRの後に実行されます。 洗浄されたライブラリは、高感度DNAバイオアナライザーで視覚化された明確なフラグメント濃縮プロファイルを示します(図2)。平均フラグメントサイズは、アダプター、シーケンシング、およびインデックスプライマーのライゲーションにより、ライブラリ調製の過程で増加します。シーケンシング後の品質管理は、Hi-C Proを介して取得され、図3に示されています。3Cのようなデータ処理と分析のために、多くの異なるバイオインフォマティクスソフトウェアアプリケーションが提案されています。その中で、HiC-Proパイプラインは最も人気のあるソリューションの1つであり、生のシーケンスデータをさまざまな解像度で最終的なコンタクトマップに処理できます55。HiC-Proは、2段階のマッピング戦略を使用して、参照ゲノム上のシーケンシングリードを調整します。次に、3C製品を再構築してフィルタリングし、情報のない接触ペアを削除し、接触マップを生成します。さらに、既知の多型のリストを使用して対立遺伝子特異的分析を実行し、異なる接触マップで2つの親対立遺伝子から来る接触を分離することができます。最近では、HiC-Proがnf-coreフレームワーク(nf-core-hic)に含まれ、拡張され、拡張性が高く再現性の高いコミュニティ駆動型パイプライン61,62を提供している。
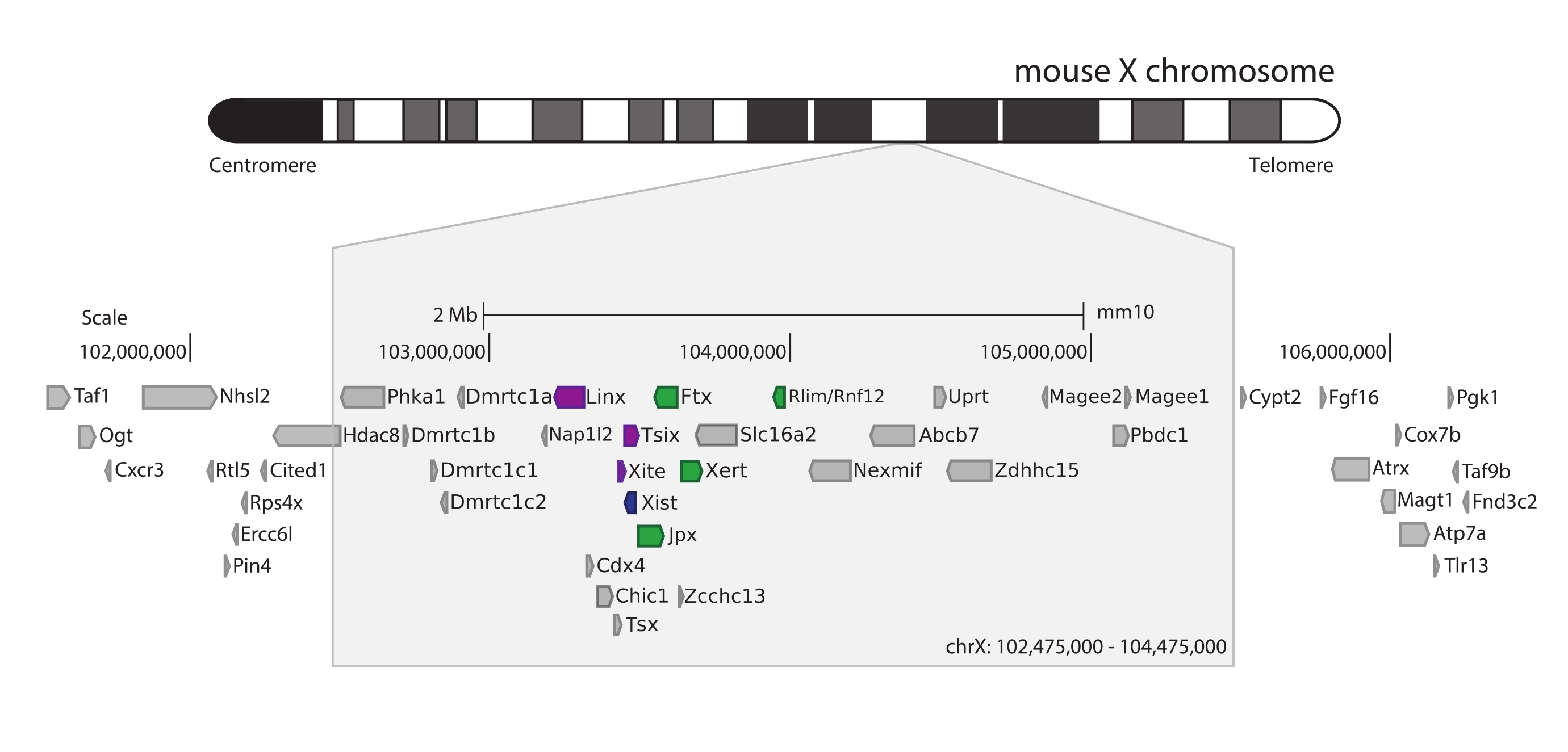
マウスXicを捕捉するために、3MbのX染色体をタイリングする28,913個のRNAプローブのアレイを設計した。この領域には、XCIの主要なプレーヤー、長いノンコーディング遺伝子Xist、およびその既知の~800 kbの調節ランドスケープが含まれます(図5)。この~800 kb領域は、Xistプロモーターとその既知のポジティブレギュレーター(すなわち、ノンコード転写産物Ftx、Jpx、およびXertおよびタンパク質コード遺伝子Rnf12)を含む1つと、Xistのネガティブシス調節因子(すなわち、そのアンチセンス転写産物Tsix、エンハンサー要素Xite、および非コード転写産物Linx)を含む隣接するTADの2つのTADに分割されます(レビュー44、 45)。
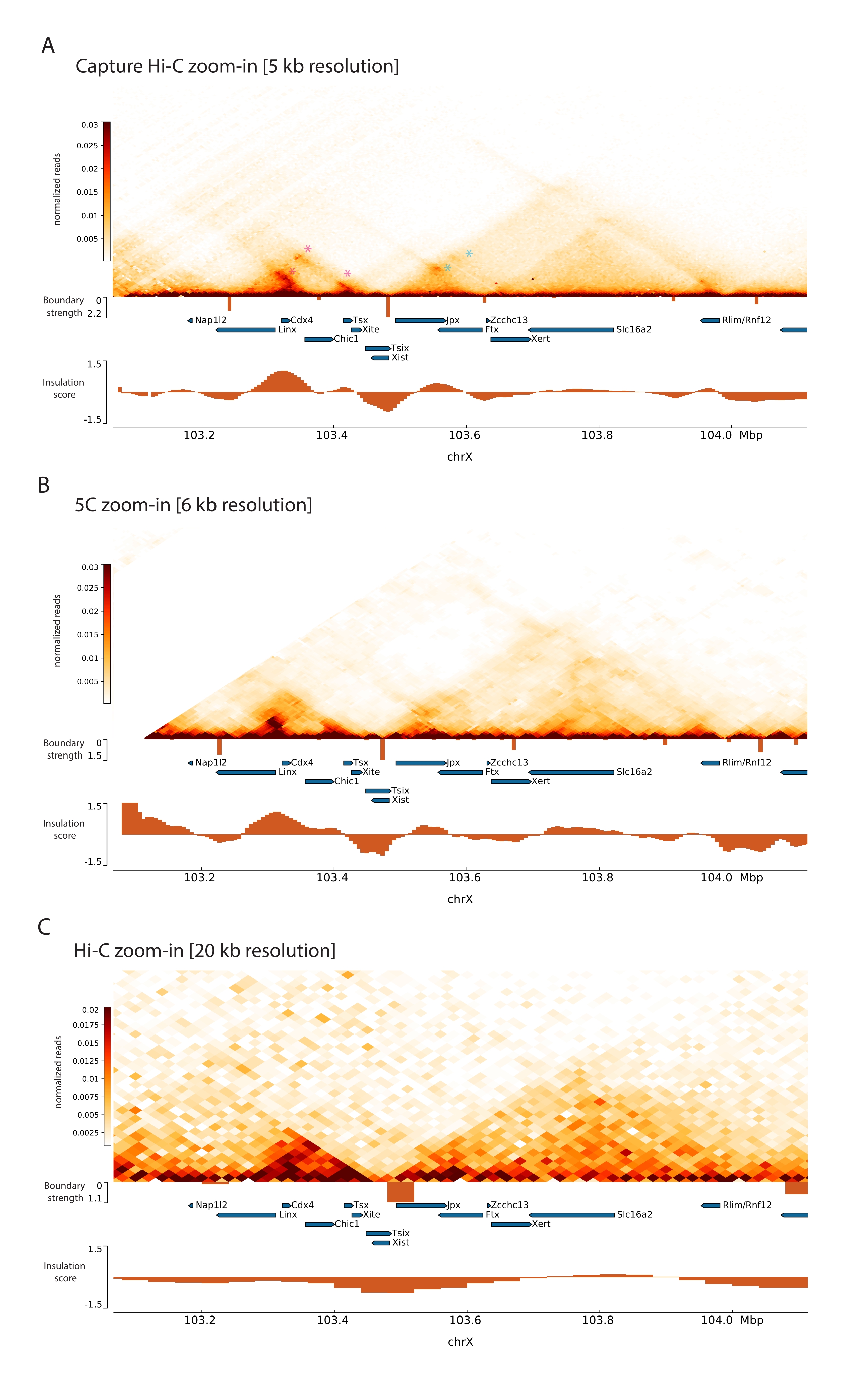
記載されたCapture Hi-CプロトコルをXicに適用することにより、この遺伝子座のトポロジ構成が前例のない分解能で得られました(図6および図7)。これは、キャプチャHi-Cプロファイルを以前に公開された5C47と比較すると特に明確です(図6および図7;別表1)およびHi-C61(図6および図7;別表1)プロファイル。例えば、サブTAD構造はより明白であり、Xistプロモーター(Xist-TAD)を含むTADは明らかに2つの小さなドメインに細分されています(図6A、青い矢印)。以前は、これは5Cプロファイルから視覚的に「推測」することしかできませんでした(図6B)が、絶縁スコアアルゴリズムを使用してこの領域の境界を検出していました。同様に、キャプチャHi-Cプロファイルの解像度により、Tsix遺伝子座(Tsix-TAD)のプロモーターを含む隣接するTAD内の2つの小さなドメイン(図6A、B)を同定できます。これは、以前は5Cでは達成されていませんでした(図6B)。注目すべきは、Capture Hi-Cおよび5Cデータからの絶縁スコアによって決定されるトポロジカル境界は、一般に、わずかに異なる場所で、異なる相対強度で検出されることです。
さらに、コンタクトループなどの他のサブTAD構造は、以前にCapture-C63で特定されたXistとFtxの間のループ(図7A)や、Capture Hi-C48の同様のプロトコルを使用して最近特定されたXistとXertの間のループ(図7B)など、キャプチャHi-Cデータからはっきりと確認できます。.Linx、Chic1、およびXite遺伝子座間のTsix-TAD内の既知の接触ホットスポットを形成するものなど、キャプチャHi-Cプロファイルの解像度が向上するため、他の接触もより正確にマッピングできます(図7A)。
図7に示すHi-Cデータと比較すると、Capture Hi-Cは分解能を4倍に向上させることができましたが、シーケンシング深度の4分の1しか必要としませんでした(つまり、126 Mの読み取り対571 M)(補足表1)。この分解能の向上により、図6と図7に示すシーケンシング深度ではHi-Cでは検出できなかったサブTADとループ相互作用の検出が可能になります。したがって、Capture Hi-C用に記述されたプロトコルは、以前のアプローチと比較して、関心のある大きなゲノム領域のはるかに詳細で高解像度の特性評価を可能にします。

図1:プローブの設計。プローブ設計に使用される戦略の概略図。 3 Mbターゲット領域にわたる各DpnII制限部位の上流および下流300 bpの領域を選択し、重なり合うビオチン化RNAプローブでタイリングしました。これらの選択された領域の1つ、chrX:102,474,805-102,475,500が表示されます。各プローブでは、40塩基以下の反復配列が許可されます。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図2:Hi-Cプレシーケンシングの品質管理をキャプチャ します。 (A)3Cテンプレートの品質管理の代表例。200 ngのDNAを1%アガロースゲルにロードしました。レーン1:1 kbのはしご。レーン2:未消化、架橋、無傷のクロマチンは、>10kbのシャープバンドとして動作します。レーン3:DpnII消化架橋クロマチンは、サイズが1 kbから3 kbの間の塗抹標本として実行されます。レーン4:最終的な3Cライブラリまたはテンプレート。消化された架橋DNA断片の自由末端は再ライゲーションされる。低分子サイズのDNA塗抹標本はほとんど検出できず、ライゲーション産物は>10kbのバンドとして検出されます。(B)高感度バイオアナライザーDNAプロファイルの代表例。左上:150 bpから700 bpの間のフラグメントサイズの分布を示す3Cライブラリのせん断に成功しました。右上:不十分なせん断3Cライブラリ。せん断されていないDNAは、断片>2 kbの広範な濃縮として検出されます。(C)左下:SPRIビーズを使用して1:1の左側サイズの選択に続いてせん断されたDNAサンプル。~300 bpの断片が濃縮されます。中央下:メーカーのプロトコルに従ったペアエンドアダプターのライゲーション後のプリキャプチャーPCRプロファイル。右下:マルチプレックスシーケンシング用のアダプター、シーケンシング、インデックス作成プライマーを含む最終的なCapture Hi-Cライブラリ。略語:bp =塩基対、FU =任意の蛍光単位。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図3:HiC-Proを使用したHi-Cシーケンシング後の品質管理のキャプチャ 。 (A)シーケンシングペアの一等航海士のリファレンスゲノム上のマッピング率の例。水色の画分は、HiC-Proによって整列され、ライゲーション接合部にまたがるリードを表します。したがって、このメトリックを使用して、実験的なライゲーションステップを検証することができます。(B)シーケンシングメイトがゲノム上で整列すると、一意に整列したリードペアのみが分析のために保持されます。(C) ダングリングエンド、セルフサークル、再ライゲーションなどの無効なペア(赤)は解析から除外されます。有効なペアの割合は、ライゲーションとプルダウン効率の良い指標です。(D)有効なペアは、染色体内/染色体間および短距離/長距離接触にさらに分けることができます。PCRアーティファクトを表す可能性が高い重複したリードペアは、分析から破棄されます。(E)対立遺伝子特異的分析のために、HiC-Proは、各親ゲノム(すなわち、C57BL / 6J x CASTEi / J)の1つまたは2つのメイトによってサポートされる対立遺伝子リードの数を報告します。母方と父方の対立遺伝子に割り当てられたリードの同じ割合が予想されます。(F)最後に、キャプチャ領域と重なる有効なペアのみを選択して、コンタクトマップを作成します。キャプチャとキャプチャのペアはターゲット領域内の接触を表し、キャプチャとレポーターのペアにはターゲット領域とオフターゲット領域の間の相互作用が含まれます。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

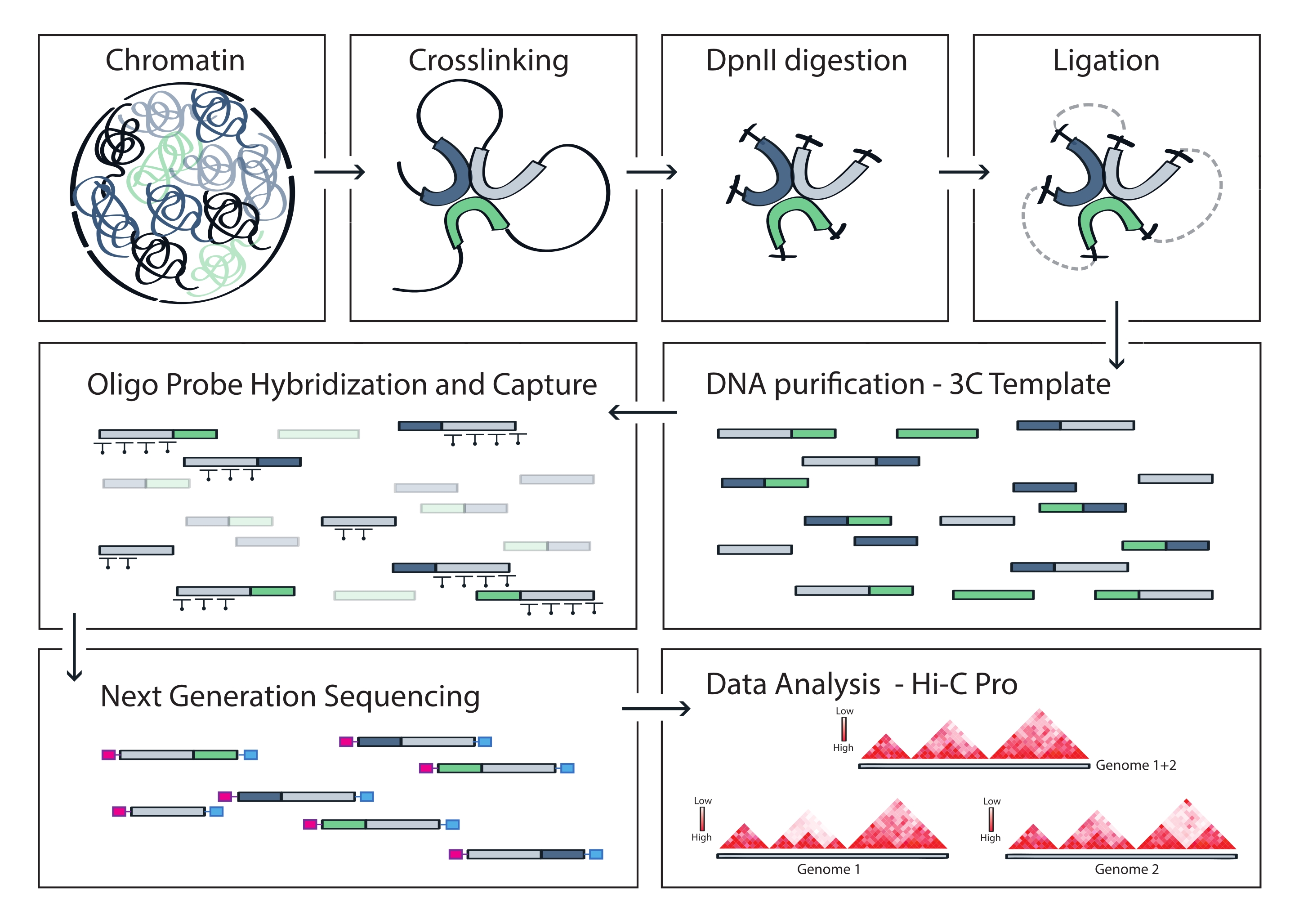
図4:キャプチャHi-Cプロトコルのワークフロー。 さまざまなプロトコルステップの概略図。ゲノムワイドな3Cテンプレートを生成するには、クロマチンを最初にホルムアルデヒドで架橋し、次にDpnII制限酵素で消化します。次に、遊離DNA末端が再ライゲーションされ、架橋が逆転し、DNAが精製されます。標的領域を包含するフラグメントを濃縮するために、ビオチン化RNAプローブのアレイを3Cテンプレートにハイブリダイズし、ストレプトアビジン媒介プルダウンによって捕捉します。キャプチャライブラリはマルチプレックスシーケンシングのために処理され、有効なライゲーションフラグメントが定量化されてターゲット全体のクロマチン接触の頻度が推測され、高解像度の相互作用マップとして視覚化されます。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図5:マウスX染色体上の Xic を包含する領域の概要。 マウスX染色体の模式図および3Mbキャプチャ領域のズームイン(ChrX:102,475,000-105,475,000)。標的領域には、XCIのマスター調節遺伝子座である Xicに対応する~800 kbのDNAが含まれています。Xicには、長いノンコーディング遺伝子、XCIの主要プレーヤーである Xist 、およびその規制環境が含まれます。Xistの正のレギュレーターは緑色で示され、負のレギュレーターは紫色で示されています。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図6:3 Mbでキャプチャされた領域全体でHi-C、5C、およびHi-C相互作用マップをキャプチャします。 (A)マウスXicを含む3 MbターゲットのHi-C相互作用マップを10 kbの解像度でキャプチャします(本研究)。(B)6kbの解像度でのAと同じターゲット領域の5C相互作用マップ(47から再処理されたデータ)。分析に含まれない反復領域は白でマスクされます。5Cデータには、独自のバイオインフォマティクス処理が必要です(47を参照)。クリーニングとアライメントの後、プライマー解像度の5Cマップは、ランニングメディアン(ウィンドウ= 30 kb、ステップ = 5)を使用してビニングされ、最終解像度6 kbに達します。(C)AおよびBと同じゲノム領域のHi-C相互作用マップを40kbの解像度で(64から再処理されたデータ)。すべてのインタラクションマップはマウスESCから生成されました。断熱材スコアは冷却ツールを使用して計算され、TAD境界での断熱材の最小値を含むヒストグラムとして表されます。TAD 境界は、マップの下に垂直線として表示されます。各線の高さは境界強度を示します。遺伝子は転写方向を指す矢印として示されています。キャプチャ Hi-C マップで排他的またはより正確に検出されたサブ TAD 境界は、Tsix および Xist TAD のサブ TAD に対してそれぞれマゼンタと青の矢印で示されます。この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図7:キャプチャされた領域内の1 MbにわたるHi-C、5C、およびHi-C相互作用マップをキャプチャします。 (A)マウスXicを含む1 Mbゲノム領域のHi-C相互作用マップを5 kbの解像度でキャプチャします(本研究)。(B)Aと同じゲノム領域の5C相互作用マップ。6 KBの解像度(47から再処理されたデータ)。分析に含まれない反復領域は白でマスクされます。注目すべきことに、5Cデータは独自のバイオインフォマティクス処理を必要とします(47を参照)。クリーニングとアライメントの後、プライマー解像度の5Cマップは、ランニングメディアン(ウィンドウ= 30 kb、ステップ = 5)を使用してビニングされ、最終解像度6 kbに達します。(C)Hi-CのAおよびBと同じゲノム領域のHi-C相互作用マップを20kbの解像度で(64から再処理されたデータ)。すべてのインタラクションマップはmESCから生成されました。断熱材スコアは冷却ツールを使用して計算され、TAD境界での断熱材の最小値を含むヒストグラムとして表されます。TAD 境界は、マップの下に垂直線として表示されます。各線の高さは境界強度を示します。遺伝子は転写方向を指す矢印として示されています。Capture Hi-C で排他的またはより正確に検出された接触ループは、Tsix および Xist TAD のループに対してそれぞれマゼンタと青のアスタリスクで示されます。この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
補足表1:この原稿で使用されているデータセットのシーケンス後の統計:キャプチャHi-C(この研究)、Hi-C64、および5C47。このファイルをダウンロードするには、ここをクリックしてください。
ディスカッション
ここでは、メガベースサイズのゲノム領域の高次構成を5〜10 kbの解像度で特徴付けるための比較的迅速で簡単なCapture Hi-Cプロトコルについて説明します。Capture Hi-Cは、ゲノムワイドな3CまたはHi-Cテンプレートから標的クロマチン相互作用を濃縮するように設計されたCapture-Cテクノロジーのファミリーに属しています。現在までに、Capture-Cアプリケーションの大部分は、ゲノム全体に散在する比較的小さな調節要素のクロマチン接触をマッピングするために利用されてきました。第1のCapture−Cプロトコルにおいて、赤血球細胞31から調製された3Cライブラリー中の>400個の予め選択されたプロモーターを捕捉するために、複数の重複RNAビオチン化プローブを使用した。その後、次世代(NG)および核滴定(NuTi)キャプチャ-Cでも同じ戦略が改善され、単一の制限部位にまたがる単一の120 bp DNAベイトと2回の連続したキャプチャーを使用して、有益なライゲーションフラグメントの濃縮を最大化することにより、>8,000プロモーターの高解像度相互作用プロファイルを達成しました32,40。.これらの戦略は、マウス胚発生、細胞分化、X染色体の不活性化、および病理学的状態における遺伝子の誤調節を含む、多くの異なる状況でシス作用要素の機能的解剖につながりました46、63、65、66、67、68、69、70、71。
Promoter Capture Hi−C(PCHi−C)において、制限フラグメント34,72のいずれかまたは両端における単一RNA 120-merビオチン化プローブのハイブリダイゼーションによって、制限フラグメントを含む>22,000個のアノテーション付きプロモーターをHi−Cライブラリーからプルダウンした。この方法では、マウス胚性幹細胞、胎児肝細胞、脂肪細胞34,35,72,73だけでなく、ヒトリンパ芽球系、造血前駆細胞、表皮角化細胞、多能性細胞など、急速に増加する細胞型における数千のプロモーターの相互作用の解剖が可能になりました37,74,75,76,77.
これらのターゲット濃縮技術と比較して、Capture Hi-Cはメガベーススケールまでの連続したゲノム領域をターゲットとし、それによって1つ以上のTADにまたがり、遺伝子の調節ランドスケープを網羅します。対象領域全体を、ターゲット内の各DpnII制限部位を包含するビオチン化プローブのアレイでタイリングする必要があります。ビオチン化アレイの3Cテンプレートへのハイブリダイゼーション、それに続くストレプトアビジンベースの捕捉、およびマルチプレックスシーケンシングのための処理は、イルミナペアエンドマルチプレックスシーケンシングのターゲットエンリッチメントシステムを使用して実行されます。プロトコル全体は、3Cライブラリの準備からNGSシーケンシングまで1週間で実行できるため高速であり、わずかな適応やカスタム固有のトラブルシューティングのみが必要です。
このプロトコルは、他の3Cベースの方法と比較して利点も提供します。5〜10 kbの解像度で相互作用マップを取得するために、100〜120 Mのペアエンドリードをシーケンスしました。比較として、ここでは571 MリードのHi-Cデータセットを使用して20 kbの解像度64 (GSM2053973)に到達し、染色体全体のHi-C22で5 kbの解像度に到達するには少なくとも10億リードが必要です。
本研究で用いた捕捉Hi−Cは、6bpカッター制限酵素47 に基づいて以前に公表された5Cよりもはるかに高い分解能に達する(補足表1)。重要なことに、5Cにおける標的相互作用を強化および増幅するために設計された戦略では、クロマチン相互作用の対立遺伝子特異的分析は不可能である。それどころか、Capture Hi−Cデータは対立遺伝子特異的にマッピングすることができ、例えばヒト細胞または遺伝的に異なるマウス系統を交配することによって誘導されたF1ハイブリッド細胞株における相同染色体の対の3D構造ランドスケープの解剖を可能にする78。対立遺伝子特異的なCapture Hi-C相互作用マップを5 kbの分解能で生成するために、SNPカバレッジを高めるために150 bpのペアエンドリードをシーケンスしました。同様の対立遺伝子特異的アプローチをヒト細胞株に適用することができ、SNPのアノテーションが利用可能である22。
重要なことに、Capture Hi-Cは一般的に高分解能を保証しながらシーケンシングコストの手頃な価格を改善しますが、カスタムメイドのビオチン化オリゴヌクレオチドの製造はこの方法の全体的なコストに影響を与えます。したがって、最適な3Cメソッドの選択は、アプリケーションによって異なり、対処されている生物学的問題と必要な解像度、および関心領域のサイズによって異なります。開発された他のCapture Hi-Cプロトコルは、ここで説明するプロトコルと主要な機能を共有しています。例えば、Capture Hi-C戦略を適用して、乳がんおよび結腸直腸がんのリスクに関連する非コード変異にまたがる~50 kbから1 Mbのゲノム領域を特徴付けました。このプロトコルでは、標的領域を3倍のカバレッジでタイリングする120量体RNAベイトをハイブリダイズすることにより、標的領域をHi-Cライブラリからプルダウンしました33、38、79。同様に、HYbrid Capture Hi-C(Hi-C 2)を使用して、最大2 Mb80の関心領域内の相互作用をターゲットにしました。どちらのプロトコルでも、ビオチンプルダウンライゲーションフラグメントに富んだHi-Cテンプレートを使用することで、当社のプロトコルと比較して、総情報リードの割合が増加しました。たとえば、ここで比較64(GSM2053973)に使用したHi-Cデータセットでは、重複を削除した後の有効なペアの割合は、図3と補足表1で説明されているように、Capture Hi-Cで得られた有効なペアの4.8倍です。ただし、ビオチン化ライゲーションフラグメントとハイブリダイズプローブの連続プルダウンにより、プロトコルが大幅に複雑になり、時間がかかると同時に、キャプチャ領域の複雑さが軽減される可能性があります。
タイリングプローブで3Cテンプレートを濃縮するための別の利用可能な方法は、マウス赤血球分化中に高い空間的および時間分解能でクロマチン構造を研究するために適用されたTiled-Cです43。Tiled-Cでは、70 bpのビオチン化プローブのパネルを使用して、2回の連続したキャプチャラウンドで大規模領域内の接触を濃縮し、標的相互作用の非常に高解像度のマップを生成します43,81。また、ダブルキャプチャエンリッチメントにより、キャプチャHi-Cと比較してプロトコルが長く複雑になります。しかし、単一の制限サイトを対象とするCapture-C戦略とは異なり、Tiled-Cでは、2回目のキャプチャはキャプチャ効率を大幅に向上させないように見えるため、おそらく省略できます43。最後に、この研究で使用されたのと同じターゲット濃縮戦略に基づく同様のタイリングアプローチが、先天性奇形の患者に記載され、トランスジェニックマウスで再設計された構造変異を含む調節ランドスケープの解剖に適用されました41,42。この場合、プローブのタイリングアレイは、DpnII制限部位41の近くではなく、ターゲット全体にわたって設計されました。それにもかかわらず、この研究は、異なる状況で大きなゲノム領域の高解像度の特性評価を達成するためのこの戦略の感度と力を強調する上で独創的でした41、42、48。
結論として、ここで説明するプロトコルは、関心のあるゲノム領域の高解像度3D特性評価のための簡単で堅牢かつ強力な戦略を表しています。このアプローチをさまざまなモデルシステム、細胞型、発生的に制御されたクロマチンランドスケープ、および健康および病理学的条件における遺伝子制御に適用することで、エピジェネティクス分野における基本的な未解決の問題の1つであるゲノムトポロジーと遺伝子制御の間の相互作用と因果関係の理解が容易になる可能性があります。さらに、Capture Hi-Cを適用して、GWAS研究によって同定されたリスク変異体の長距離相互作用と高次クロマチンフォールディングをマッピングすることは、さまざまな状況でヒト疾患に関連するノンコーディングゲノム遺伝子座の機能的関連性を明らかにし、それによって潜在的に根底にある病因のプロセスへの新しい洞察を提供する可能性があります。
開示事項
Kai Hauschulzは、Agilent Technologies - Diagnostic and Genomics Groupのフィールドアプリケーションサイエンティストです。他のすべての著者は、競合する利益を宣言しません。
謝辞
ハード研究所での作業は、欧州研究会議の上級研究者賞(XPRESS - AdG671027)によってサポートされました。A.L.は、欧州連合のマリー・スクウォドフスカ・キュリー・アクションズ個人フェローシップ(IF-838408)によってサポートされています。A.H.は、マリー・スクウォドフスカ・キュリー・グラント契約813327の下で、ITN革新的で学際的なネットワーククロムデザインによってサポートされています。著者らは、有益な技術的アドバイスを提供してくれたDaniel Ibrahim(MPI for Molecular Genetics、Berlin)、Institut Curie(パリ)のNGSプラットフォーム、およびEMBLのVladimir BenesとGenomics Core Facility(ハイデルベルク)にサポートと支援を提供してくれたことに感謝している。
資料
| Name | Company | Catalog Number | Comments |
| 10x PBS pH 7.4 | Gibco | 10010-023 | |
| 37% (vol/vol) paraformaldehyde solution | Electron Microscopy Sciences | 15686 | single use glass-vials; do not reuse |
| 50 mL PP conical tube | Falcon | 352070 | |
| Agarose | Sigma | A9539-500g | |
| Bioanalyzer | Agilent | G2939BA | |
| Cell Scrapers - 25 cm Handle and 3.0 cm Blade | Falcon | 353089 | |
| CHIR99021 | Axon Medchem BV | Axon 1386 | |
| cOmplete Mini, Protease inhibitor cocktail (EDTA-free) | Merck | 11836170001 | |
| Countess Cell Counting Chamber Slides | Invitrogen | C10228 | |
| Countess II FL | Invitrogen | ZGEXSCCOUNTESS2FL | Automated cell counter |
| Covaris S2 | Covaris | 500217 | Sonicator |
| DNA LoBind tube, 1.5 mL | Eppendorf | 30108051 | |
| DpnII (50000 units/mL) | New England Biolabs | R0543M | |
| Dulbecco's Modified Eagle Medium (DMEM) | Merck | D6429 | |
| Ethanol (100%) | Merck | 1.00983.2500 | |
| Fetal Bovine Serum (FBS) | Thermo Scientific | 10270106 | |
| gelatine from porcine skin | Sigma | G1890 | |
| GeneRuler 1 kb Plus DNA Ladder | Thermo Scientific | SM0313 | |
| GlycoBlue | Thermo Scientific | AM9516 | Coprecipitant |
| High-Sensitivity Bioanlayzer chips | Agilent | 5067-4626 | |
| Large Cooling Centrifuge 5920 R | Eppendorf | 5948000018 | |
| leukaemia inhibitory factor (LIF) | Merck | ESG1107 | |
| Liquiport | KNF | NF300 | Benchtop aspiration system |
| Low-binding filter tips | Biozym | VT0260U, VT0240, VT0220, VT0200U | |
| Molecular biology grade water | Merck | W3500-6x500ML | |
| Next Seq 500 | Illumina | SY-415-1001 | |
| Next Seq 500 High Output v2 Kit (300 cycles) | Illumina | FC-404-2004 | |
| Nonidet P40 Substitute (NP40) | Merck | 11332473001 | |
| PD0325901 | Axon Medchem BV | Axon 1408 | |
| Protease inhibitor cocktail (EDTA-free) | Merck | 11873580001 | |
| Proteinase K - recombinant, PCR-grade (20 mg/mL) | Thermo Scientific | EO0491 | |
| Qubit 2.0 | Thermo Scientific | Q32871 | |
| Qubit assay tubes | Thermo Scientific | Q32856 | |
| Qubit dsDNA High Sensitivity kit | Thermo Scientific | Q32851 | |
| RNase A (10 mg/mL) | Thermo Scientific | EN0531 | |
| Sodium acetate pH 5.2 (3M) | Merck | S7899 | |
| speed vacuum concentrator | Eppendorf | EP5305000100-1EA | |
| Agencourt AMPureXP | Beckman Coulter | A63881 | SPRI beads |
| SureSelect Target Enrichment Box 1 | Agilent | 5190-8645 | |
| SureSelect Target Enrichment Kit ILM Indexing Hyb Module Box 2 | Agilent | 5190-4455 | |
| SureSelect XT Library Prep Kit ILM | Agilent | 5500-0132 | |
| T4 ligase (30 units/µL) | Thermo Scientific | EL0013 | |
| table-top Centrifuge 5427 R | Eppendorf | 5409000012 | |
| Triton-X-100 (500 mL) | Merck | X100-500ML | |
| Trypan Blue | Invitrogen | T10282 | |
| Trypsine | Thermo Scientific | 25300054 | |
| UltraPure Glycine | Thermo Scientific | 15527013 | |
| β-mercaptoethanol | Thermo Scientific | 31350010 |
参考文献
- Ibrahim, D. M., Mundlos, S. The role of 3D chromatin domains in gene regulation: a multi-facetted view on genome organization. Current Opinion in Genetics & Development. 61, 1-8 (2020).
- Bolt, C. C., Duboule, D. The regulatory landscapes of developmental genes. Development. 147 (3), (2020).
- Glaser, J., Mundlos, S. 3D or not 3D: Shaping the genome during development. Cold Spring Harbor Perspectives in Biology. 14 (5), 040188(2021).
- Denker, A., De Laat, W. The second decade of 3C technologies: detailed insights into nuclear organization. Genes & Development. 30 (12), 1357-1382 (2016).
- Kempfer, R., Pombo, A. Methods for mapping 3D chromosome architecture. Nature Reviews Genetics. 21 (4), 207-226 (2020).
- McCord, R. P., Kaplan, N., Giorgetti, L. Chromosome conformation capture and beyond: Toward an integrative view of chromosome structure and function. Molecular Cell. 77 (4), 688-708 (2020).
- Jerkovic, I., Cavalli, G. Understanding 3D genome organization by multidisciplinary methods. Nature ReviewsMolecular Cell Biology. 22 (8), 511-528 (2021).
- Hsieh, T. -H. S., et al. Mapping nucleosome resolution chromosome folding in yeast by Micro-C. Cell. 162 (1), 108-119 (2015).
- Krietenstein, N., et al. Ultrastructural details of mammalian chromosome architecture. Molecular Cell. 78 (3), 554-565 (2020).
- Dekker, J., Rippe, K., Dekker, M., Kleckner, N. Capturing chromosome conformation. Science. 295 (5558), 1306-1311 (2002).
- Naumova, N., Smith, E. M., Zhan, Y., Dekker, J. Analysis of long-range chromatin interactions using Chromosome Conformation Capture. Methods. 58 (3), 192-203 (2012).
- Simonis, M., et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nature Genetics. 38 (11), 1348-1354 (2006).
- Zhao, Z., et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra-and interchromosomal interactions. Nature Genetics. 38 (11), 1341-1347 (2006).
- Würtele, H., Chartrand, P. Genome-wide scanning of HoxB1-associated loci in mouse ES cells using an open-ended Chromosome Conformation Capture methodology. Chromosome Research. 14 (5), 477-495 (2006).
- De Wit, E., De Laat, W. A decade of 3C technologies: insights into nuclear organization. Genes & Development. 26 (1), 11-24 (2012).
- Dostie, J., et al. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Research. 16 (10), 1299-1309 (2006).
- Splinter, E., et al. The inactive X chromosome adopts a unique three-dimensional conformation that is dependent on Xist RNA. Genes & Development. 25 (13), 1371-1383 (2011).
- Ferraiuolo, M. A., Sanyal, A., Naumova, N., Dekker, J., Dostie, J. From cells to chromatin: capturing snapshots of genome organization with 5C technology. Methods. 58 (3), 255-267 (2012).
- Kim, J. H., et al. 5C-ID: Increased resolution Chromosome-Conformation-Capture-Carbon-Copy with in situ 3C and double alternating primer design. Methods. 142, 39-46 (2018).
- Lieberman-Aiden, E., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326 (5950), 289-293 (2009).
- Zhang, Y., et al. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell. 148 (5), 908-921 (2012).
- Rao, S. S. P., et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 159 (7), 1665-1680 (2014).
- Dixon, J. R., et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 485 (7398), 376-380 (2012).
- Nora, E. P., et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 485 (7398), 381-385 (2012).
- Krefting, J., Andrade-Navarro, M. A., Ibn-Salem, J. Evolutionary stability of topologically associating domains is associated with conserved gene regulation. BMC Biology. 16 (1), 87(2018).
- Galupa, R., Heard, E. Topologically associating domains in chromosome architecture and gene regulatory landscapes during development, disease, and evolution. Cold Spring Harbor Symposia on Quantitative Biology. 82, 267-278 (2017).
- Tena, J. J., Santos-Pereira, J. M. Topologically associating domains and regulatory landscapes in development, evolution and disease. Frontiers in Cell and Developmental Biology. 9, 702787(2021).
- Lupiáñez, D. G., Spielmann, M., Mundlos, S. Breaking TADs: How alterations of chromatin domains result in disease. Trends in Genetics. 32 (4), 225-237 (2016).
- Davidson, I. F., Peters, J. -M. Genome folding through loop extrusion by SMC complexes. Nature Reviews Molecular Cell Biology. 22 (7), 445-464 (2021).
- Schmitt, A. D., Hu, M., Ren, B. Genome-wide mapping and analysis of chromosome architecture. Nature Reviews Molecular Cell Biology. 17 (12), 743-755 (2016).
- Hughes, J. R., et al. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nature Genetics. 46 (2), 205-212 (2014).
- Davies, J. O. J., et al. Multiplexed analysis of chromosome conformation at vastly improved sensitivity. Nature Methods. 13 (1), 74-80 (2016).
- Jäger, R., et al. Capture Hi-C identifies the chromatin interactome of colorectal cancer risk loci. Nature Communications. 6, 6178(2015).
- Schoenfelder, S., et al. The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Research. 25 (4), 582-597 (2015).
- Sahlén, P., et al. Genome-wide mapping of promoter-anchored interactions with close to single-enhancer resolution. Genome Biology. 16, 156(2015).
- Joshi, O., et al. Dynamic reorganization of extremely long-range promoter-promoter interactions between two states of pluripotency. Cell Stem Cell. 17 (6), 748-757 (2015).
- Mifsud, B., et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nature Genetics. 47 (6), 598-606 (2015).
- Dryden, N. H., et al. Unbiased analysis of potential targets of breast cancer susceptibility loci by Capture Hi-C. Genome Research. 24 (11), 1854-1868 (2014).
- Oudelaar, A. M., Davies, J. O. J., Downes, D. J., Higgs, D. R., Hughes, J. R. Robust detection of chromosomal interactions from small numbers of cells using low-input Capture-C. Nucleic Acids Research. 45 (22), 184(2017).
- Oudelaar, A. M., et al. Single-allele chromatin interactions identify regulatory hubs in dynamic compartmentalized domains. Nature Genetics. 50 (12), 1744-1751 (2018).
- Franke, M., et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature. 538 (7624), 265-269 (2016).
- Despang, A., et al. Functional dissection of the Sox9-Kcnj2 locus identifies nonessential and instructive roles of TAD architecture. Nature Genetics. 51 (8), 1263-1271 (2019).
- Oudelaar, A. M., et al. Dynamics of the 4D genome during in vivo lineage specification and differentiation. Nature Communications. 11 (1), 1-12 (2020).
- Galupa, R., Heard, E. X-chromosome inactivation: A crossroads between chromosome architecture and gene regulation. Annual Review of Genetics. 52, 535-566 (2018).
- Loda, A., Collombet, S., Heard, E. Gene regulation in time and space during X-chromosome inactivation. Nature Reviews. Molecular Cell Biology. 23 (4), 231-249 (2022).
- van Bemmel, J. G., et al. The bipartite TAD organization of the X-inactivation center ensures opposing developmental regulation of Tsix and Xist. Nature Genetics. 51 (6), 1024-1034 (2019).
- Galupa, R., et al. A conserved noncoding locus regulates random monoallelic Xist expression across a topological boundary. Molecular Cell. 77 (2), 352-367 (2020).
- Gjaltema, R. A. F., et al. Distal and proximal cis-regulatory elements sense X chromosome dosage and developmental state at the Xist locus. Molecular Cell. 82 (1), 190-208 (2022).
- Galupa, R., et al. Inversion of a topological domain leads to restricted changes in its gene expression and affects inter-domain communication. Development. 149 (9), (2022).
- Savarese, F., Flahndorfer, K., Jaenisch, R., Busslinger, M., Wutz, A. Hematopoietic precursor cells transiently reestablish permissiveness for X inactivation. Molecular and Cellular Biology. 26 (19), 7167-7177 (2006).
- Schulz, E. G., et al. The two active X chromosomes in female ESCs block exit from the pluripotent state by modulating the ESC signaling network. Cell Stem Cell. 14 (2), 203-216 (2014).
- Gnirke, A., et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nature Biotechnology. 27 (2), 182-189 (2009).
- Akgol Oksuz, B., et al. Systematic evaluation of chromosome conformation capture assays. Nature Methods. 18 (9), 1046-1055 (2021).
- Piccinini, F., Tesei, A., Arienti, C., Bevilacqua, A. Cell counting and viability assessment of 2D and 3D Cell cultures: Expected reliability of the trypan blue assay. Biological Procedures Online. 19 (1), 8(2017).
- Servant, N., et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biology. 16, 259(2015).
- Abdennur, N., Mirny, L. A. Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics. 36 (1), 311-316 (2020).
- Imakaev, M., et al. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nature Methods. 9 (10), 999-1003 (2012).
- Forcato, M., et al. Comparison of computational methods for Hi-C data analysis. Nature Methods. 14 (7), 679-685 (2017).
- Venev, S., et al. open2c/cooltools: v0.4.1. , (2021).
- Wages, J. M. NUCLEIC ACIDS | Immunoassays. Encyclopedia of Analytical Science. , Elsevier. 408-417 (2005).
- Ewels, P. A., et al. The nf-core framework for community-curated bioinformatics pipelines. Nature Biotechnology. 38 (3), 276-278 (2020).
- Servant, N., Peltzer, A. nf-core/hic: Initial release of nf-core/hic. Zenodo. , (2019).
- Furlan, G., et al. The Ftx noncoding locus controls X chromosome inactivation independently of its RNA products. Molecular Cell. 70 (3), 462-472 (2018).
- Giorgetti, L., et al. Structural organization of the inactive X chromosome in the mouse. Nature. 535 (7613), 575-579 (2016).
- Simon, C. S., et al. Functional characterisation of cis-regulatory elements governing dynamic Eomes expression in the early mouse embryo. Development. 144 (7), 1249-1260 (2017).
- Williams, R. M., et al. Reconstruction of the global neural crest gene regulatory network in vivo. Developmental Cell. 51 (2), 255-276 (2019).
- Godfrey, L., et al. DOT1L inhibition reveals a distinct subset of enhancers dependent on H3K79 methylation. Nature Communications. 10 (1), 2803(2019).
- Hanssen, L. L. P., et al. Tissue-specific CTCF-cohesin-mediated chromatin architecture delimits enhancer interactions and function in vivo. Nature Cell Biology. 19 (8), 952-961 (2017).
- Larke, M. S. C., et al. Enhancers predominantly regulate gene expression during differentiation via transcription initiation. Molecular Cell. 81 (5), 983-997 (2021).
- Oudelaar, A. M., et al. A revised model for promoter competition based on multi-way chromatin interactions at the α-globin locus. Nature Communications. 10 (1), 5412(2019).
- Long, H. K., et al. Loss of extreme long-range enhancers in human neural crest drives a craniofacial disorder. Cell Stem Cell. 27 (5), 765-783 (2020).
- Schoenfelder, S., Javierre, B. -M., Furlan-Magaril, M., Wingett, S. W., Fraser, P. Promoter Capture Hi-C: High-resolution, genome-wide profiling of promoter interactions. Journal of Visualized Experiments. (136), e57320(2018).
- Siersbæk, R., et al. Dynamic rewiring of promoter-anchored chromatin loops during adipocyte differentiation. Molecular Cell. 66 (3), 420-435 (2017).
- Rubin, A. J., et al. Lineage-specific dynamic and pre-established enhancer-promoter contacts cooperate in terminal differentiation. Nature Genetics. 49 (10), 1522-1528 (2017).
- Freire-Pritchett, P., et al. Global reorganisation of cis-regulatory units upon lineage commitment of human embryonic stem cells. eLife. 6, 21926(2017).
- Javierre, B. M., et al. Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell. 167 (5), 1369-1384 (2016).
- Miguel-Escalada, I., et al. Human pancreatic islet three-dimensional chromatin architecture provides insights into the genetics of type 2 diabetes. Nature Genetics. 51 (7), 1137-1148 (2019).
- Keane, T. M., et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 477 (7364), 289-294 (2011).
- Baxter, J. S., et al. Capture Hi-C identifies putative target genes at 33 breast cancer risk loci. Nature Communications. 9 (1), 1028(2018).
- Sanborn, A. L., et al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proceedings of the National Academy of Sciences. 112 (47), 6456-6465 (2015).
- Owens, D. D. G., et al. Dynamic Runx1 chromatin boundaries affect gene expression in hematopoietic development. Nature Communications. 13 (1), 773(2022).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved